

Design, Synthesis, and Biological Evaluation of Novel Tetrahydroacridin Hybrids with Sulfur-Inserted Linkers as Potential Multitarget Agents for Alzheimer’s Disease

 and
and
Abstract
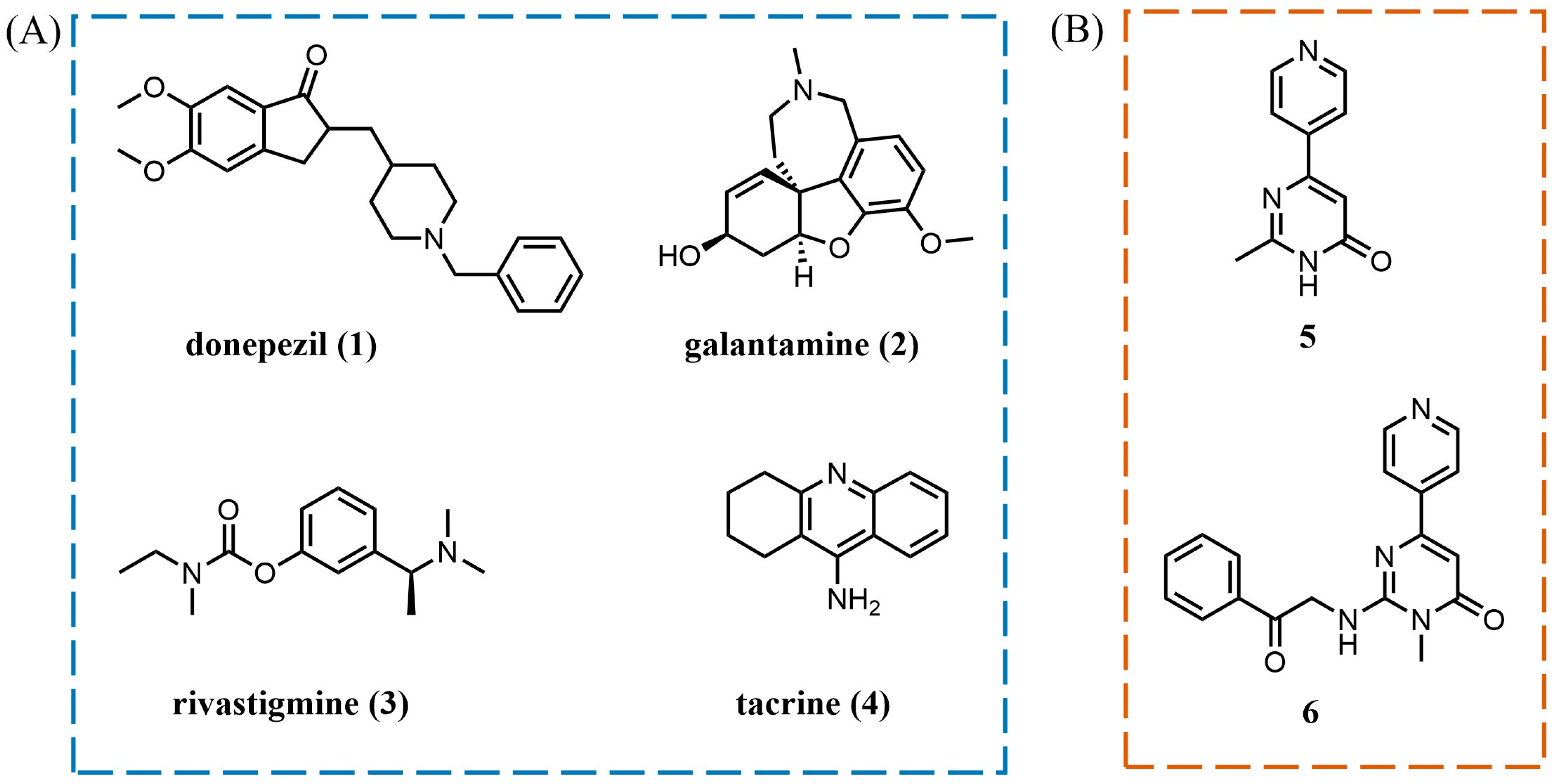
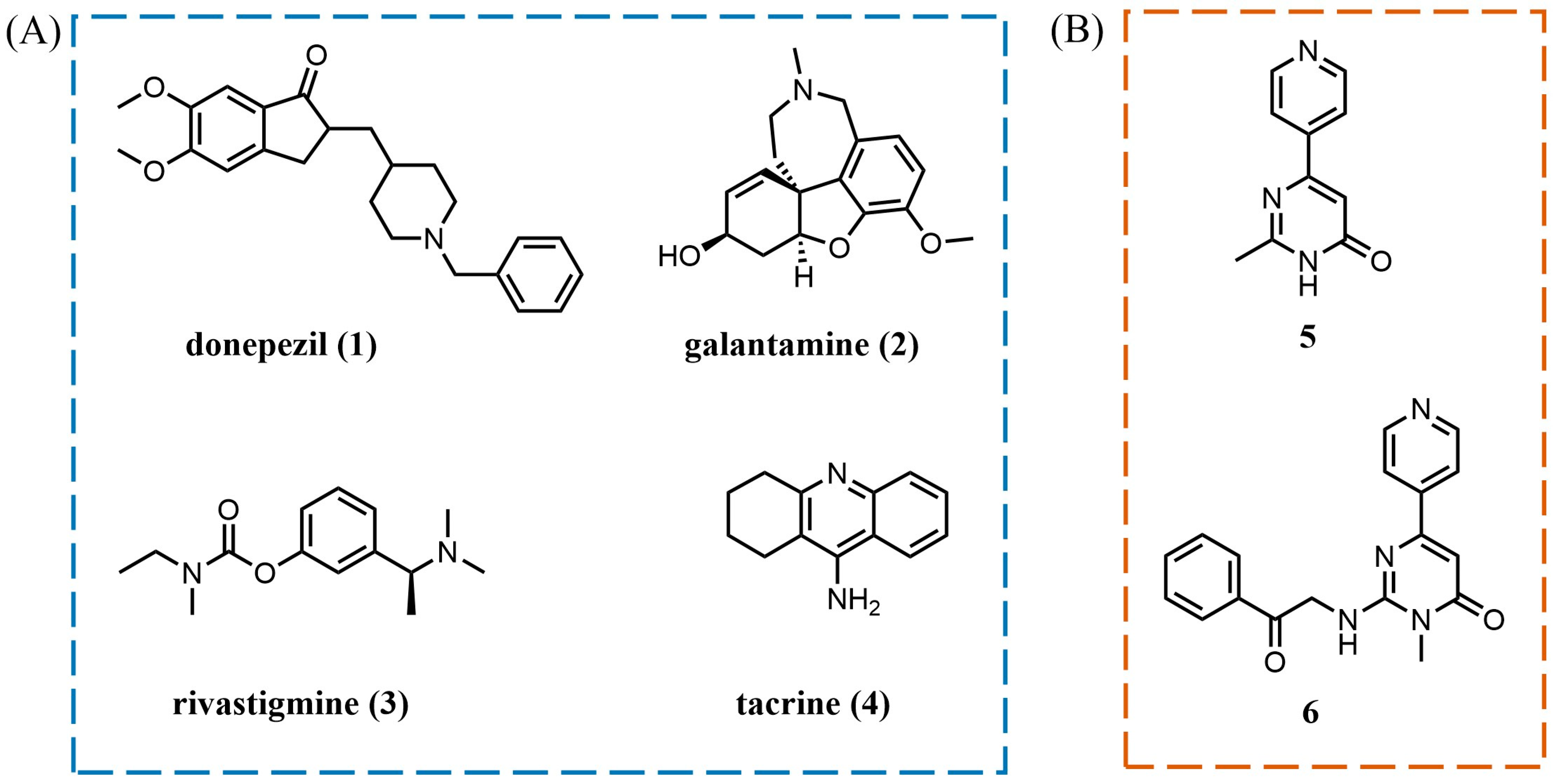
:1. Introduction
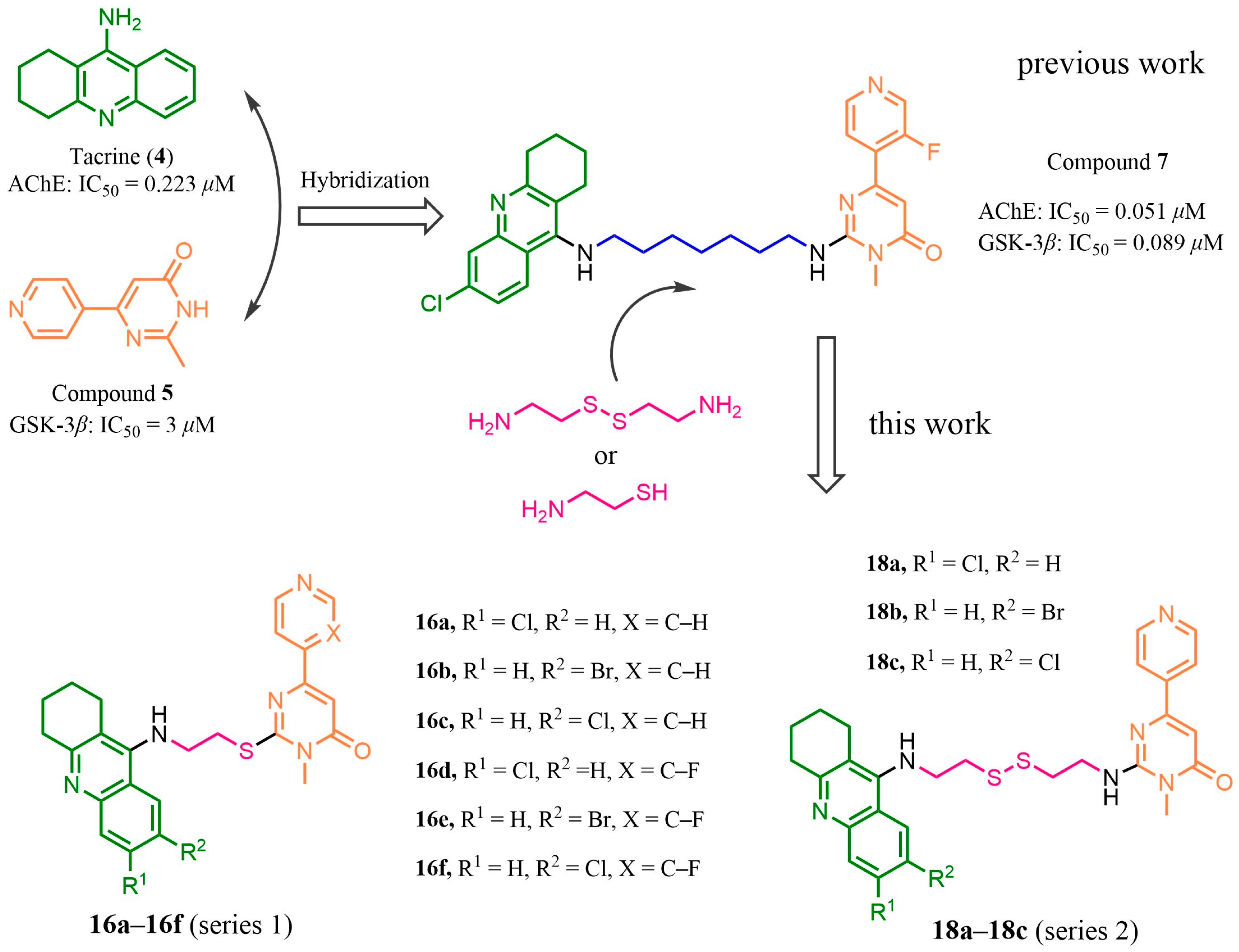
2. Results and Discussion
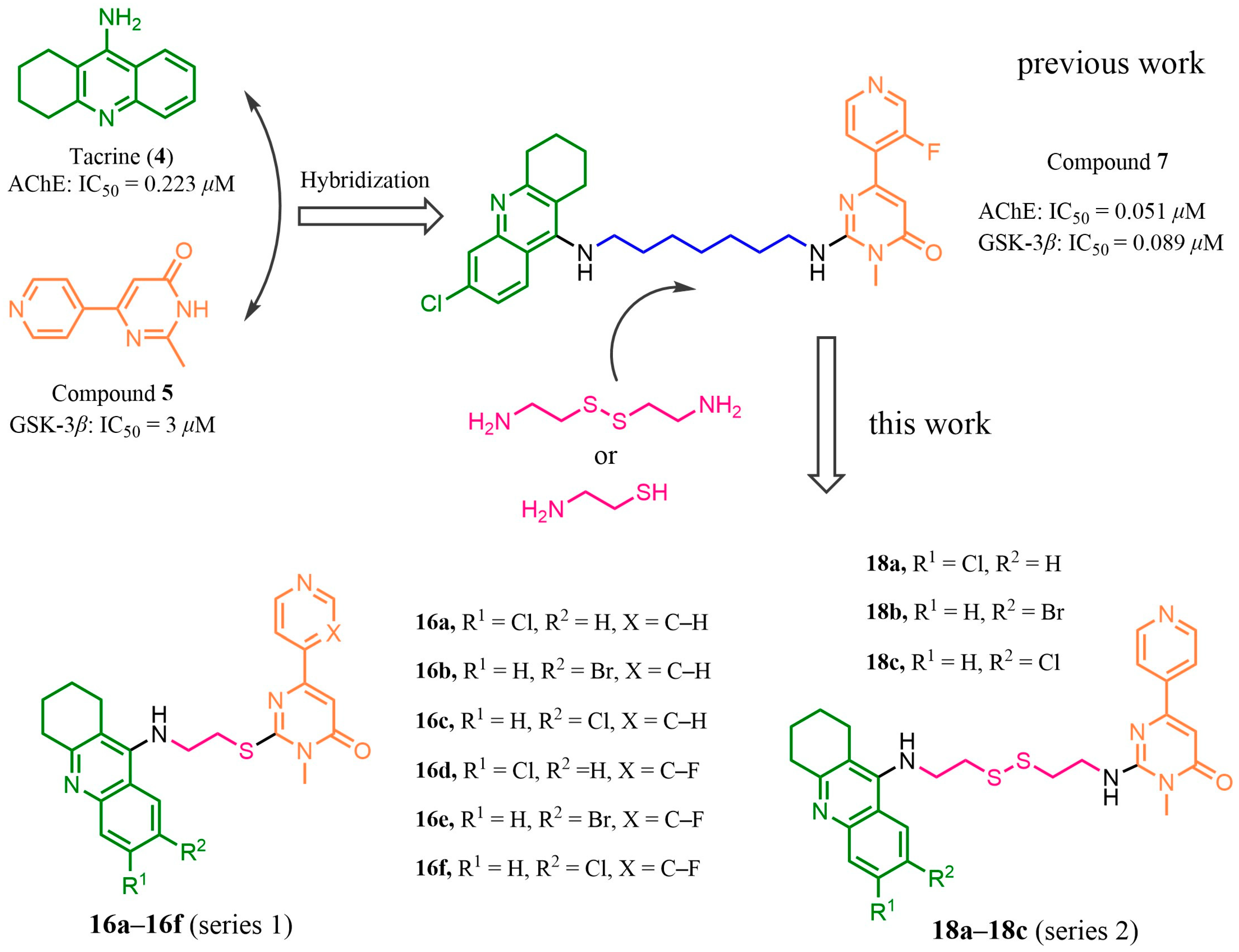
2.1. Design
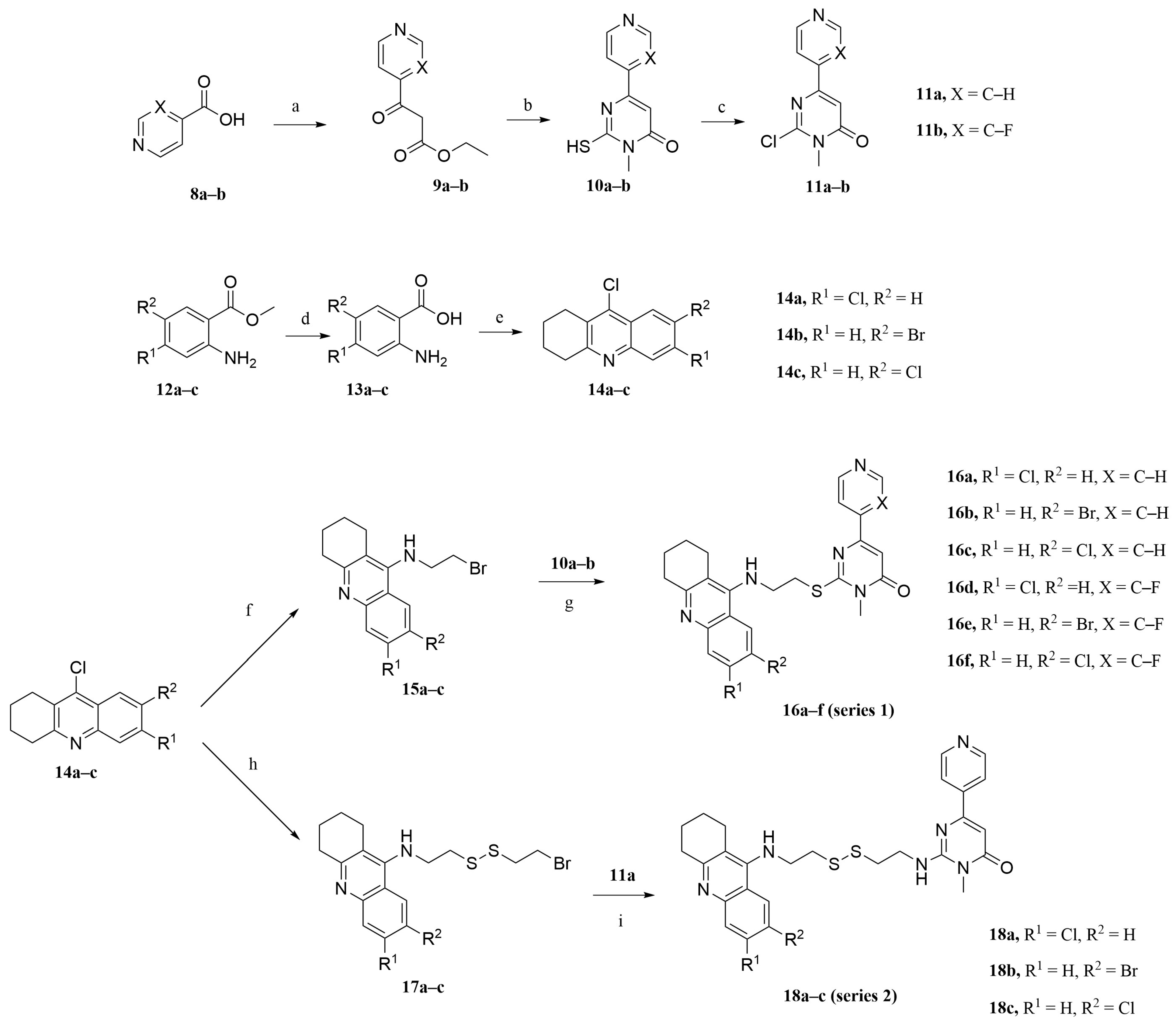
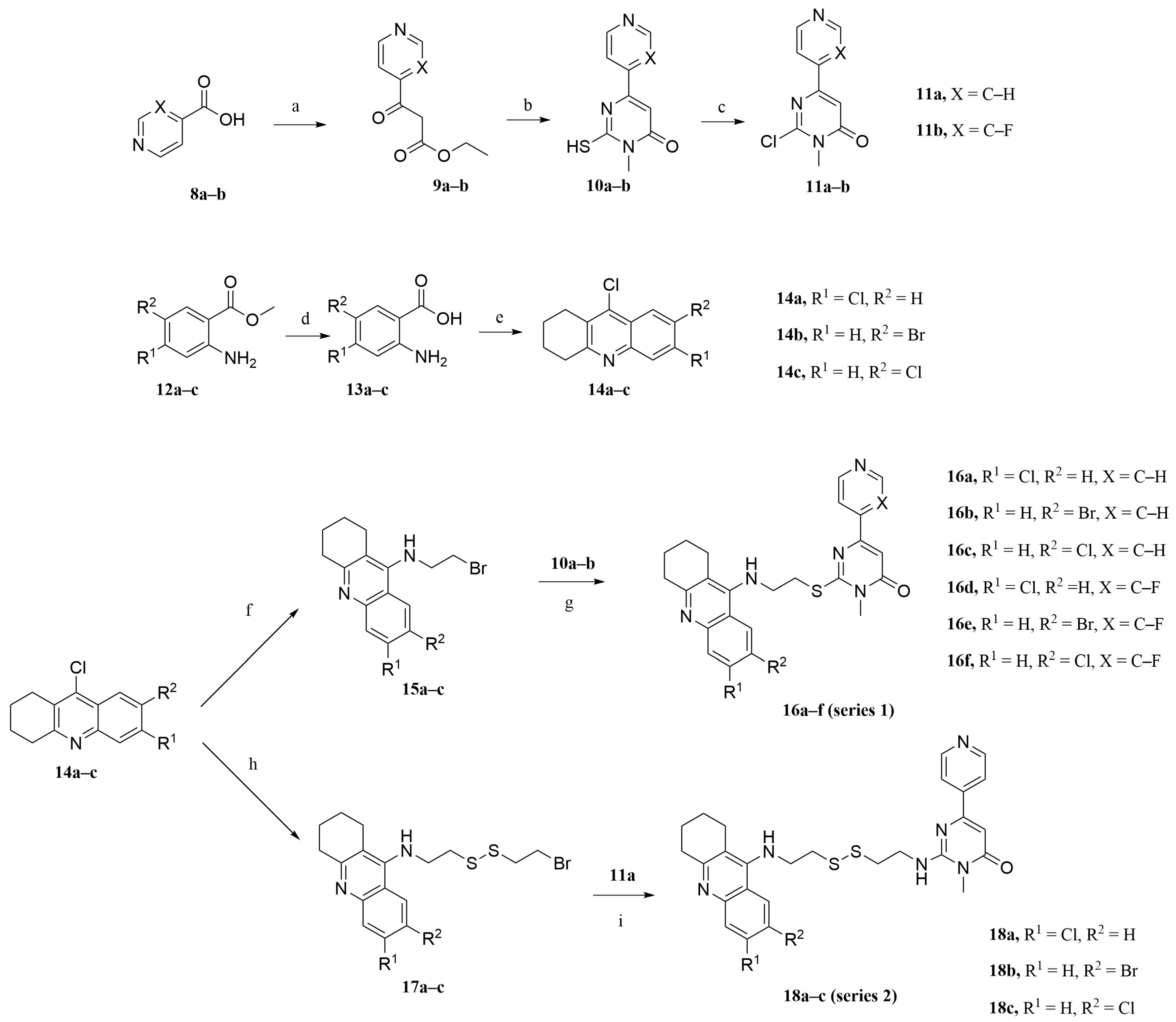
2.2. Synthesis
2.3. In Vitro Inhibitory Activities against AChE and GSK-3β
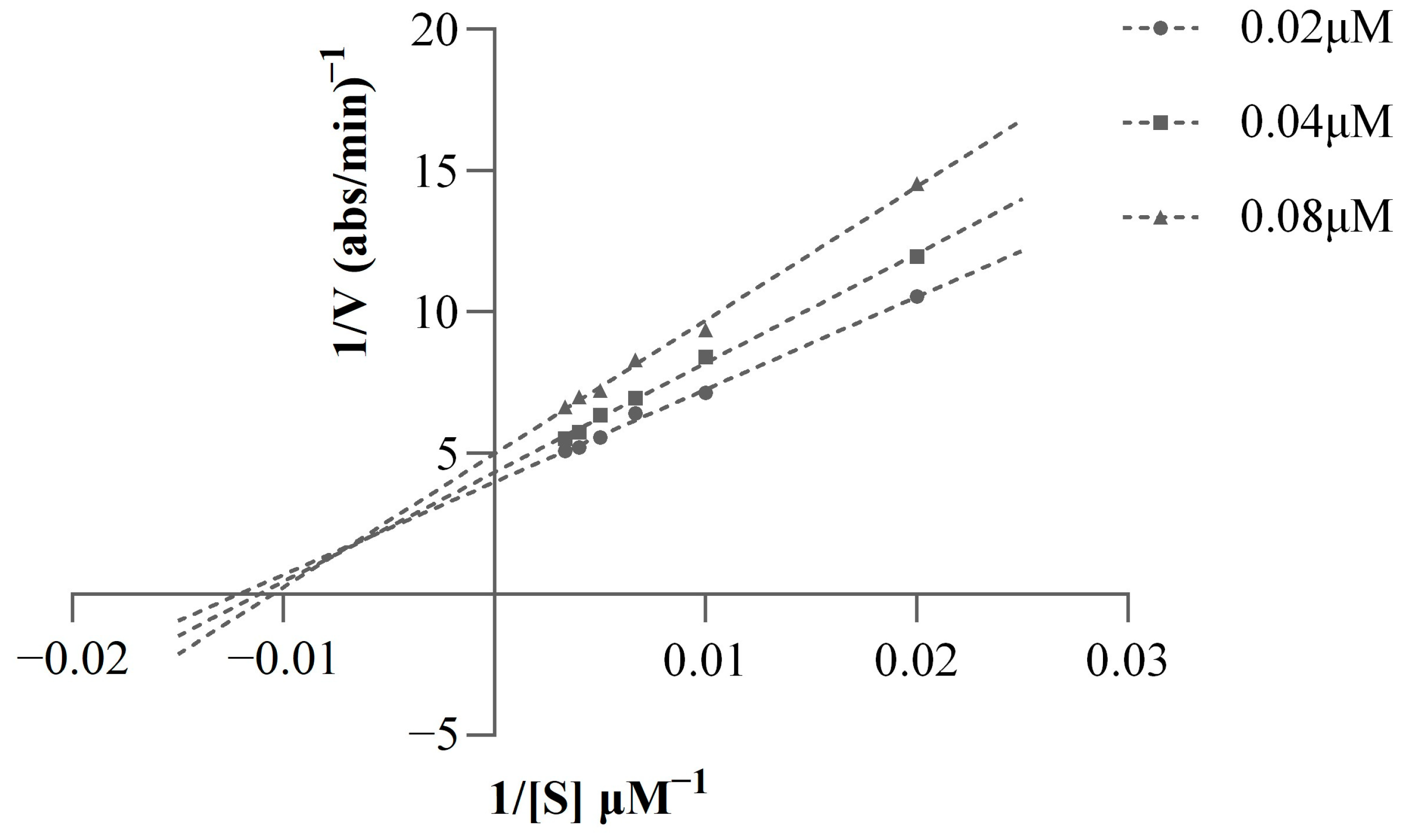
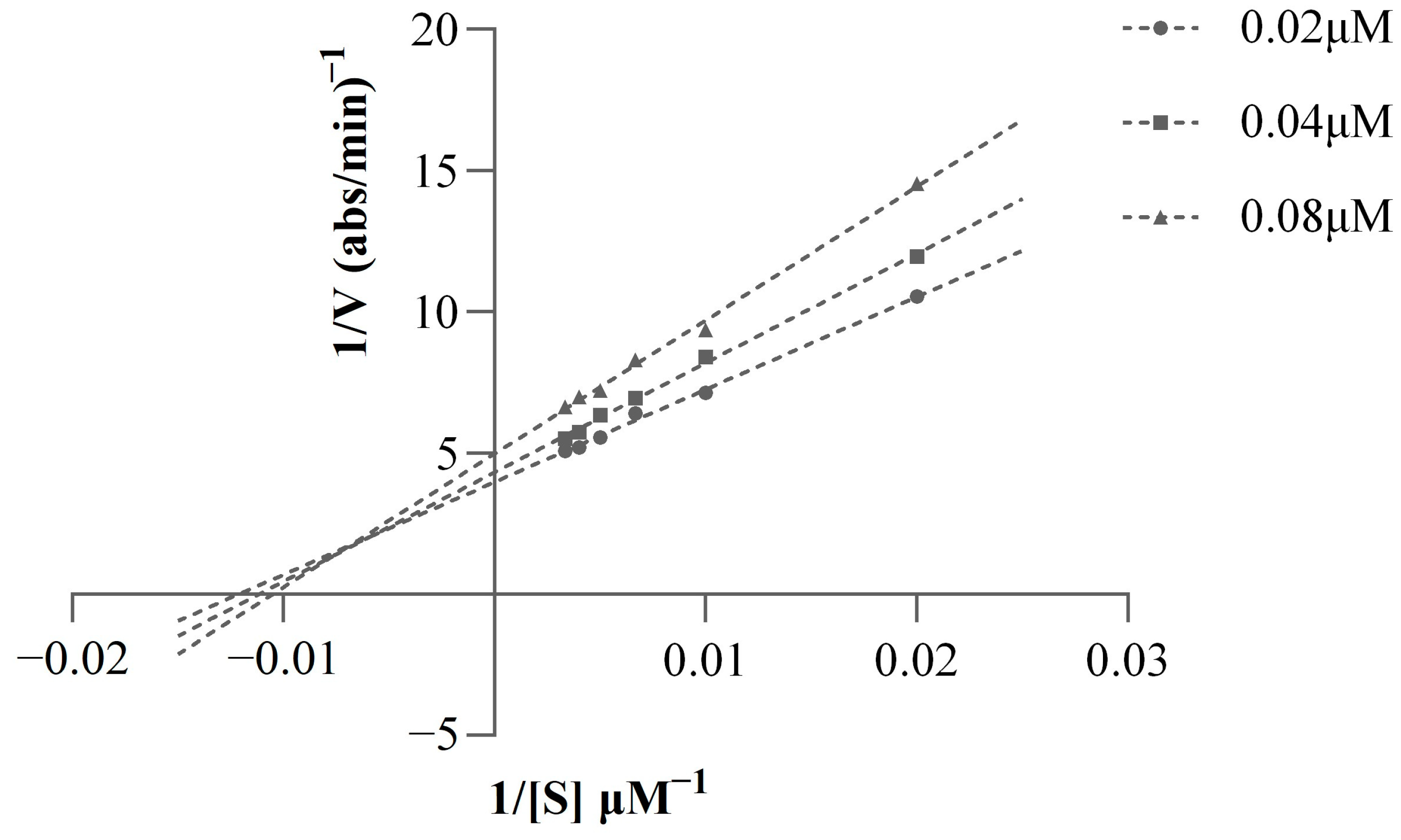
2.4. Kinetic Study of AChE Inhibition
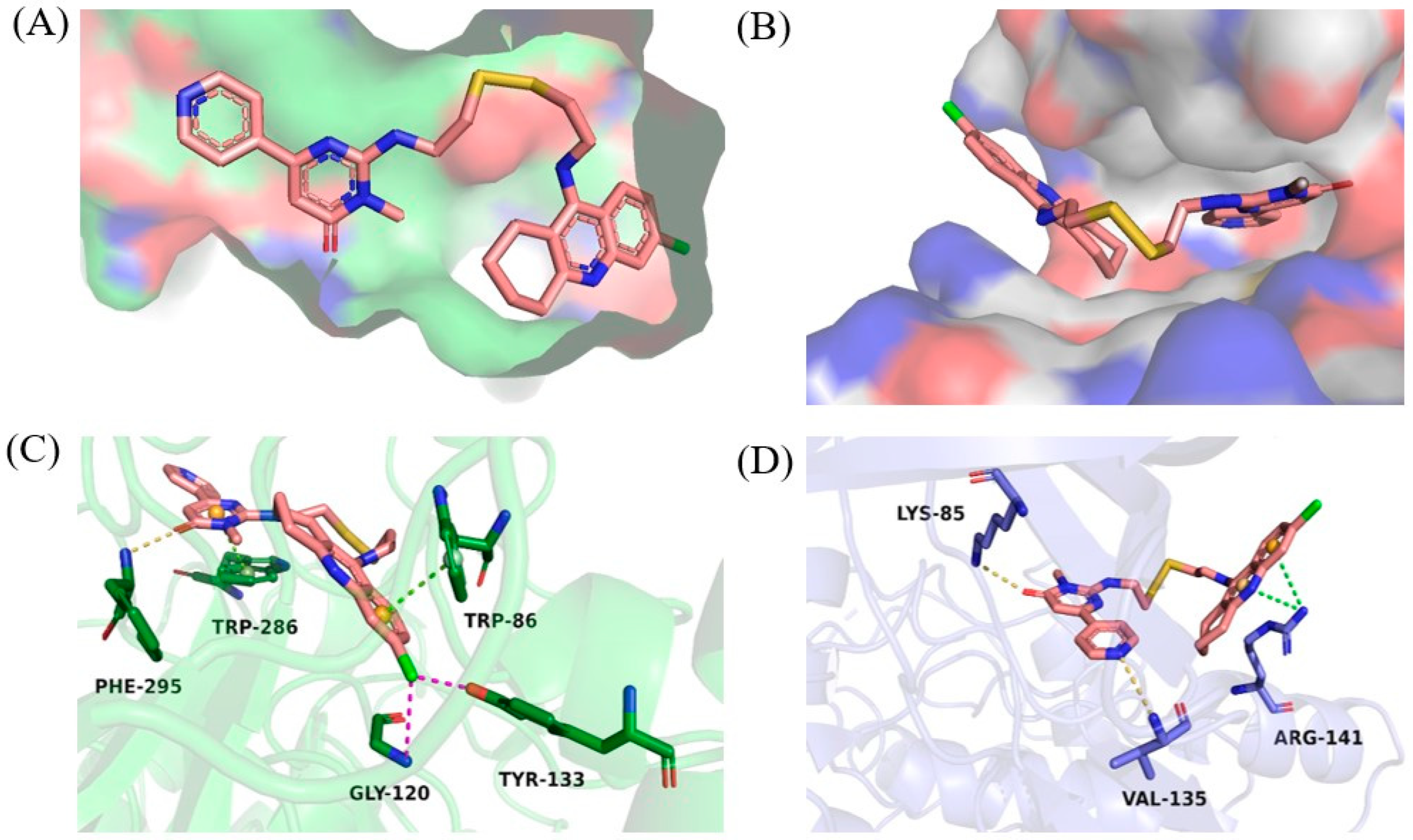
2.5. Molecular Modeling Studies
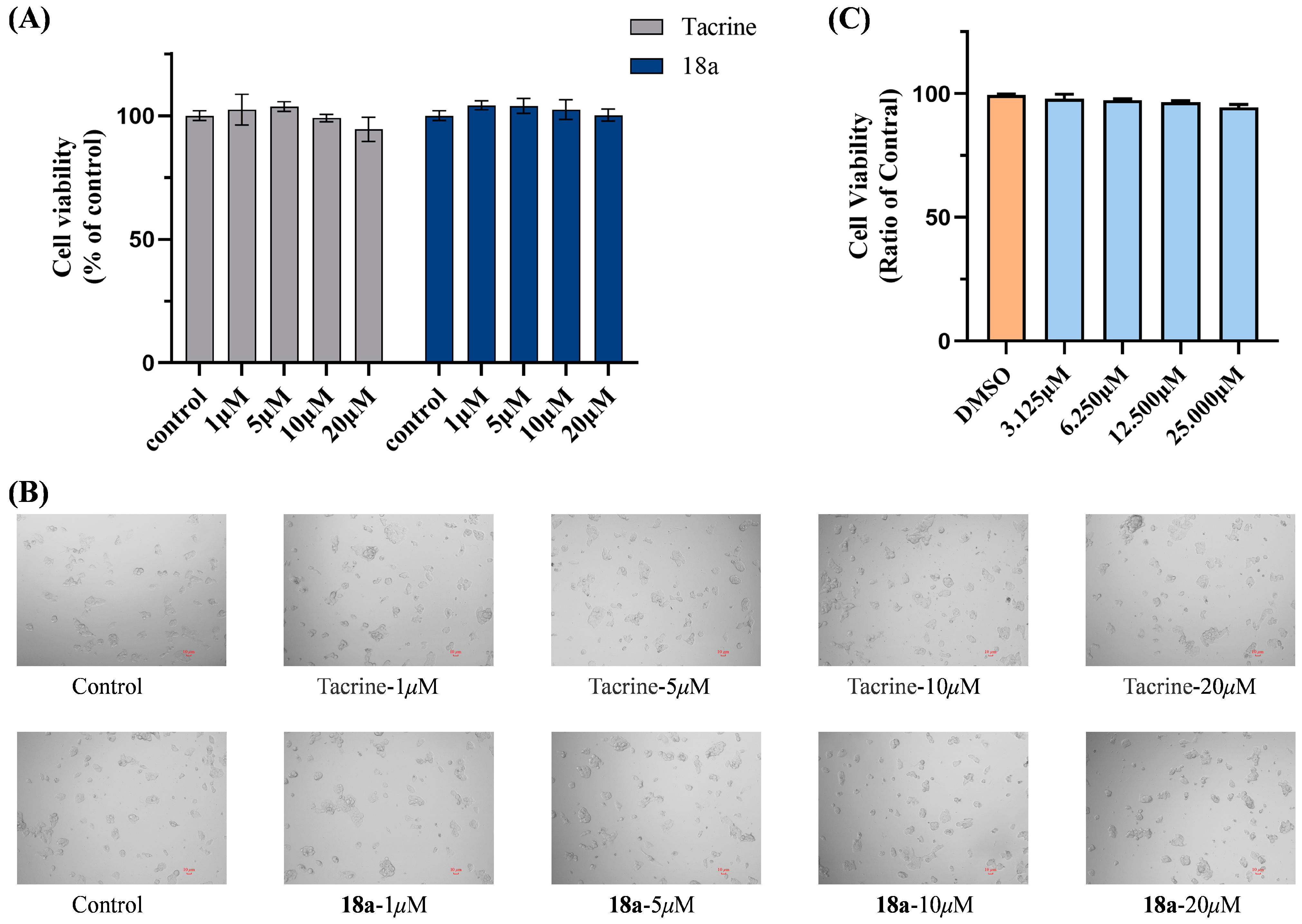
2.6. Cytotoxicity Bioassays in HepG2 and SH-SY5Y Cell Lines
2.7. Theoretical Prediction of the ADME Properties
3. Materials and Methods
3.1. Chemistry
3.2. Synthetic Procedure
3.2.1. Preparation of Intermediates 9a–9b, 10a–10b, and 11a–11b
3.2.2. Preparation of Intermediates 13a–13c and 14a–14c
3.2.3. Preparation of Intermediates 15a–15c
3.2.4. General Procedures for the Synthesis of Target Compounds 16a–16f
3.2.5. Preparation of Intermediates 17a–17c
3.2.6. General Procedures for the Synthesis of Target Compounds 18a–18c
3.3. Characterization Details
3.3.1. 2-((2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (16a)
3.3.2. 2-((2-((7-Bromo-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (16b)
3.3.3. 2-((2-((7-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (16c)
3.3.4. 2-((2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-6-(3-fluoropyridin-4-yl)-3-methylpyrimidin-4(3H)-one (16d)
3.3.5. 2-((2-((7-Bromo-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-6-(3-fluoropyridin-4-yl)-3-methylpyrimidin-4(3H)-one (16e)
3.3.6. 2-((2-((7-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)thio)-6-(3-fluoropyridin-4-yl)-3-methylpyrimidin-4(3H)-one (16f)
3.3.7. 2-((2-((2-((6-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)disulfanyl)ethyl)amino)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (18a)
3.3.8. 2-((2-((2-((7-Bromo-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)disulfanyl)ethyl)amino)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (18b)
3.3.9. 2-((2-((2-((7-Chloro-1,2,3,4-tetrahydroacridin-9-yl)amino)ethyl)disulfanyl)ethyl)amino)-3-methyl-6-(pyridin-4-yl)pyrimidin-4(3H)-one (18c)
3.4. AChE Inhibition
3.5. GSK-3β Inhibition
3.6. Kinetic Study of AChE Inhibition
3.7. Cell Lines and Cell Culture
3.8. Cytotoxicity Bioassays in HepG2 and SH-SY5Y Cell Lines
3.9. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beato, A.; Gori, A.; Boucherle, B.; Peuchmaur, M.; Haudecoeur, R. β-Carboline as a Privileged Scaffold for Multitarget Strategies in Alzheimer’s Disease Therapy. J. Med. Chem. 2021, 64, 1392–1422. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali. A Review on Alzheimer’s Disease Pathophysiology and Its Management: An Update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Soeda, Y.; Yoshikawa, M.; Almeida, O.F.X.; Sumioka, A.; Maeda, S.; Osada, H.; Kondoh, Y.; Saito, A.; Miyasaka, T.; Kimura, T.; et al. Toxic Tau Oligomer Formation Blocked by Capping of Cysteine Residues with 1,2-Dihydroxybenzene Groups. Nat. Commun. 2015, 6, 10216. [Google Scholar] [CrossRef] [PubMed]
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s Disease; a Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.W.; Mohamed, K.O.; Sayed, H.S.; Mahmoud, Z. Recent Modifications of Anti-Dementia Agents Focusing on Tacrine and/or Donepezil Analogs. Med. Chem. 2023, 19, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Tewari, D.; Mamun, A.A.; Mathew, B.; Aleya, L.; Barreto, G.E.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Revisiting the Role of Brain and Peripheral Aβ in the Pathogenesis of Alzheimer’s Disease. J. Neurol. Sci. 2020, 416, 116974. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, A.; Fayaz, F.; Wakode, S.; Pottoo, F.H. Biological Signatures of Alzheimer’s Disease. Curr. Top. Med. Chem. 2020, 20, 770–781. [Google Scholar] [CrossRef]
- Lim, J.W.; Kim, S.K.; Choi, S.Y.; Kim, D.H.; Gadhe, C.G.; Lee, H.N.; Kim, H.-J.; Kim, J.; Cho, S.J.; Hwang, H.; et al. Identification of Crizotinib Derivatives as Potent SHIP2 Inhibitors for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2018, 157, 405–422. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Li, X.; Li, T.; Zhang, P.; Li, X.; Lu, L.; Sun, Y.; Zhang, B.; Allen, S.; White, L.; Phillips, J.; et al. Discovery of Novel Hybrids Containing Clioquinol−1-Benzyl-1,2,3,6-Tetrahydropyridine as Multi-Target-Directed Ligands (MTDLs) against Alzheimer’s Disease. Eur. J. Med. Chem. 2022, 244, 114841. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Holtzman, D.M. Biomarker Modeling of Alzheimer’s Disease. Neuron 2013, 80, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. Author Correction: History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable Drug Discovery of Multi-Target-Directed Ligands for Alzheimer’s Disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Li, Y.; Liu, R.; Pang, X.; Li, C.; Yang, R.; He, Y.; Lian, W.; Liu, A.-L.; Du, G.-H. Discovery of Multitarget-Directed Ligands against Alzheimer’s Disease through Systematic Prediction of Chemical–Protein Interactions. J. Chem. Inf. Model. 2015, 55, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Gehlot, P.; Kumar, S.; Kumar Vyas, V.; Singh Choudhary, B.; Sharma, M.; Malik, R. Guanidine-Based β Amyloid Precursor Protein Cleavage Enzyme 1 (BACE-1) Inhibitors for the Alzheimer’s Disease (AD): A Review. Bioorg. Med. Chem. 2022, 74, 117047. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-Inflammatory Effects of Flavonoids in Neurodegenerative Disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Jalili-Baleh, L.; Babaei, E.; Abdpour, S.; Nasir Abbas Bukhari, S.; Foroumadi, A.; Ramazani, A.; Sharifzadeh, M.; Abdollahi, M.; Khoobi, M. A Review on Flavonoid-Based Scaffolds as Multi-Target-Directed Ligands (MTDLs) for Alzheimer’s Disease. Eur. J. Med. Chem. 2018, 152, 570–589. [Google Scholar] [CrossRef] [PubMed]
- McHardy, S.F.; Wang, H.-Y.L.; McCowen, S.V.; Valdez, M.C. Recent Advances in Acetylcholinesterase Inhibitors and Reactivators: An Update on the Patent Literature (2012–2015). Expert Opin. Ther. Pat. 2017, 27, 455–476. [Google Scholar] [CrossRef] [PubMed]
- González, J.F.; Alcántara, A.R.; Doadrio, A.L.; Sánchez-Montero, J.M. Developments with Multi-Target Drugs for Alzheimer’s Disease: An Overview of the Current Discovery Approaches. Expert Opin. Drug Discov. 2019, 14, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-Target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-Y.; Chen, T.-K.; Zhou, J.-T.; He, S.-Y.; Yang, H.-Y.; Chen, Y.; Qu, W.; Feng, F.; Sun, H.-P. Dual GSK-3β/AChE Inhibitors as a New Strategy for Multitargeting Anti-Alzheimer’s Disease Drug Discovery. ACS Med. Chem. Lett. 2018, 9, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wu, L.; Liu, W.; Tian, L.; Chen, H.; Wu, Z.; Wang, N.; Liu, X.; Qiu, J.; Feng, X.; et al. Design, Synthesis and Biological Evaluation of Novel Coumarin Derivatives as Multifunctional Ligands for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2022, 242, 114689. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, X.; Liu, W.; Gao, Y.; Wu, L.; Huang, Y.; Chen, H.; Li, D.; Zhou, L.; Wang, N.; et al. Discovery of Novel β-Carboline Derivatives as Selective AChE Inhibitors with GSK-3β Inhibitory Property for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2022, 229, 114095. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, J.; Wang, Y.; Chen, L.; Duan, Y.; Huang, J.; Liu, C.; Chen, Y.; Liu, W.; Sun, H.; et al. Rational Design and Biological Evaluation of a New Class of Thiazolopyridyl Tetrahydroacridines as Cholinesterase and GSK-3 Dual Inhibitors for Alzheimer’s Disease. Eur. J. Med. Chem. 2020, 207, 112751. [Google Scholar] [CrossRef]
- Liu, W.; Tian, L.; Wu, L.; Chen, H.; Wang, N.; Liu, X.; Zhao, C.; Wu, Z.; Jiang, X.; Wu, Q.; et al. Discovery of Novel β-Carboline-1,2,3-Triazole Hybrids as AChE/GSK-3β Dual Inhibitors for Alzheimer’s Disease Treatment. Bioorg. Chem. 2022, 129, 106168. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The Cholinergic System in the Pathophysiology and Treatment of Alzheimer’s Disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, X.; Chen, H.; Wang, K.; Xue, T.; Mi, J.; Ban, Y.; Zhu, G.; Zhou, Y.; Dong, W.; et al. Development of the “Hidden” Multi-Target-Directed Ligands by AChE/BuChE for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2023, 251, 115253. [Google Scholar] [CrossRef]
- Chen, Y.-G. Research Progress in the Pathogenesis of Alzheimer’s Disease. Chin. Med. J. 2018, 131, 1618. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Wang, K.; Dong, J.; Tang, L. Alzheimer’s Disease: Updated Multi-Targets Therapeutics Are in Clinical and in Progress. Eur. J. Med. Chem. 2022, 238, 114464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Wang, Y.; Li, X.; Wang, S.; Wang, Z. Recent Advance on Carbamate-Based Cholinesterase Inhibitors as Potential Multifunctional Agents against Alzheimer’s Disease. Eur. J. Med. Chem. 2022, 240, 114606. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, Z.; Cai, H.; Xu, S.; Liu, J.; Jiang, J.; Yao, H.; Wu, X.; Xu, J. Design, Synthesis, Biological Evaluation and Docking Study of 4-Isochromanone Hybrids Bearing N-Benzyl Pyridinium Moiety as Dual Binding Site Acetylcholinesterase Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5212–5216. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of Acetylcholinesterase Inhibitors in Alzheimer’s Disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Li, X.; Li, T.; Zhan, F.; Cheng, F.; Lu, L.; Zhang, B.; Li, J.; Hu, Z.; Zhou, S.; Jia, Y.; et al. Design, Synthesis, and Biological Evaluation of Novel Chromanone Derivatives as Multifunctional Agents for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2022, 13, 3488–3501. [Google Scholar] [CrossRef] [PubMed]
- Amenta, F.; Parnetti, L.; Gallai, V.; Wallin, A. Treatment of Cognitive Dysfunction Associated with Alzheimer’s Disease with Cholinergic Precursors. Ineffective Treatments or Inappropriate Approaches? Mech. Ageing Dev. 2001, 122, 2025–2040. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Augustinsson, K.-B.; Nachmansohn, D. Distinction between Acetylcholine-Esterase and Other Choline Ester-Splitting Enzymes. Science 1949, 110, 98–99. [Google Scholar] [CrossRef]
- Massoulié, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef] [PubMed]
- Massoulie, J.; Bon, S. The Molecular Forms of Cholinesterase and Acetylcholinesterase in Vertebrates. Annu. Rev. Neurosci. 1982, 5, 57–106. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.L.; Luk, W.K.W.; Bi, C.W.C.; Liu, E.Y.L.; Wu, K.Q.Y.; Yao, P.; Dong, T.T.X.; Tsim, K.W.K. Erythropoietin Regulates the Expression of Dimeric Form of Acetylcholinesterase during Differentiation of Erythroblast. J. Neurochem. 2018, 146, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jia, Y.; Li, J.; Zhang, P.; Li, T.; Lu, L.; Yao, H.; Liu, J.; Zhu, Z.; Xu, J. Novel and Potent Acetylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease from Natural (±)-7,8-Dihydroxy-3-Methyl-Isochroman-4-One. Molecules 2022, 27, 3090. [Google Scholar] [CrossRef] [PubMed]
- Ghanei-Nasab, S.; Khoobi, M.; Hadizadeh, F.; Marjani, A.; Moradi, A.; Nadri, H.; Emami, S.; Foroumadi, A.; Shafiee, A. Synthesis and Anticholinesterase Activity of Coumarin-3-Carboxamides Bearing Tryptamine Moiety. Eur. J. Med. Chem. 2016, 121, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s Disease Therapy with Acetylcholinesterase Inhibitors and Memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [PubMed]
- Hung, S.Y.; Fu, W.M. Drug Candidates in Clinical Trials for Alzheimer’s Disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Liu, H.; Shi, X.; Ai, Y.; Liu, Q.; Cheng, Y. The Efficacy and Safety of Alzheimer’s Disease Therapies: An Updated Umbrella Review. J. Alzheimers Dis. 2022, 85, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget Compounds Bearing Tacrine- and Donepezil-like Structural and Functional Motifs for the Potential Treatment of Alzheimer’s Disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Brion, J.-P. Developmental Expression and Localization of Glycogen Synthase Kinase-3β in Rat Brain. J. Chem. Neuroanat. 1999, 16, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Martín, M.; Fuster-Matanzo, A.; Teixeira, C.M.; Jurado-Arjona, J.; Ulloa, F.; deFelipe, J.; Rábano, A.; Hernández, F.; Soriano, E.; Ávila, J. GSK-3β Overexpression Causes Reversible Alterations on Postsynaptic Densities and Dendritic Morphology of Hippocampal Granule Neurons In Vivo. Mol. Psychiatry 2013, 18, 451–460. [Google Scholar] [CrossRef]
- Engel, T.; Gómez-Sintes, R.; Alves, M.; Jimenez-Mateos, E.M.; Fernández-Nogales, M.; Sanz-Rodriguez, A.; Morgan, J.; Beamer, E.; Rodríguez-Matellán, A.; Dunleavy, M.; et al. Bi-Directional Genetic Modulation of GSK-3β Exacerbates Hippocampal Neuropathology in Experimental Status Epilepticus. Cell Death Dis. 2018, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-B.; Shaw, P.-C.; Wong, C.-C.; Wan, D.C.-C. Expression of Glycogen Synthase Kinase-3 Isoforms in Mouse Tissues and Their Transcription in the Brain. J. Chem. Neuroanat. 2002, 23, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xiong, M.; Zhang, Z. The Role of Glycogen Synthase Kinase 3β in Neurodegenerative Diseases. Front. Mol. Neurosci. 2023, 16, 1209703. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Drouin-Ouellet, J.; Tirolo, C.; Pulvirenti, A.; Giugno, R.; Testa, N.; Caniglia, S.; Serapide, M.F.; Cisbani, G.; Barker, R.A.; et al. GSK-3β-Induced Tau Pathology Drives Hippocampal Neuronal Cell Death in Huntington’s Disease: Involvement of Astrocyte–Neuron Interactions. Cell Death Dis. 2016, 7, e2206. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; George, J.L.; Wills, J.; Duka, V.; Shah, K.; Lee, Y.-C.; Rodriguez, O.; Simkins, T.; Winter, M.; Moechars, D.; et al. GSK-3β Dysregulation Contributes to Parkinson’s-like Pathophysiology with Associated Region-Specific Phosphorylation and Accumulation of Tau and α-Synuclein. Cell Death Differ. 2015, 22, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Wandosell, F.; Hernández, F. Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease Pathogenesis and Glycogen Synthase Kinase-3 Inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Luna-Medina, R.; Alonso-Gil, S.; Sanz-SanCristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perez-Castillo, A. Glycogen Synthase Kinase 3 Inhibition Promotes Adult Hippocampal Neurogenesis in Vitro and in Vivo. ACS Chem. Neurosci. 2012, 3, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T.; Banach-Kasper, E.; Gralec, K. GSK-3β at the Intersection of Neuronal Plasticity and Neurodegeneration. Neural. Plast. 2019, 2019, e4209475. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A. Cholinergic Modulation of Amyloid Precursor Protein Processing with Emphasis on M1 Muscarinic Receptor: Perspectives and Challenges in Treatment of Alzheimer’s Disease. J. Neurochem. 2012, 120 (Suppl. 1), 22–33. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Gómez, M.-Á.; Llorens-Álvarez, E.; Alom, J.; del Ser, T.; Avila, J.; Sáez-Valero, J.; García-Ayllón, M.-S. Tau Phosphorylation by Glycogen Synthase Kinase 3β Modulates Enzyme Acetylcholinesterase Expression. J. Neurochem. 2021, 157, 2091–2105. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Jin, Q.; Wu, J.; Zhang, X.-J. GSK3beta Mediates the Induced Expression of Synaptic Acetylcholinesterase during Apoptosis. J. Neurochem. 2008, 104, 409–419. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Tian, L.; Gao, Y.; Liu, W.; Chen, H.; Jiang, X.; Xu, Z.; Ding, H.; Zhao, Q. Design, Synthesis and Biological Evaluation of Harmine Derivatives as Potent GSK-3β/DYRK1A Dual Inhibitors for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2021, 222, 113554. [Google Scholar] [CrossRef] [PubMed]
- Oukoloff, K.; Coquelle, N.; Bartolini, M.; Naldi, M.; Le Guevel, R.; Bach, S.; Josselin, B.; Ruchaud, S.; Catto, M.; Pisani, L.; et al. Design, Biological Evaluation and X-Ray Crystallography of Nanomolar Multifunctional Ligands Targeting Simultaneously Acetylcholinesterase and Glycogen Synthase Kinase-3. Eur. J. Med. Chem. 2019, 168, 58–77. [Google Scholar] [CrossRef] [PubMed]
- Uehara, F.; Shoda, A.; Aritomo, K.; Fukunaga, K.; Watanabe, K.; Ando, R.; Shinoda, M.; Ueno, H.; Kubodera, H.; Sunada, S.; et al. 6-(4-Pyridyl)Pyrimidin-4(3H)-Ones as CNS Penetrant Glycogen Synthase Kinase-3β Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6928–6932. [Google Scholar] [CrossRef] [PubMed]
- Shuai, W.; Li, W.; Yin, Y.; Yang, L.; Xu, F.; Xu, S.; Yao, H.; Zhu, Z.; Xu, J. Design, Synthesis and Molecular Modeling of Isothiochromanone Derivatives as Acetylcholinesterase Inhibitors. Future Med. Chem. 2019, 11, 2687–2699. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-Based Multi-Functional Cholinesterase Inhibitors for Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Grishchenko, M.V.; Makhaeva, G.F.; Burgart, Y.V.; Rudakova, E.V.; Boltneva, N.P.; Kovaleva, N.V.; Serebryakova, O.G.; Lushchekina, S.V.; Astakhova, T.Y.; Zhilina, E.F.; et al. Conjugates of Tacrine with Salicylamide as Promising Multitarget Agents for Alzheimer’s Disease. ChemMedChem 2022, 17, e202200080. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Uras, G.; Zhang, P.; Xu, S.; Yin, Y.; Liu, J.; Qin, S.; Li, X.; Allen, S.; Bai, R.; et al. Discovery of Novel Tacrine–Pyrimidone Hybrids as Potent Dual AChE/GSK-3 Inhibitors for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2021, 64, 7483–7506. [Google Scholar] [CrossRef] [PubMed]
- Minarini, A.; Milelli, A.; Tumiatti, V.; Rosini, M.; Simoni, E.; Bolognesi, M.L.; Andrisano, V.; Bartolini, M.; Motori, E.; Angeloni, C.; et al. Cystamine-Tacrine Dimer: A New Multi-Target-Directed Ligand as Potential Therapeutic Agent for Alzheimer’s Disease Treatment. Neuropharmacology 2012, 62, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Sivaprakasam, P.; Han, X.; Civiello, R.L.; Jacutin-Porte, S.; Kish, K.; Pokross, M.; Lewis, H.A.; Ahmed, N.; Szapiel, N.; Newitt, J.A.; et al. Discovery of New Acylaminopyridines as GSK-3 Inhibitors by a Structure Guided in-Depth Exploration of Chemical Space around a Pyrrolopyridinone Core. Bioorg. Med. Chem. Lett. 2015, 25, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, W.; Huang, Y.; Liu, M.; Tian, C.; Lu, H.; Jia, H.; Xu, Z.; Ding, H.; Zhao, Q. Facile Synthesis of C1-Substituted β-Carbolines as CDK4 Inhibitors for the Treatment of Cancer. Bioorg. Chem. 2022, 121, 105659. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | X | IC50 (μM) ± SEM a | |
|---|---|---|---|---|---|
| AChE b | GSK-3β c | ||||
| 16a | Cl | H | C-H | 2.30 ± 0.14 | 15.10 ± 0.90 |
| 16b | H | Br | C-H | 20.00 ± 1.50 | 38.50 ± 0.70 |
| 16c | H | Cl | C-H | 10.30 ± 0.70 | 2.28 ± 0.07 |
| 16d | Cl | H | C-F | 1.89 ± 0.21 | 1.69 ± 0.09 |
| 16e | H | Br | C-F | 15.30 ± 0.90 | 0.94 ± 0.02 |
| 16f | H | Cl | C-F | 16.50 ± 0.80 | 2.44 ± 0.06 |
| 18a | Cl | H | 0.047 ± 0.002 | 0.93 ± 0.08 | |
| 18b | H | Br | 2.13 ± 0.11 | 0.37 ± 0.02 | |
| 18c | H | Cl | 2.24 ± 0.13 | 0.42 ± 0.03 | |
| Tacrine | 0.229 ± 0.01 | ||||
| AR-A014418 d | 0.222 ± 0.005 | ||||
| Compound | MW a | logP a | tPSA(Å2) a | HBD a | HBA a | Rotor a | Vio a |
|---|---|---|---|---|---|---|---|
| 16a | 478.01 | 4.48 | 98.00 | 1 | 4 | 6 | 0 |
| 16b | 522.46 | 4.59 | 98.00 | 1 | 4 | 6 | 1 |
| 16c | 478.01 | 4.49 | 98.00 | 1 | 4 | 6 | 0 |
| 16d | 496.00 | 4.80 | 98.00 | 1 | 5 | 6 | 0 |
| 16e | 540.45 | 4.82 | 98.00 | 1 | 5 | 6 | 1 |
| 16f | 496.00 | 4.73 | 98.00 | 1 | 5 | 6 | 0 |
| 18a | 553.14 | 4.60 | 135.33 | 2 | 4 | 10 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Ze, X.; Qin, S.; Zhang, B.; Li, X.; Gong, Q.; Zhang, H.; Zhu, Z.; Xu, J. Design, Synthesis, and Biological Evaluation of Novel Tetrahydroacridin Hybrids with Sulfur-Inserted Linkers as Potential Multitarget Agents for Alzheimer’s Disease. Molecules 2024, 29, 1782. https://doi.org/10.3390/molecules29081782
Wu X, Ze X, Qin S, Zhang B, Li X, Gong Q, Zhang H, Zhu Z, Xu J. Design, Synthesis, and Biological Evaluation of Novel Tetrahydroacridin Hybrids with Sulfur-Inserted Linkers as Potential Multitarget Agents for Alzheimer’s Disease. Molecules. 2024; 29(8):1782. https://doi.org/10.3390/molecules29081782
Chicago/Turabian StyleWu, Xiuyuan, Xiaotong Ze, Shuai Qin, Beiyu Zhang, Xinnan Li, Qi Gong, Haiyan Zhang, Zheying Zhu, and Jinyi Xu. 2024. "Design, Synthesis, and Biological Evaluation of Novel Tetrahydroacridin Hybrids with Sulfur-Inserted Linkers as Potential Multitarget Agents for Alzheimer’s Disease" Molecules 29, no. 8: 1782. https://doi.org/10.3390/molecules29081782
APA StyleWu, X., Ze, X., Qin, S., Zhang, B., Li, X., Gong, Q., Zhang, H., Zhu, Z., & Xu, J. (2024). Design, Synthesis, and Biological Evaluation of Novel Tetrahydroacridin Hybrids with Sulfur-Inserted Linkers as Potential Multitarget Agents for Alzheimer’s Disease. Molecules, 29(8), 1782. https://doi.org/10.3390/molecules29081782