
BiMPADR: A Deep Learning Framework for Predicting Adverse Drug Reactions in New Drugs

Abstract
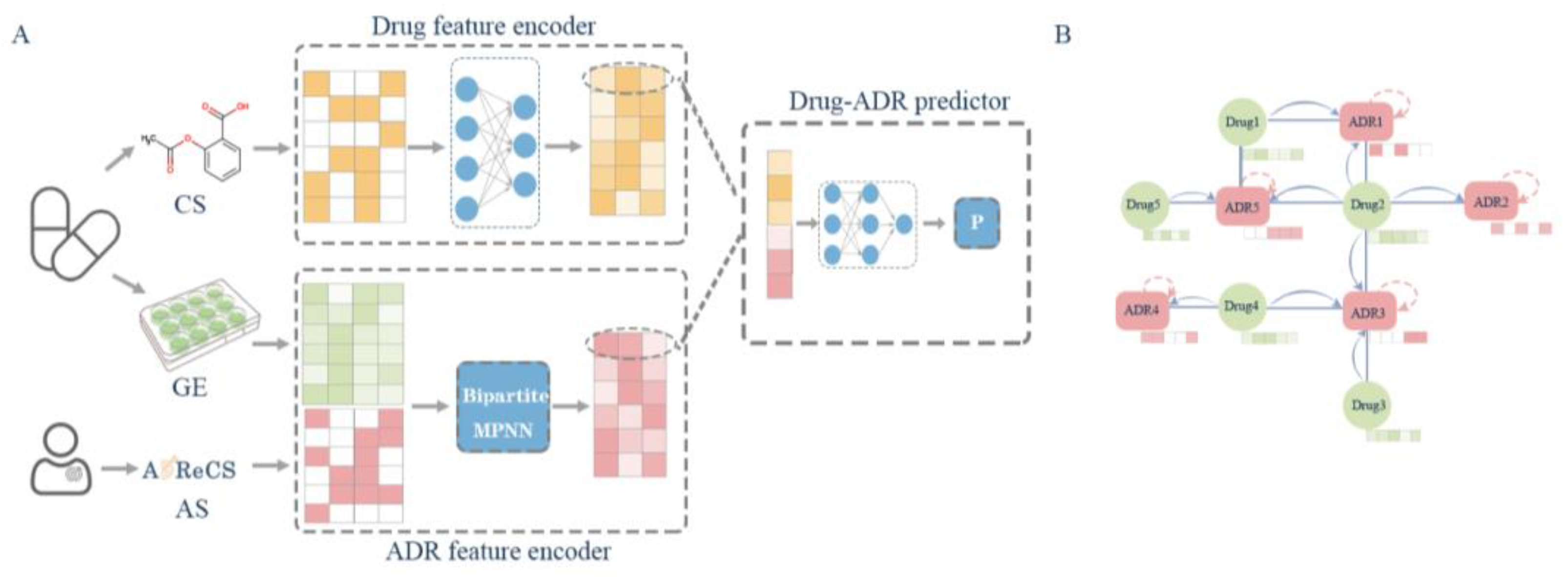
:1. Introduction
2. Results and Discussion
2.1. Performance on Different Datasets
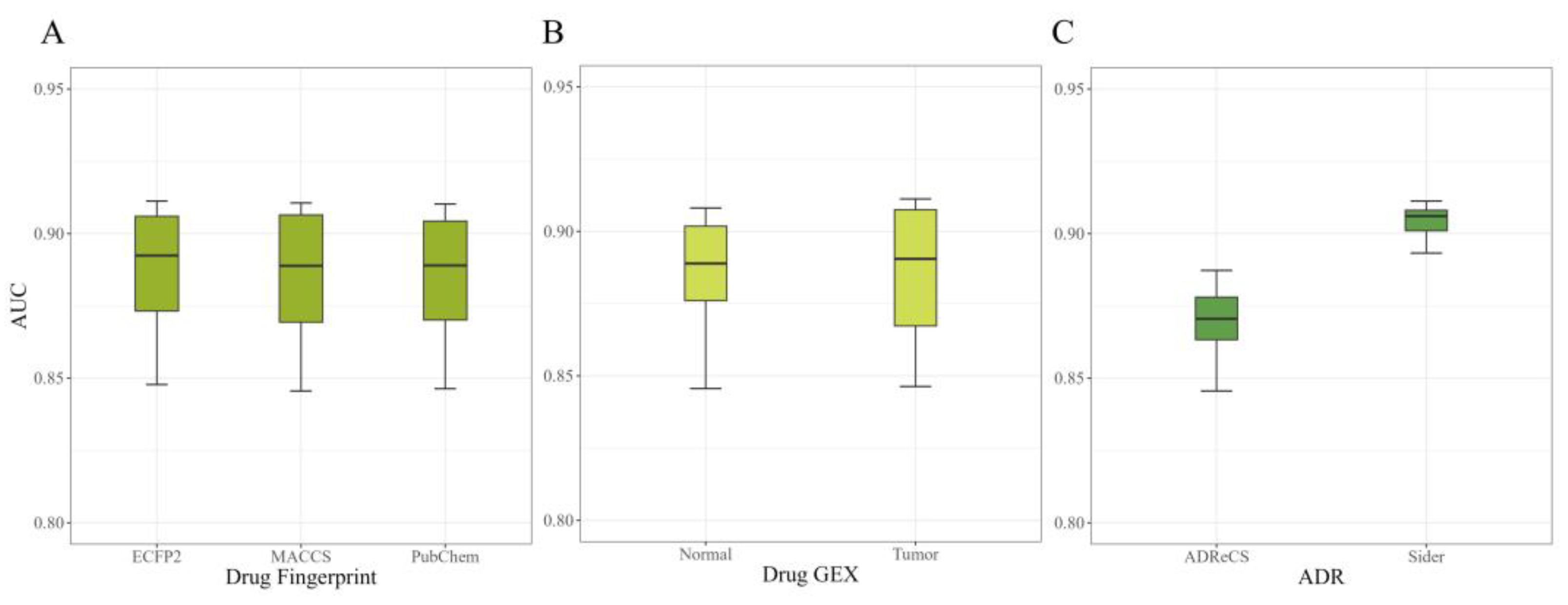
2.1.1. Performance on Different Fingerprints
2.1.2. Performance on Different GE
2.1.3. Performance on ADR Selection
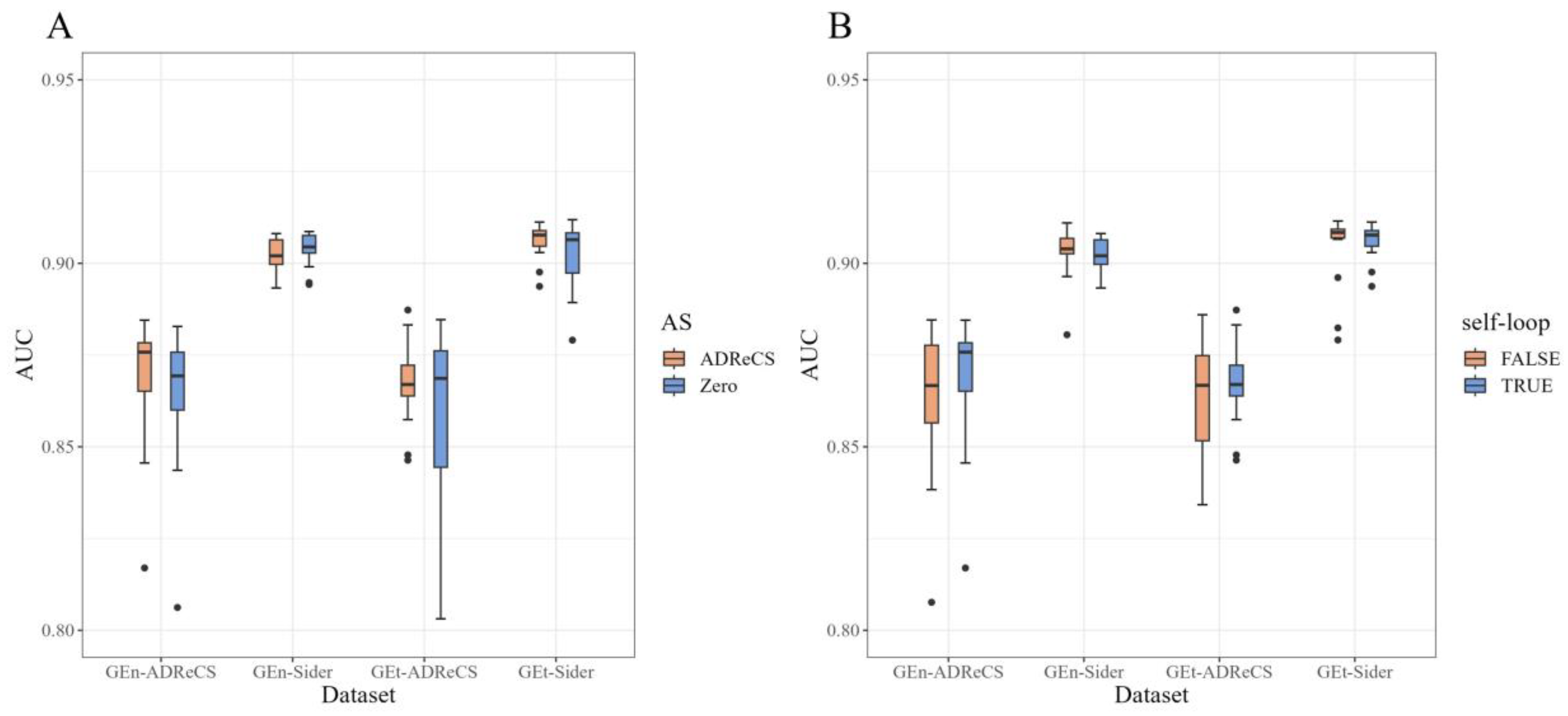
2.2. Ablation Study
- The first variant involved replacing the initial feature vectors of adverse reactions with zero vectors, completely excluding the use of ADR–gene association information.
- The second variant maintained the same input as the original model but only utilized this information during the computation of attention coefficients in the binary network information propagation, without incorporating the adverse reaction initial features in the information update function, denoted as . The difference in this process lies in the addition of a self-loop, where the original method is set to TRUE, while the ablation experiments are set to FALSE.
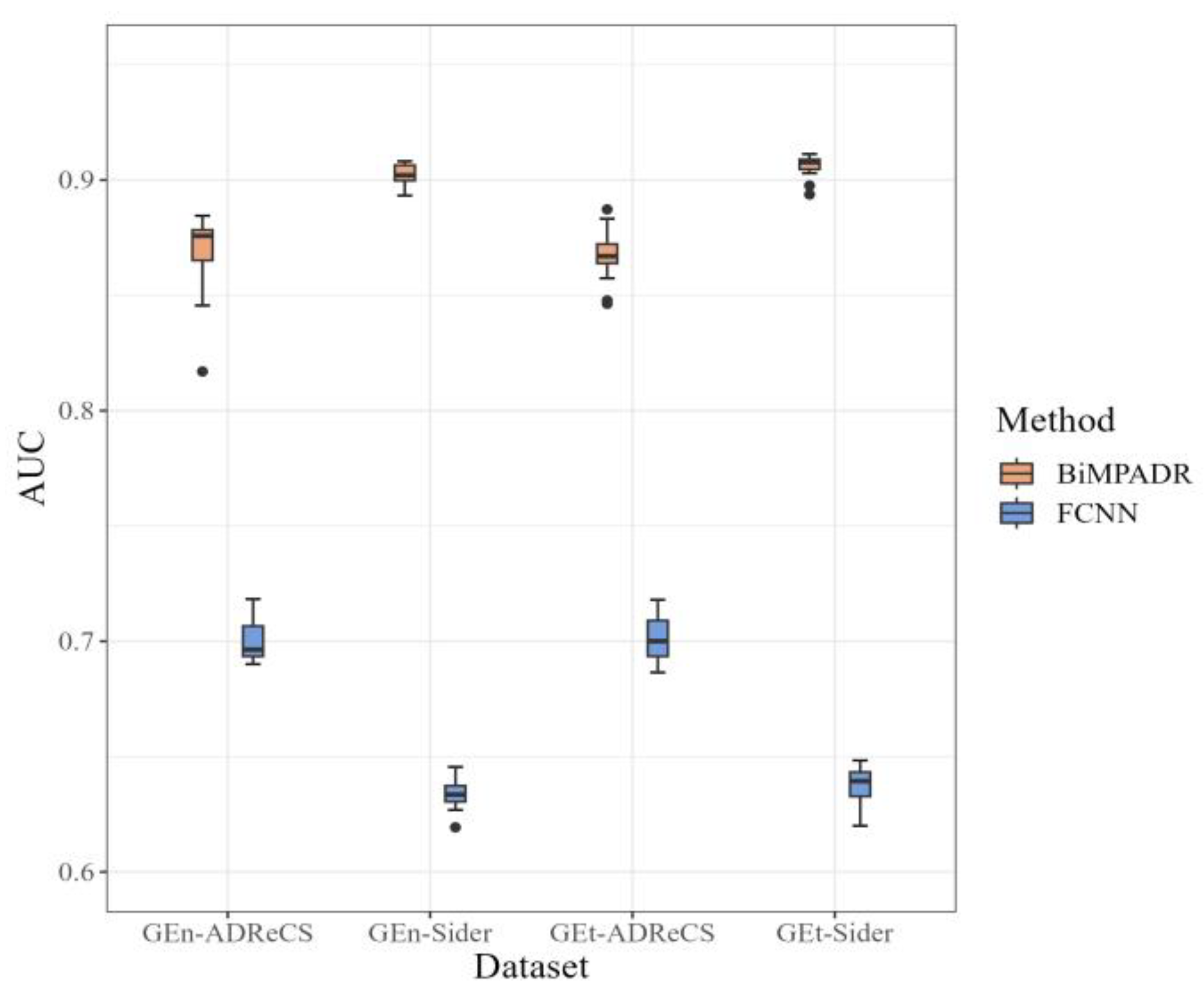
2.3. Performance of BiMPADR Compared with State-of-the-Art Methods
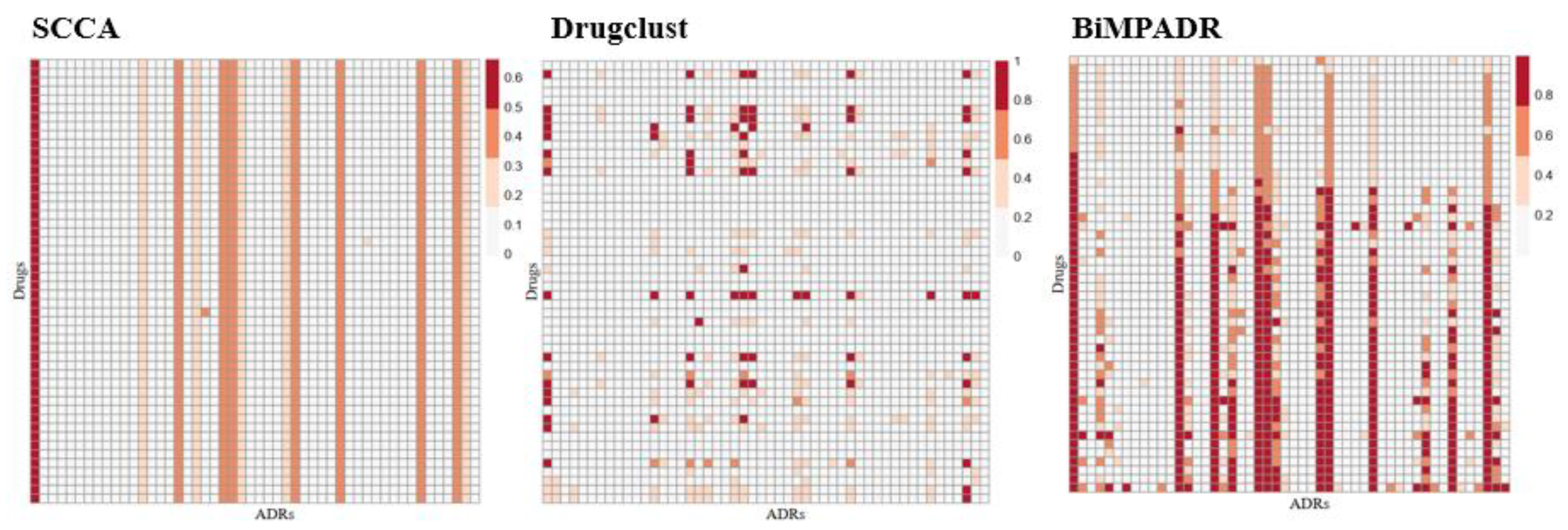
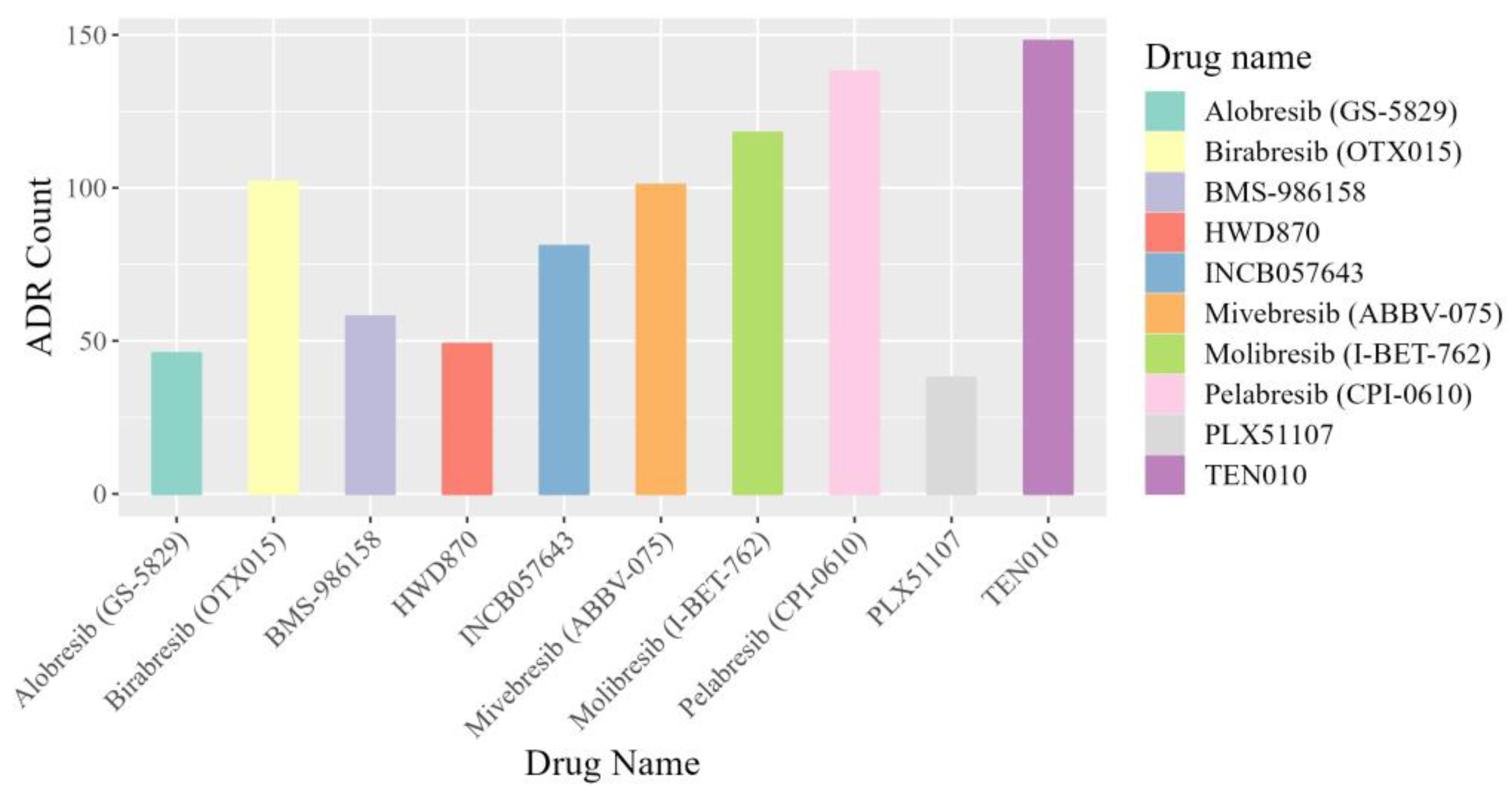
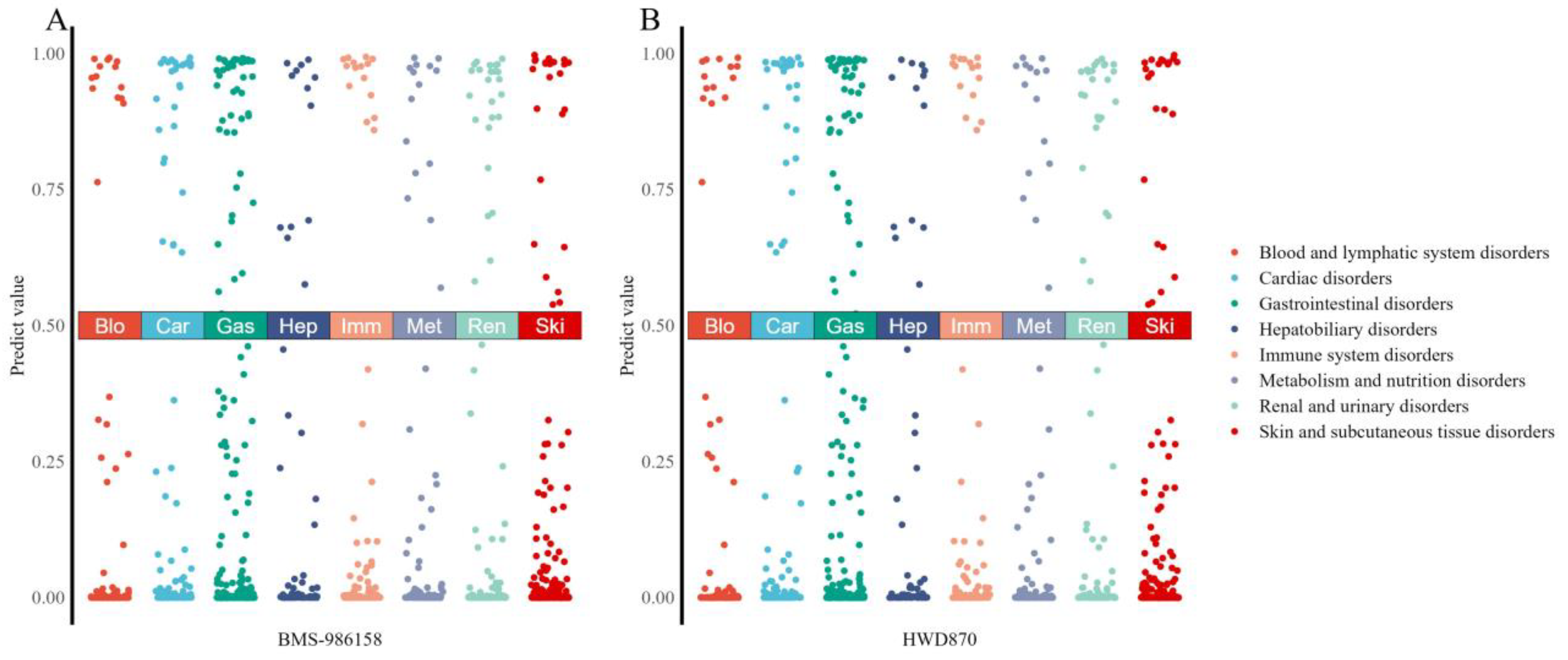
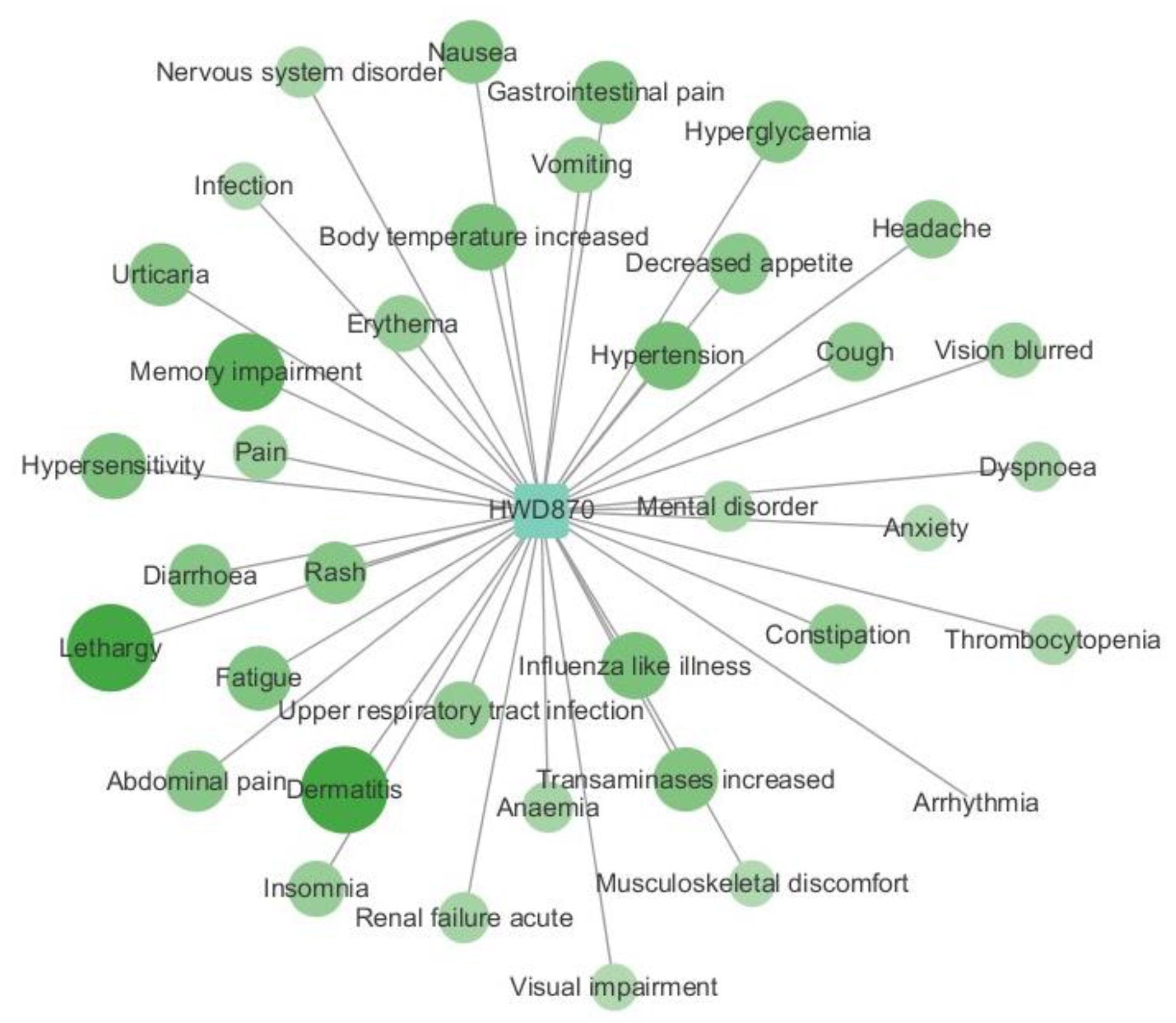
2.4. Case Study
3. Materials and Methods
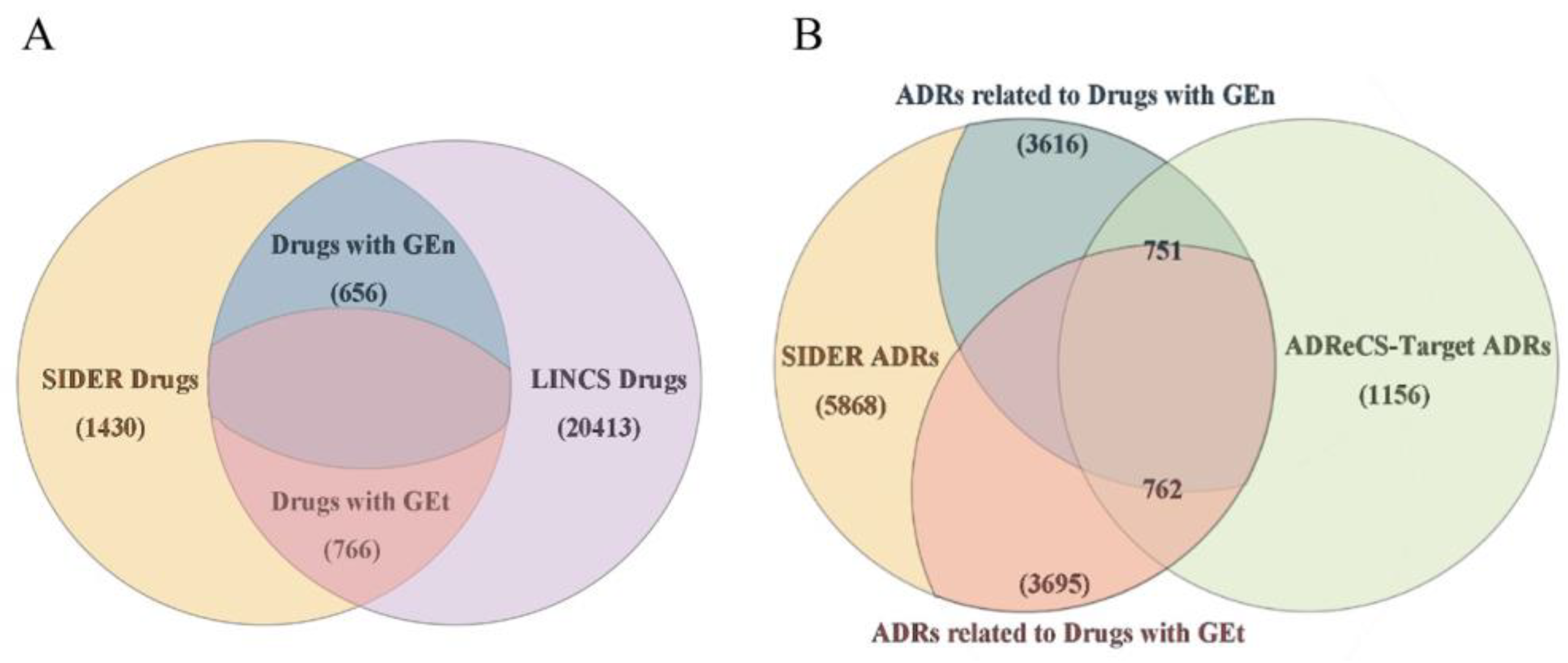
3.1. Datasets
3.2. Methods
3.2.1. MPNNs
3.2.2. Overall Schema of the Deep Learning Network
3.2.3. MPNN Layer with ADR Embedding Vector
3.3. Experimental Setting
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nebeker, J.R.; Barach, P.; Samore, M.H. Clarifying adverse drug events: A clinician’s guide to terminology, documentation, and reporting. Ann. Intern. Med. 2004, 140, 795–801. [Google Scholar] [CrossRef]
- Pirmohamed, M.; James, S.; Meakin, S.; Green, C.; Scott, A.K.; Walley, T.J.; Farrar, K.; Park, B.K.; Breckenridge, A.M. Adverse drug reactions as cause of admission to hospital: Prospective analysis of 18 820 patients. BMJ (Clin. Res. Ed.) 2004, 329, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Cocos, A.; Fiks, A.G.; Masino, A.J. Deep learning for pharmacovigilance: Recurrent neural network architectures for labeling adverse drug reactions in Twitter posts. J. Am. Med. Inform. Assoc. JAMIA 2017, 24, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.H.; Burrows, A.D.; Anderson, R.L. Can preclinical drug development help to predict adverse events in clinical trials? Drug discovery today 2022, 27, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Gurwitz, J.H.; Field, T.S.; Avorn, J.; McCormick, D.; Jain, S.; Eckler, M.; Benser, M.; Edmondson, A.C.; Bates, D.W. Incidence and preventability of adverse drug events in nursing homes. Am. J. Med. 2000, 109, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Batel Marques, F.; Penedones, A.; Mendes, D.; Alves, C. A systematic review of observational studies evaluating costs of adverse drug reactions. Clin. Outcomes Res. CEOR 2016, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Ernst, F.R.; Grizzle, A.J. Drug-related morbidity and mortality: Updating the cost-of-illness model. J. Am. Pharm. Assoc. 2001, 41, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Trial watch: Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- da Silva, B.A.; Krishnamurthy, M. The alarming reality of medication error: A patient case and review of Pennsylvania and National data. J. Community Hosp. Intern. Med. Perspect. 2016, 6, 31758. [Google Scholar] [CrossRef]
- Tatonetti, N.P. The Next Generation of Drug Safety Science: Coupling Detection, Corroboration, and Validation to Discover Novel Drug Effects and Drug-Drug Interactions. Clin. Pharmacol. Ther. 2018, 103, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Voskens, C.J.; Goldinger, S.M.; Loquai, C.; Robert, C.; Kaehler, K.C.; Berking, C.; Bergmann, T.; Bockmeyer, C.L.; Eigentler, T.; Fluck, M.; et al. The price of tumor control: An analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS ONE 2013, 8, e53745. [Google Scholar] [CrossRef] [PubMed]
- Cami, A.; Arnold, A.; Manzi, S.; Reis, B. Predicting adverse drug events using pharmacological network models. Sci. Transl. Med. 2011, 3, 114ra127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zou, H.; Luo, L.; Liu, Q.; Wu, W.; Xiao, W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing 2016, 173, 979–987. [Google Scholar] [CrossRef]
- Galeano, D.; Paccanaro, A. A recommender system approach for predicting drug side effects. In Proceedings of the 2018 International Joint Conference on Neural Networks (IJCNN), Rio de Janeiro, Brazil, 8–13 July 2018; pp. 1–8. [Google Scholar]
- Lin, J.; Kuang, Q.; Li, Y.; Zhang, Y.; Sun, J.; Ding, Z.; Li, M. Prediction of adverse drug reactions by a network based external link prediction method. Anal. Methods 2013, 5, 6120–6127. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Pauwels, E.; Kotera, M. Drug side-effect prediction based on the integration of chemical and biological spaces. J. Chem. Inf. Model. 2012, 52, 3284–3292. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wu, Y.; Chen, Y.; Sun, J.; Zhao, Z.; Chen, X.-w.; Matheny, M.E.; Xu, H. Large-scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J. Am. Med. Inform. Assoc. 2012, 19, e28–e35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, F.; Luo, L.; Zhang, J. Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinform. 2015, 16, 365. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug-side effect association via semisupervised model and multiple kernel learning. IEEE J. Biomed. Health Inform. 2018, 23, 2619–2632. [Google Scholar] [CrossRef]
- Pauwels, E.; Stoven, V.; Yamanishi, Y. Predicting drug side-effect profiles: A chemical fragment-based approach. BMC Bioinform. 2011, 12, 169. [Google Scholar] [CrossRef]
- Niu, S.-Y.; Xin, M.-Y.; Luo, J.; Liu, M.-Y.; Jiang, Z.-R. Dsep: A tool implementing novel method to predict side effects of drugs. J. Comput. Biol. 2015, 22, 1108–1117. [Google Scholar] [CrossRef]
- Dimitri, G.M.; Lió, P. DrugClust: A machine learning approach for drugs side effects prediction. Comput. Biol. Chem. 2017, 68, 204–210. [Google Scholar] [CrossRef]
- Xuan, P.; Wang, M.; Liu, Y.; Wang, D.; Zhang, T.; Nakaguchi, T. Integrating specific and common topologies of heterogeneous graphs and pairwise attributes for drug-related side effect prediction. Brief. Bioinform. 2022, 23, bbac126. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, K.H.; Song, M.; Lee, D. Building the process-drug–side effect network to discover the relationship between biological Processes and side effects. BMC Bioinform. 2011, 12, S2. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Meyer, U.A. Induction of drug metabolism: The role of nuclear receptors. Pharmacol. Rev. 2003, 55, 649–673. [Google Scholar] [CrossRef]
- Toyoshiba, H.; Sawada, H.; Naeshiro, I.; Horinouchi, A. Similar compounds searching system by using the gene expression microarray database. Toxicol. Lett. 2009, 186, 52–57. [Google Scholar] [CrossRef]
- Babcock, J.J.; Du, F.; Xu, K.; Wheelan, S.J.; Li, M. Integrated analysis of drug-induced gene expression profiles predicts novel hERG inhibitors. PLoS ONE 2013, 8, e69513. [Google Scholar] [CrossRef]
- Zhang, J.D.; Sach-Peltason, L.; Kramer, C.; Wang, K.; Ebeling, M. Multiscale modelling of drug mechanism and safety. Drug Discov. Today 2020, 25, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-H.; He, Q.-S.; Liu, K.; Cheng, J.; Zhong, M.-D.; Chen, L.-S.; Yao, L.-X.; Ji, Z.-L. ADReCS-Target: Target profiles for aiding drug safety research and application. Nucleic Acids Res. 2018, 46, D911–D917. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Message passing neural networks. In Machine Learning Meets Quantum Physics; Springer: Cham, Switzerland, 2020; pp. 199–214. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Ruby, U.; Yendapalli, V. Binary cross entropy with deep learning technique for image classification. Int. J. Adv. Trends Comput. Sci. Eng. 2020, 9, 5393–5397. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Yin, M.; Guo, Y.; Hu, R.; Cai, W.L.; Li, Y.; Pei, S.; Sun, H.; Peng, C.; Li, J.; Ye, R. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020, 11, 1833. [Google Scholar] [CrossRef] [PubMed]
- Bonazzoli, E.; Predolini, F.; Cocco, E.; Bellone, S.; Altwerger, G.; Menderes, G.; Zammataro, L.; Bianchi, A.; Pettinella, F.; Riccio, F. Inhibition of BET bromodomain proteins with GS-5829 and GS-626510 in uterine serous carcinoma, a biologically aggressive variant of endometrial cancer. Clin. Cancer Res. 2018, 24, 4845–4853. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Maduskuie, T.; Burn, T.; Diamond-Fosbenner, S.; Falahatpisheh, N.; Volgina, A.; Zolotarjova, N.; Wen, X.; Feldman, P.; Rupar, M. Preclinical characterization of the potent and selective BET inhibitor INCB057643 in models of hematologic malignancies. Cancer Res. 2017, 77, 5071. [Google Scholar] [CrossRef]
- Faivre, E.J.; Wilcox, D.M.; Hessler, P.; Uziel, T.; Tapang, P.; Magoc, T.; Albert, D.H.; Fang, G.; Rosenberg, S.; McDaniel, K. ABBV-075, a novel BET family inhibitor, disrupts critical transcription programs that drive prostate cancer growth to induce potent anti-tumor activity in vitro and in vivo. Cancer Res. 2016, 76, 4694. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Gehling, V.S.; Hewitt, M.C.; Vaswani, R.G.; Côté, A.; Leblanc, Y.; Nasveschuk, C.G.; Bellon, S.; Bergeron, L.; Campbell, R. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.; Iwata, K.; Ooike, S.; Sugahara, K.; Nakamura, H.; Daibata, M. Development of the BET bromodomain inhibitor OTX015. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics, Boston, MA, USA, 19–23 October 2013. [Google Scholar]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Firle, K.; Szymansky, A.; Witthauer, M.; Dorado-Garcia, H.; Toedling, J.; Schoenbeck, K.; Henssen, A.; Hertwig, F.; Eggert, A.; Schulte, J. Preclinical evaluation of BET-bromodomain inhibitor TEN-010 as monotherapy and combination therapy in MYC-driven neuroblastoma. Ann. Oncol. 2018, 29, iii12–iii13. [Google Scholar] [CrossRef]
- Ozer, H.G.; El-Gamal, D.; Powell, B.; Hing, Z.A.; Blachly, J.S.; Harrington, B.; Mitchell, S.; Grieselhuber, N.R.; Williams, K.; Lai, T.-H. BRD4 profiling identifies critical chronic lymphocytic leukemia oncogenic circuits and reveals sensitivity to PLX51107, a novel structurally distinct BET inhibitor. Cancer Discov. 2018, 8, 458–477. [Google Scholar] [CrossRef]
- Hilton, J.; Cristea, M.; Postel-Vinay, S.; Baldini, C.; Voskoboynik, M.; Edenfield, W.; Shapiro, G.I.; Cheng, M.L.; Vuky, J.; Corr, B. BMS-986158, a small molecule inhibitor of the bromodomain and extraterminal domain proteins, in patients with selected advanced solid tumors: Results from a phase 1/2a trial. Cancers 2022, 14, 4079. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Desai, P.; Lee, S.; Ritchie, E.K.; Winer, E.S.; DeMario, M.; Brennan, B.; Nüesch, E.; Chesne, E.; Brennan, L. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 2021, 62, 1740–1748. [Google Scholar] [CrossRef]
- Senapati, J.; Fiskus, W.C.; Daver, N.; Wilson, N.R.; Ravandi, F.; Garcia-Manero, G.; Kadia, T.; DiNardo, C.D.; Jabbour, E.; Burger, J. Phase I Results of Bromodomain and Extra-Terminal Inhibitor PLX51107 in Combination with Azacitidine in Patients with Relapsed/Refractory Myeloid Malignancies. Clin. Cancer Res. 2023, 29, 4352–4360. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.A.; Supko, J.G.; Maris, M.B.; Flinn, I.W.; Goy, A.; Younes, A.; Bobba, S.; Senderowicz, A.M.; Efuni, S.; Rippley, R. A phase I study of pelabresib (CPI-0610), a small-molecule inhibitor of BET proteins, in patients with relapsed or refractory lymphoma. Cancer Res. Commun. 2022, 2, 795–805. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; LoRusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N., Jr.; Chen, X.; Hu, B.; Freise, K.J. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | CS | Train | Test | External Validation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUC | Precision | ACC | AUC | Precision | ACC | AUC | Precision | ACC | ||
| GEn-ADReCS | ECFP2 | 0.948 ± 0.015 | 0.839 ± 0.032 | 0.877 ± 0.017 | 0.873 ± 0.018 | 0.796 ± 0.034 | 0.802 ± 0.015 | 0.861 ± 0.026 | 0.177 ± 0.028 | 0.77 ± 0.053 |
| MACCS | 0.958 ± 0.007 | 0.844 ± 0.016 | 0.889 ± 0.009 | 0.879 ± 0.019 | 0.798 ± 0.028 | 0.808 ± 0.015 | 0.871 ± 0.016 | 0.178 ± 0.017 | 0.774 ± 0.033 | |
| PubChem | 0.97 ± 0.008 | 0.869 ± 0.017 | 0.907 ± 0.013 | 0.894 ± 0.01 | 0.815 ± 0.019 | 0.819 ± 0.007 | 0.874 ± 0.007 | 0.193 ± 0.012 | 0.802 ± 0.019 | |
| GEn-SIDER | ECFP2 | 0.975 ± 0.012 | 0.89 ± 0.027 | 0.923 ± 0.025 | 0.898 ± 0.009 | 0.853 ± 0.011 | 0.831 ± 0.012 | 0.903 ± 0.003 | 0.109 ± 0.007 | 0.849 ± 0.013 |
| MACCS | 0.983 ± 0.01 | 0.898 ± 0.028 | 0.937 ± 0.021 | 0.906 ± 0.006 | 0.852 ± 0.017 | 0.84 ± 0.003 | 0.903 ± 0.007 | 0.106 ± 0.013 | 0.842 ± 0.024 | |
| PubChem | 0.98 ± 0.011 | 0.892 ± 0.034 | 0.928 ± 0.027 | 0.909 ± 0.013 | 0.847 ± 0.003 | 0.84 ± 0.015 | 0.902 ± 0.003 | 0.105 ± 0.005 | 0.844 ± 0.01 | |
| GEt-ADReCS | ECFP2 | 0.95 ± 0.024 | 0.852 ± 0.03 | 0.882 ± 0.032 | 0.878 ± 0.019 | 0.807 ± 0.027 | 0.803 ± 0.023 | 0.872 ± 0.015 | 0.188 ± 0.015 | 0.805 ± 0.015 |
| MACCS | 0.96 ± 0.014 | 0.842 ± 0.032 | 0.888 ± 0.022 | 0.877 ± 0.012 | 0.788 ± 0.029 | 0.798 ± 0.017 | 0.868 ± 0.01 | 0.168 ± 0.02 | 0.768 ± 0.042 | |
| PubChem | 0.966 ± 0.011 | 0.873 ± 0.029 | 0.908 ± 0.018 | 0.877 ± 0.013 | 0.813 ± 0.019 | 0.801 ± 0.019 | 0.863 ± 0.01 | 0.189 ± 0.019 | 0.808 ± 0.029 | |
| GEt-SIDER | ECFP2 | 0.982 ± 0.007 | 0.897 ± 0.024 | 0.934 ± 0.017 | 0.913 ± 0.008 | 0.849 ± 0.02 | 0.842 ± 0.009 | 0.907 ± 0.005 | 0.107 ± 0.013 | 0.85 ± 0.023 |
| MACCS | 0.989 ± 0.005 | 0.917 ± 0.014 | 0.951 ± 0.01 | 0.91 ± 0.006 | 0.86 ± 0.01 | 0.842 ± 0.008 | 0.905 ± 0.007 | 0.11 ± 0.006 | 0.859 ± 0.012 | |
| PubChem | 0.99 ± 0.005 | 0.918 ± 0.016 | 0.951 ± 0.012 | 0.91 ± 0.005 | 0.865 ± 0.013 | 0.837 ± 0.011 | 0.907 ± 0.002 | 0.114 ± 0.008 | 0.864 ± 0.013 | |
| Dataset | Train | Test | External Validation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | Precision | ACC | AUC | Precision | ACC | AUC | Precision | ACC | |
| GEn-ADReCS | 0.953 ± 0.02 | 0.851 ± 0.033 | 0.887 ± 0.03 | 0.878 ± 0.018 | 0.804 ± 0.02 | 0.808 ± 0.017 | 0.864 ± 0.019 | 0.184 ± 0.018 | 0.789 ± 0.025 |
| GEn-SIDER | 0.984 ± 0.012 | 0.906 ± 0.034 | 0.939 ± 0.026 | 0.904 ± 0.009 | 0.855 ± 0.016 | 0.836 ± 0.006 | 0.904 ± 0.005 | 0.109 ± 0.01 | 0.849 ± 0.018 |
| GEt-ADReCS | 0.937 ± 0.026 | 0.823 ± 0.043 | 0.864 ± 0.032 | 0.871 ± 0.022 | 0.785 ± 0.035 | 0.8 ± 0.018 | 0.858 ± 0.026 | 0.167 ± 0.027 | 0.765 ± 0.052 |
| GEt-SIDER | 0.98 ± 0.017 | 0.897 ± 0.023 | 0.933 ± 0.026 | 0.911 ± 0.012 | 0.849 ± 0.012 | 0.843 ± 0.011 | 0.902 ± 0.01 | 0.103 ± 0.008 | 0.845 ± 0.016 |
| Dataset | Train | Test | External Validation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | Precision | ACC | AUC | Precision | ACC | AUC | Precision | ACC | |
| GEn-SIDER | 0.978 ± 0.016 | 0.892 ± 0.028 | 0.927 ± 0.027 | 0.906 ± 0.009 | 0.852 ± 0.015 | 0.839 ± 0.007 | 0.903 ± 0.007 | 0.108 ± 0.01 | 0.848 ± 0.018 |
| GEn-ADReCS | 0.953 ± 0.027 | 0.851 ± 0.04 | 0.888 ± 0.037 | 0.875 ± 0.019 | 0.801 ± 0.023 | 0.805 ± 0.016 | 0.863 ± 0.02 | 0.182 ± 0.019 | 0.785 ± 0.03 |
| GEt-SIDER | 0.982 ± 0.012 | 0.903 ± 0.033 | 0.937 ± 0.022 | 0.914 ± 0.01 | 0.856 ± 0.025 | 0.844 ± 0.007 | 0.904 ± 0.01 | 0.11 ± 0.018 | 0.854 ± 0.029 |
| GEt-ADReCS | 0.951 ± 0.018 | 0.847 ± 0.031 | 0.886 ± 0.026 | 0.878 ± 0.014 | 0.803 ± 0.02 | 0.81 ± 0.012 | 0.864 ± 0.015 | 0.179 ± 0.017 | 0.788 ± 0.026 |
| Dataset | Train | Test | External Validation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | Precision | ACC | AUC | Precision | ACC | AUC | Precision | ACC | |
| GEn-SIDER | 0.802 ± 0.011 | 0.719 ± 0.009 | 0.716 ± 0.008 | 0.649 ± 0.023 | 0.659 ± 0.03 | 0.608 ± 0.02 | 0.634 ± 0.007 | 0.038 ± 0.003 | 0.755 ± 0.032 |
| GEn-ADReCS | 0.877 ± 0.016 | 0.753 ± 0.024 | 0.775 ± 0.015 | 0.716 ± 0.01 | 0.667 ± 0.014 | 0.643 ± 0.01 | 0.7 ± 0.009 | 0.103 ± 0.005 | 0.712 ± 0.033 |
| GEt-SIDER | 0.798 ± 0.011 | 0.718 ± 0.012 | 0.713 ± 0.008 | 0.651 ± 0.019 | 0.67 ± 0.034 | 0.606 ± 0.016 | 0.638 ± 0.008 | 0.039 ± 0.003 | 0.771 ± 0.041 |
| GEt-ADReCS | 0.879 ± 0.019 | 0.755 ± 0.018 | 0.777 ± 0.015 | 0.717 ± 0.012 | 0.67 ± 0.017 | 0.642 ± 0.01 | 0.701 ± 0.01 | 0.1 ± 0.006 | 0.712 ± 0.037 |
| Dataset | Method | AUC | Precision | ACC |
|---|---|---|---|---|
| GEn-SIDER | DrugClust | 0.6044 ± 0.0111 | 0.1877 ± 0.0177 | 0.9644 ± 0.003 |
| SCCA | 0.9131 ± 0.0002 | 0.0392 ± 0.0008 | 0.4814 ± 0.0121 | |
| BiMPADR | 0.902 ± 0.003 | 0.105 ± 0.005 | 0.844 ± 0.01 | |
| GEn-Adrecs | DrugClust | 0.615 ± 0.0169 | 0.2415 ± 0.0243 | 0.913 ± 0.0086 |
| SCCA | 0.8891 ± 0.0005 | 0.1091 ± 0.0014 | 0.5468 ± 0.0066 | |
| BiMPADR | 0.874 ± 0.007 | 0.193 ± 0.012 | 0.802 ± 0.019 | |
| GEt-SIDER | DrugClust | 0.6335 ± 0.0169 | 0.2087 ± 0.0283 | 0.9662 ± 0.0017 |
| SCCA | 0.9137 ± 0.0005 | 0.0381 ± 0.0009 | 0.4736 ± 0.0128 | |
| BiMPADR | 0.907 ± 0.002 | 0.114 ± 0.008 | 0.864 ± 0.013 | |
| GEt-Adrecs | DrugClust | 0.651 ± 0.0202 | 0.2498 ± 0.0195 | 0.9125 ± 0.0042 |
| SCCA | 0.8897 ± 0.0004 | 0.1061 ± 0.0005 | 0.5485 ± 0.0022 | |
| BiMPADR | 0.863 ± 0.01 | 0.189 ± 0.019 | 0.808 ± 0.029 |
| Drug Name | ADR Name | Pred Value | NCT Number |
|---|---|---|---|
| BMS-986158 | Transaminases increased | 0.998 | NCT02419417 |
| Rhabdomyolysis | 0.998 | ||
| Dermatitis | 0.997 | NCT02419417 | |
| Intermittent claudication | 0.997 | NCT02419417 | |
| Hypertriglyceridaemia | 0.997 | ||
| Hyperglycaemia | 0.996 | NCT02419417 | |
| Hyperlipidaemia | 0.996 | ||
| Upper respiratory tract infection | 0.996 | NCT02419417 | |
| Influenza-like illness | 0.996 | NCT02419417 | |
| Gastroenteritis | 0.995 | NCT02419417 |
| Drug Name | ADR Name | Pred Value | NCT Number |
|---|---|---|---|
| BMS-986158 | Anemia | 0.991 | NCT02419417 |
| Leukopenia | 0.983 | NCT02419417 | |
| Lymphopenia | 0.689 | NCT02419417 | |
| Neutropenia | 0.985 | NCT02419417 | |
| Thrombocytopenia | 0.991 | NCT02419417 |
| Dataset | Number of Drugs | Number of ADRs | Number of Drugs in External Dataset |
|---|---|---|---|
| GEn-SIDER | 656 | 3616 | 774 |
| GEn-ADReCS | 656 | 751 | 774 |
| GEt-SIDER | 766 | 3695 | 664 |
| GEt-ADReCS | 766 | 762 | 664 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zhang, L.; Wang, L.; Ji, J.; He, J.; Zheng, X.; Cao, L.; Li, K. BiMPADR: A Deep Learning Framework for Predicting Adverse Drug Reactions in New Drugs. Molecules 2024, 29, 1784. https://doi.org/10.3390/molecules29081784
Li S, Zhang L, Wang L, Ji J, He J, Zheng X, Cao L, Li K. BiMPADR: A Deep Learning Framework for Predicting Adverse Drug Reactions in New Drugs. Molecules. 2024; 29(8):1784. https://doi.org/10.3390/molecules29081784
Chicago/Turabian StyleLi, Shuang, Liuchao Zhang, Liuying Wang, Jianxin Ji, Jia He, Xiaohan Zheng, Lei Cao, and Kang Li. 2024. "BiMPADR: A Deep Learning Framework for Predicting Adverse Drug Reactions in New Drugs" Molecules 29, no. 8: 1784. https://doi.org/10.3390/molecules29081784