
Anticancer Effects of Abietane Diterpene 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus grandidentatus and Its Semi-Synthetic Analogs: An In Silico Computational Approach

 ,
,  ,
,  ,
,  , , , ,
, , , ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
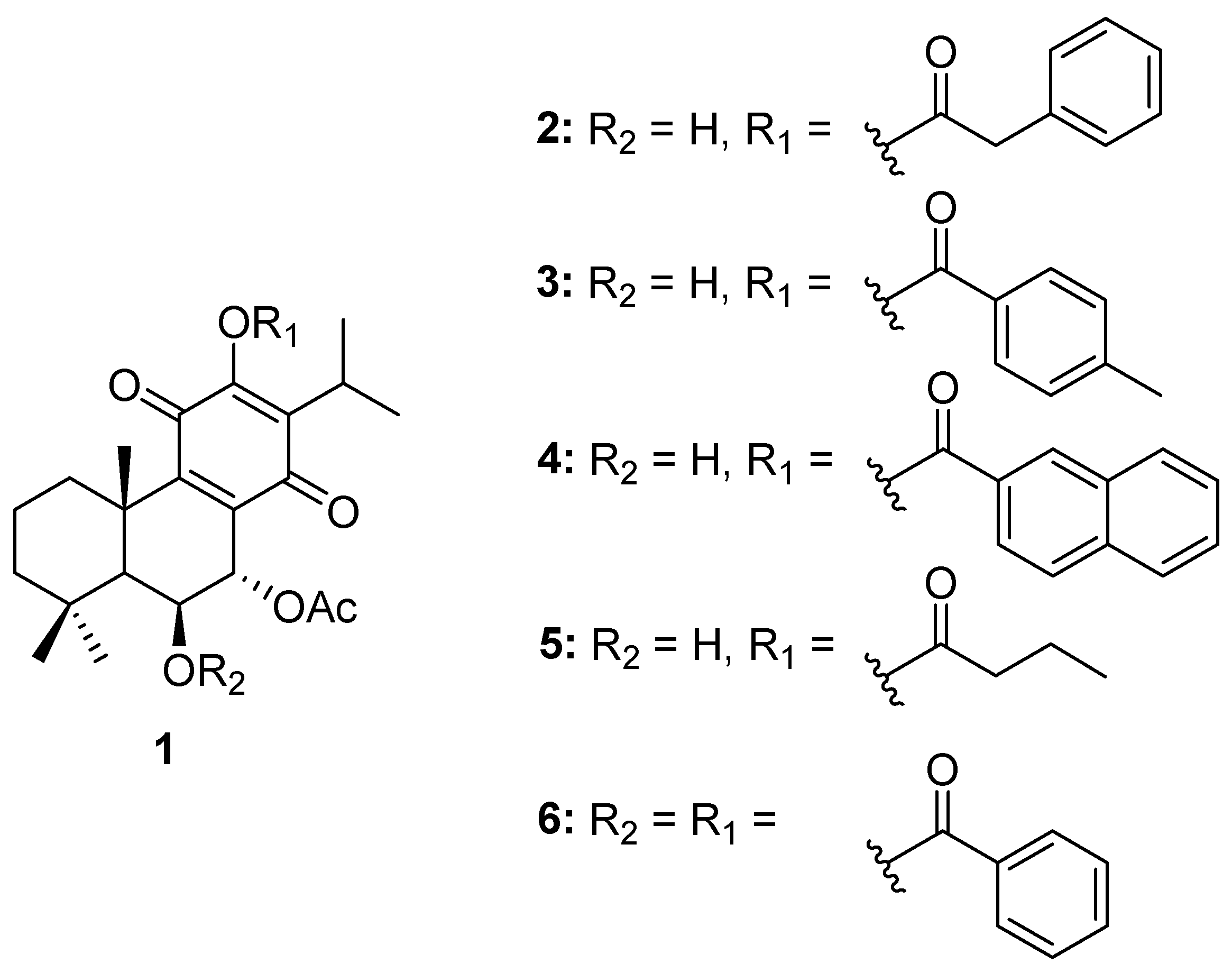
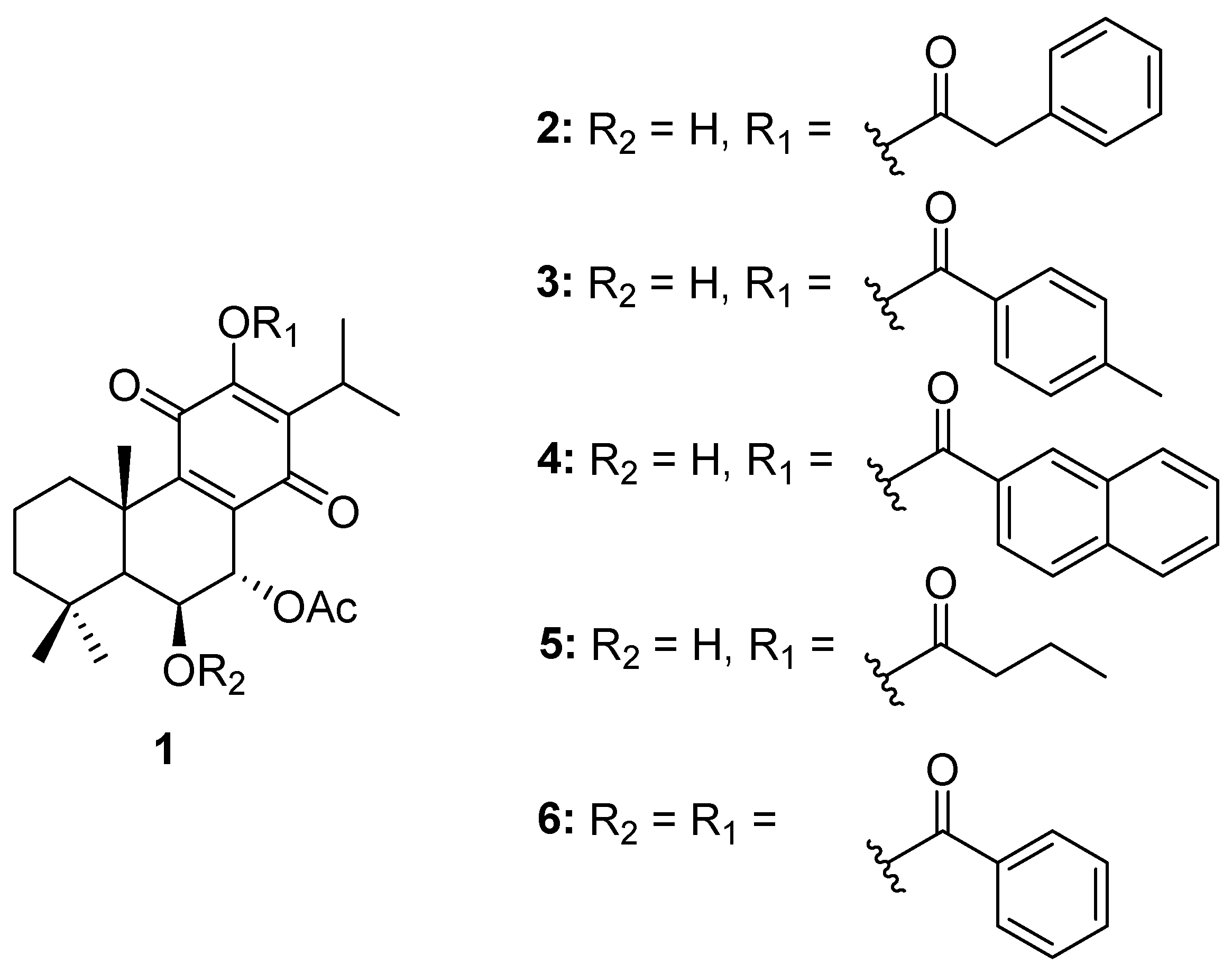
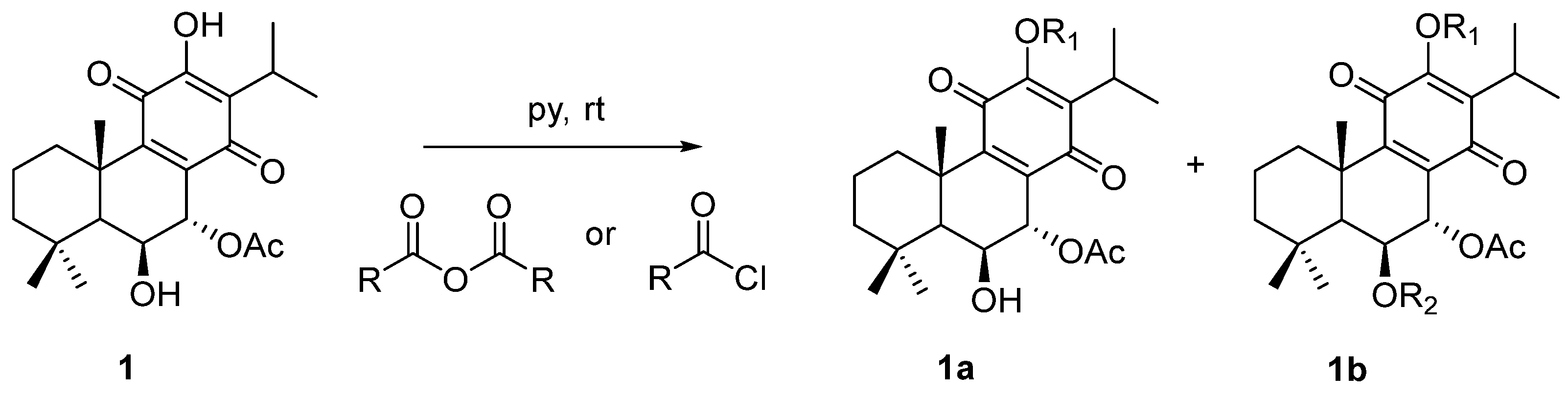
2.1. Compounds
2.2. ADMET and Drug-Likeness Analysis Results
2.3. Toxicity Prediction Results
2.4. Antineoplastic and Anticarcinogenic Activity Results
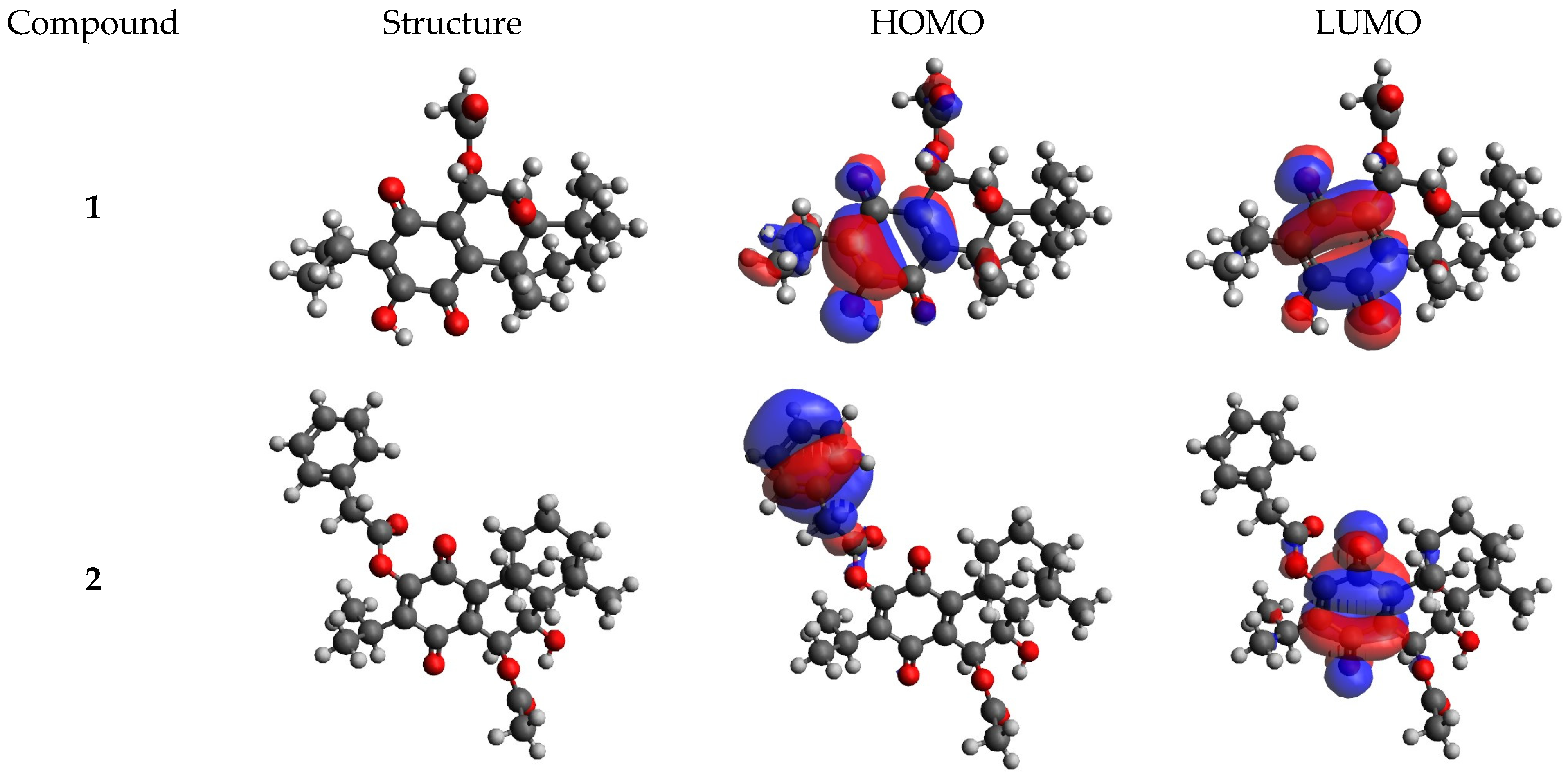
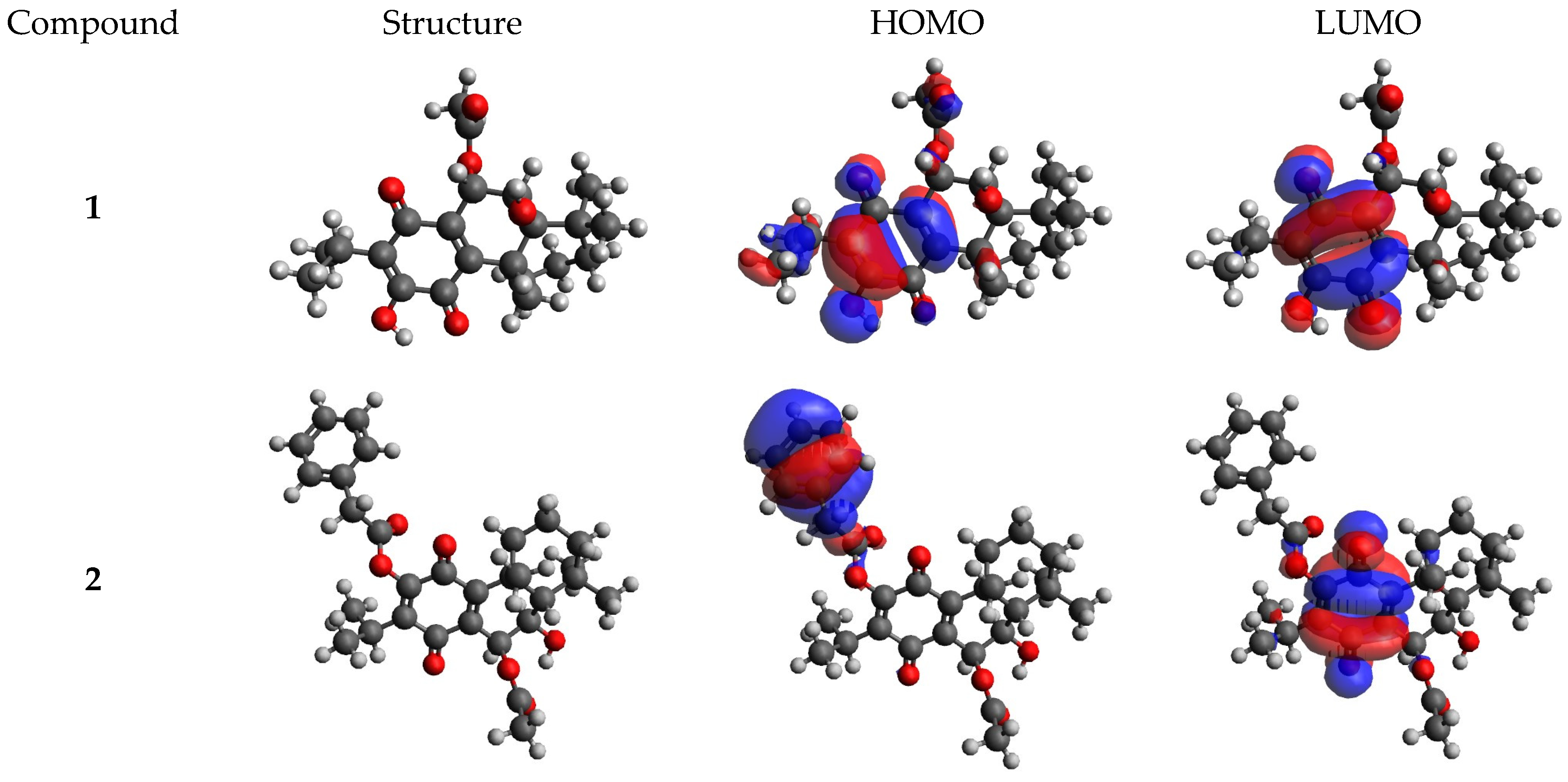
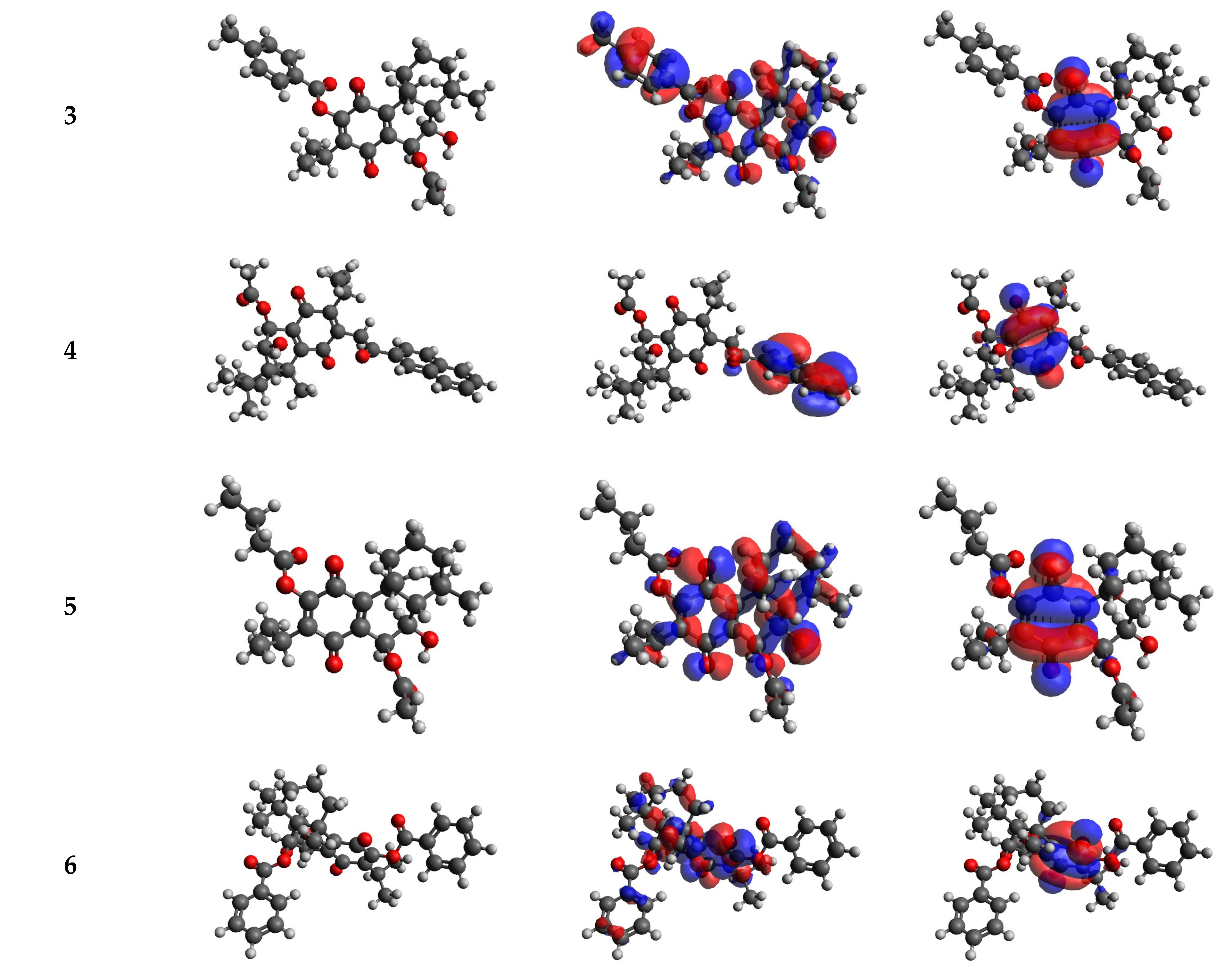
2.5. DFT Calculations Results
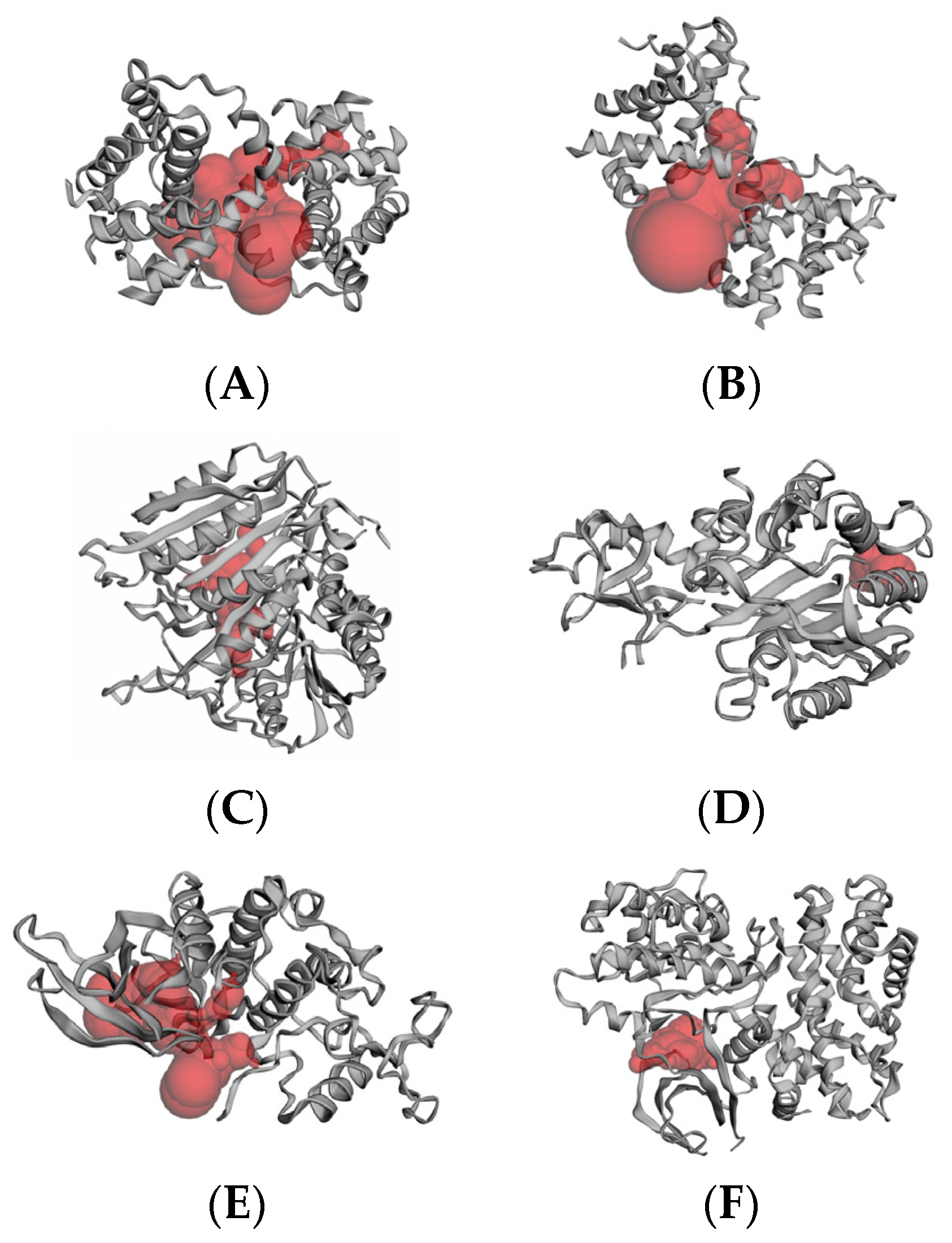
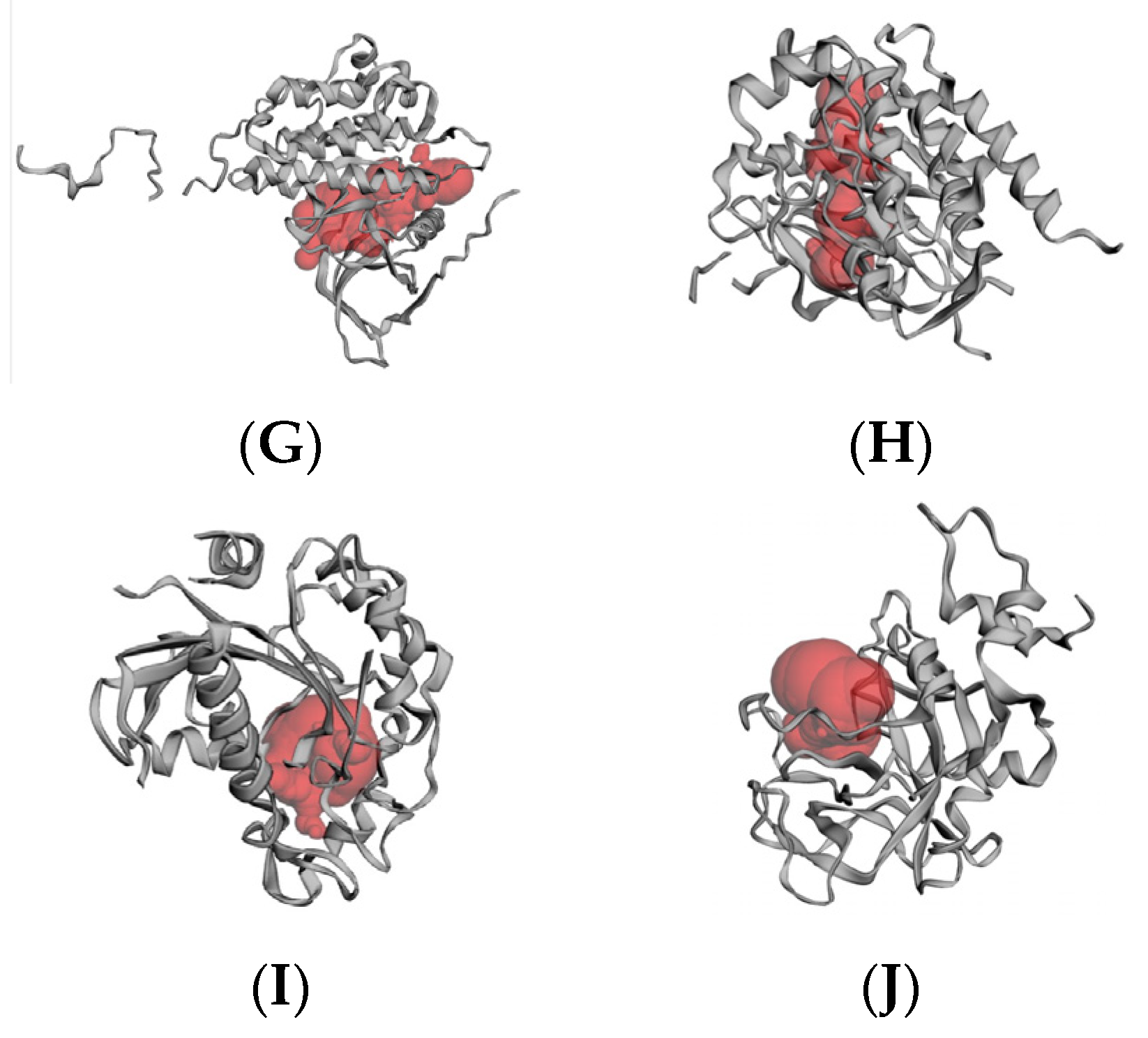
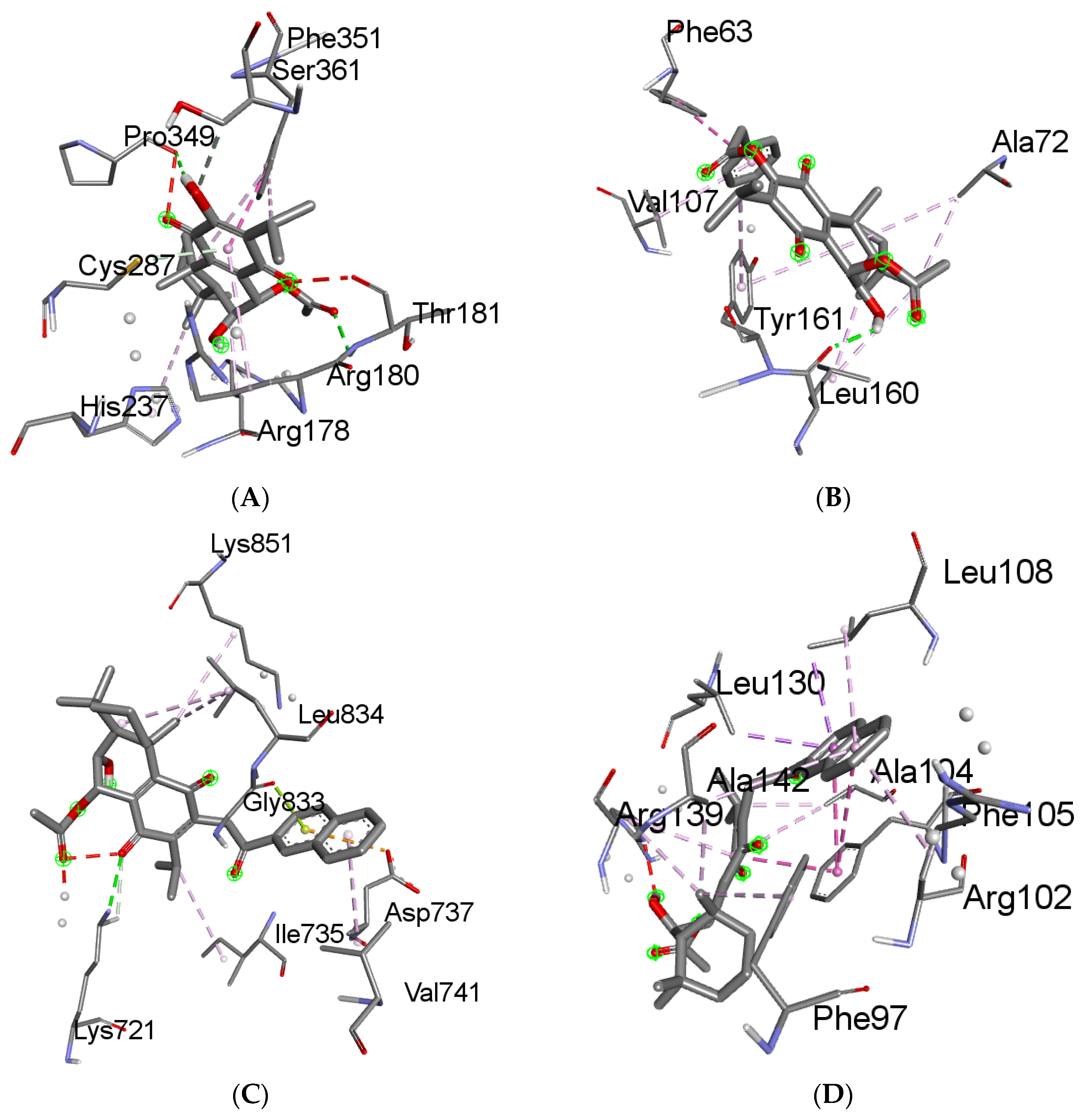
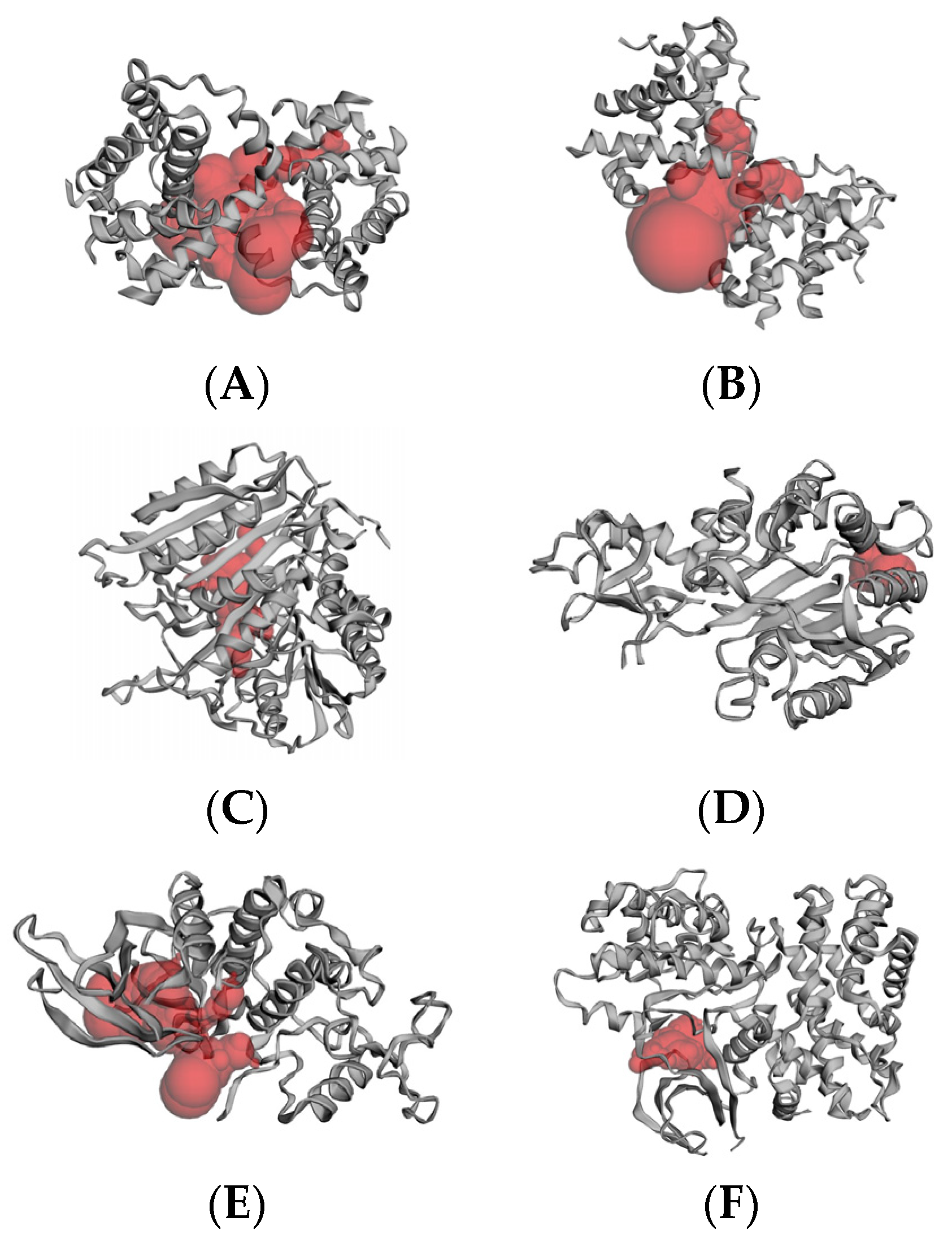
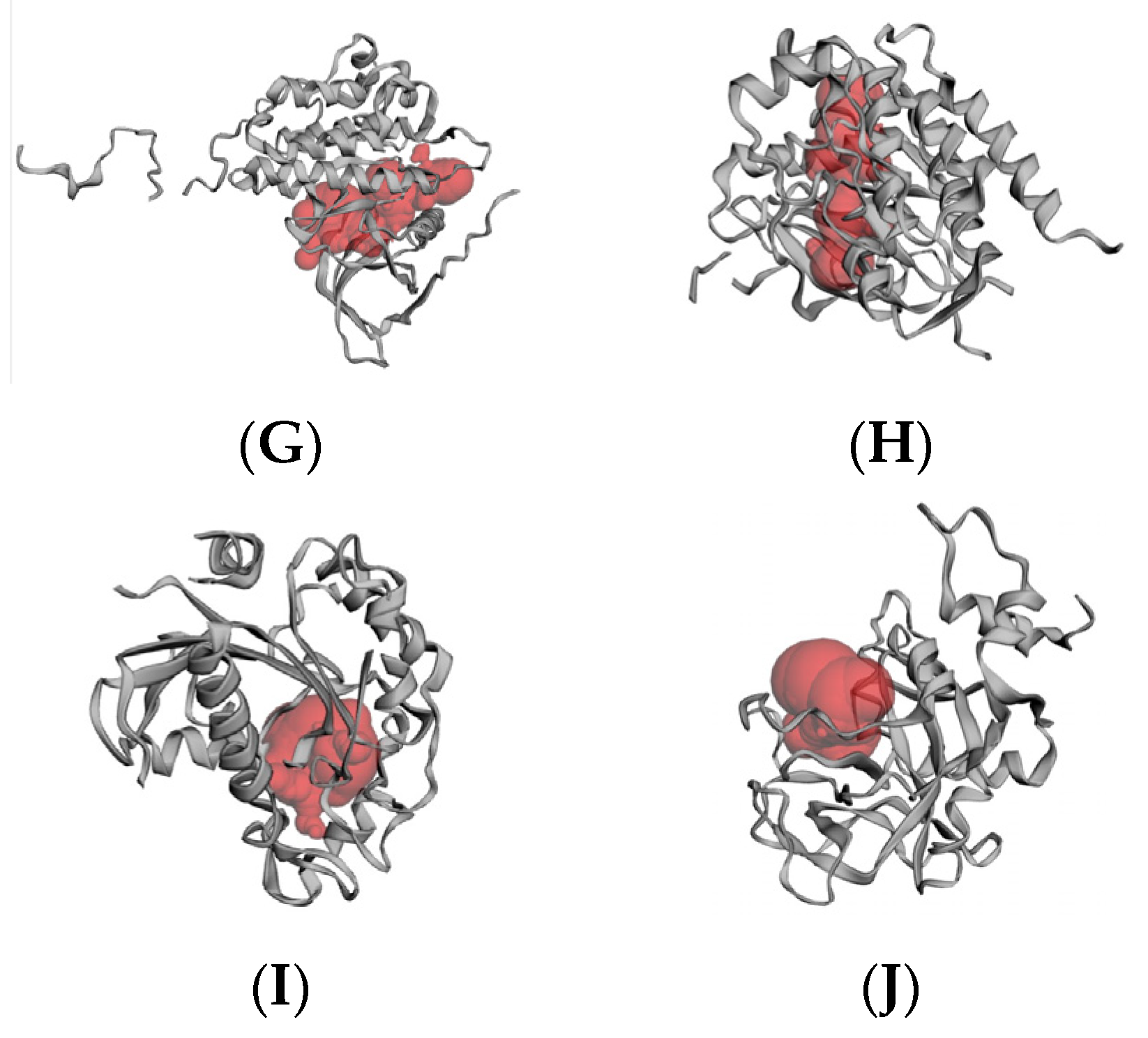
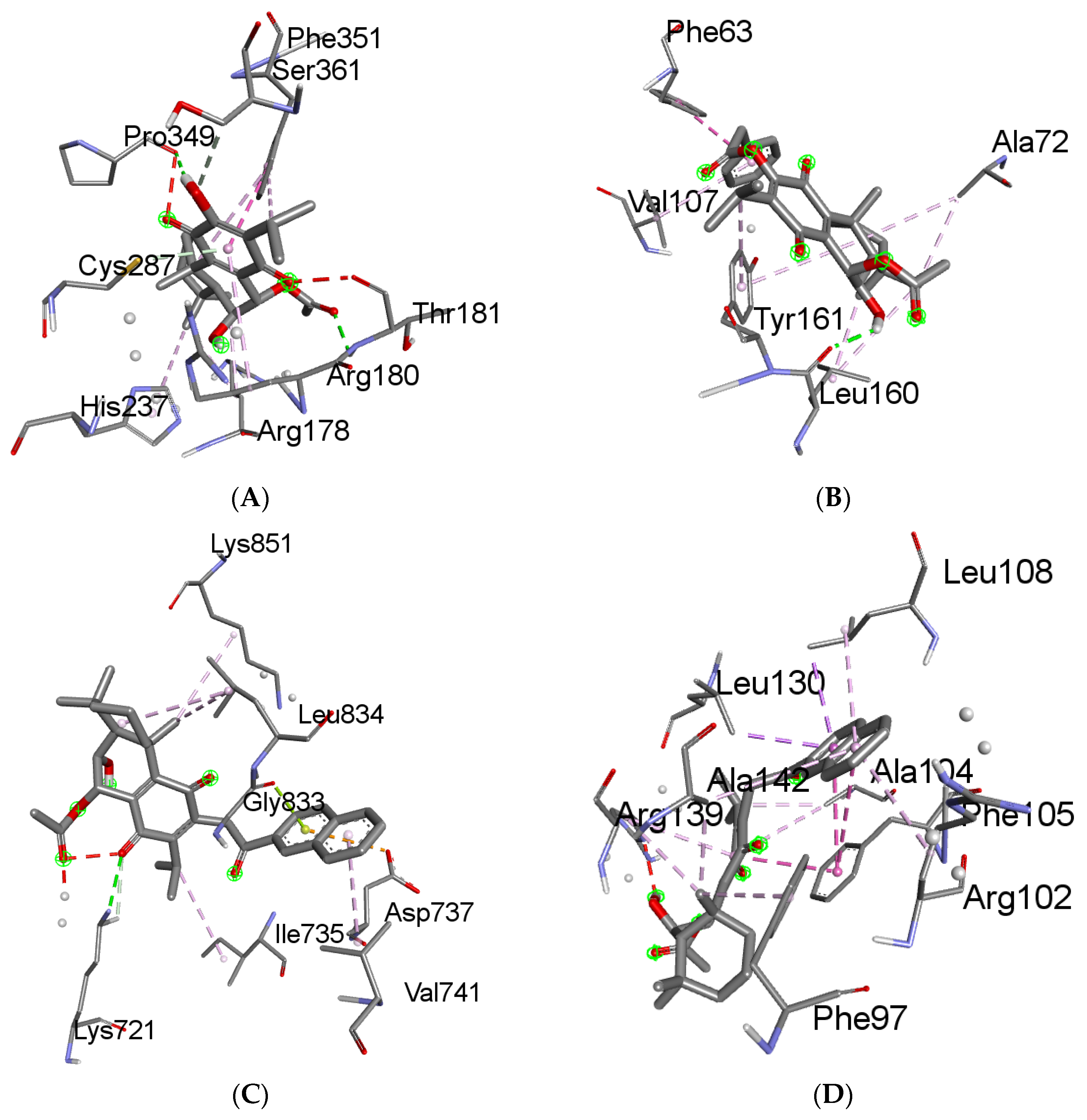
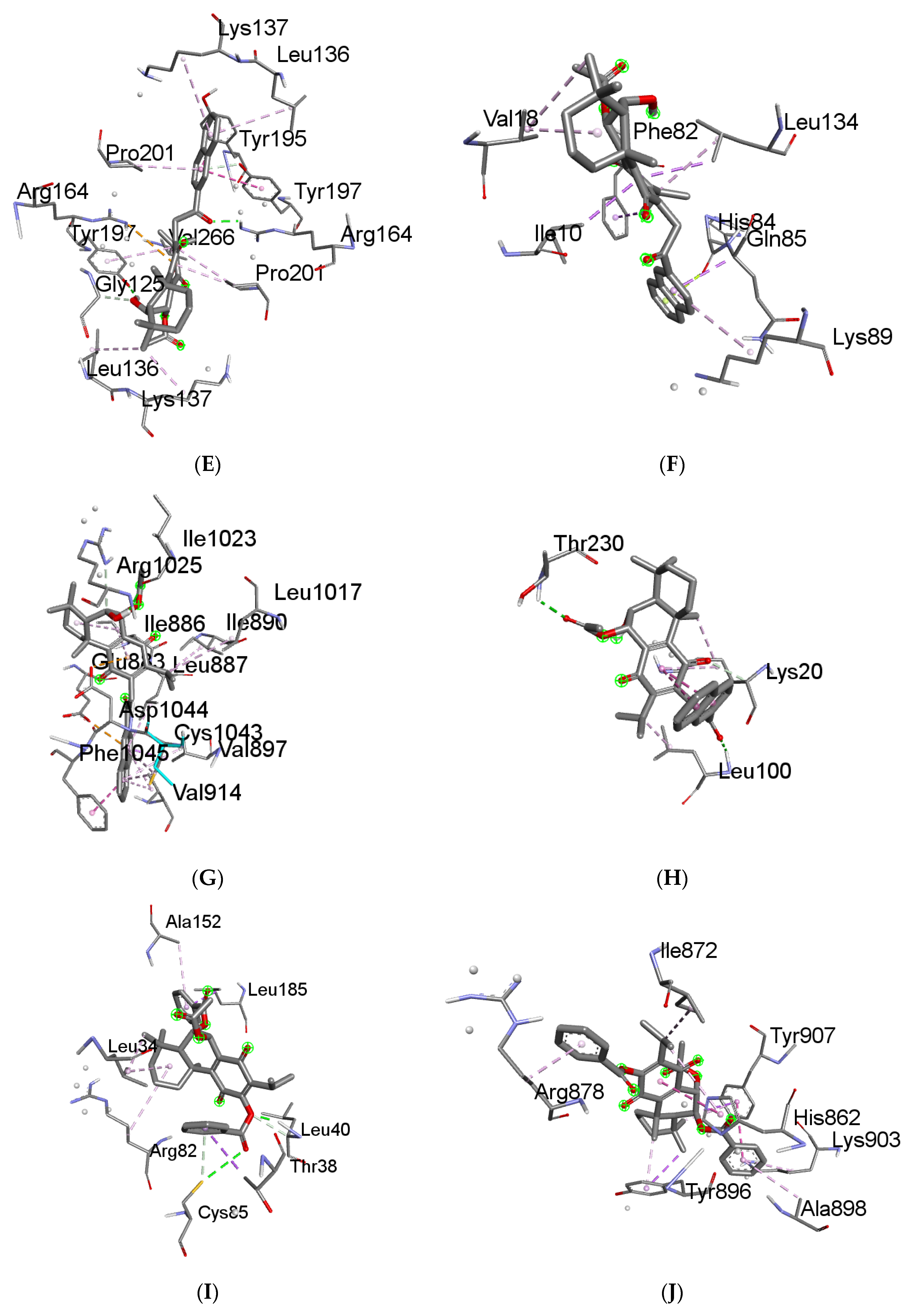
2.6. Molecular Docking Results
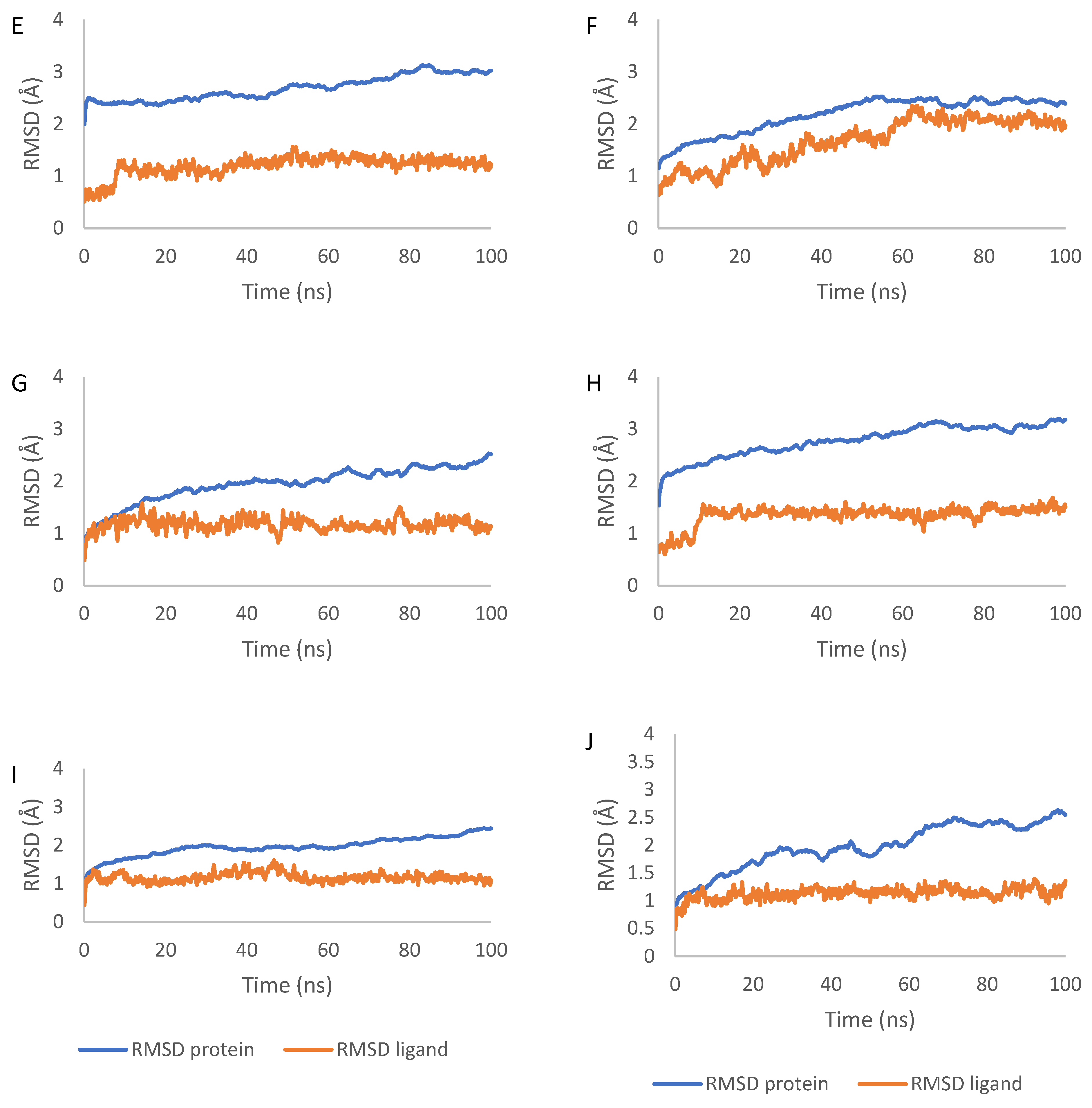
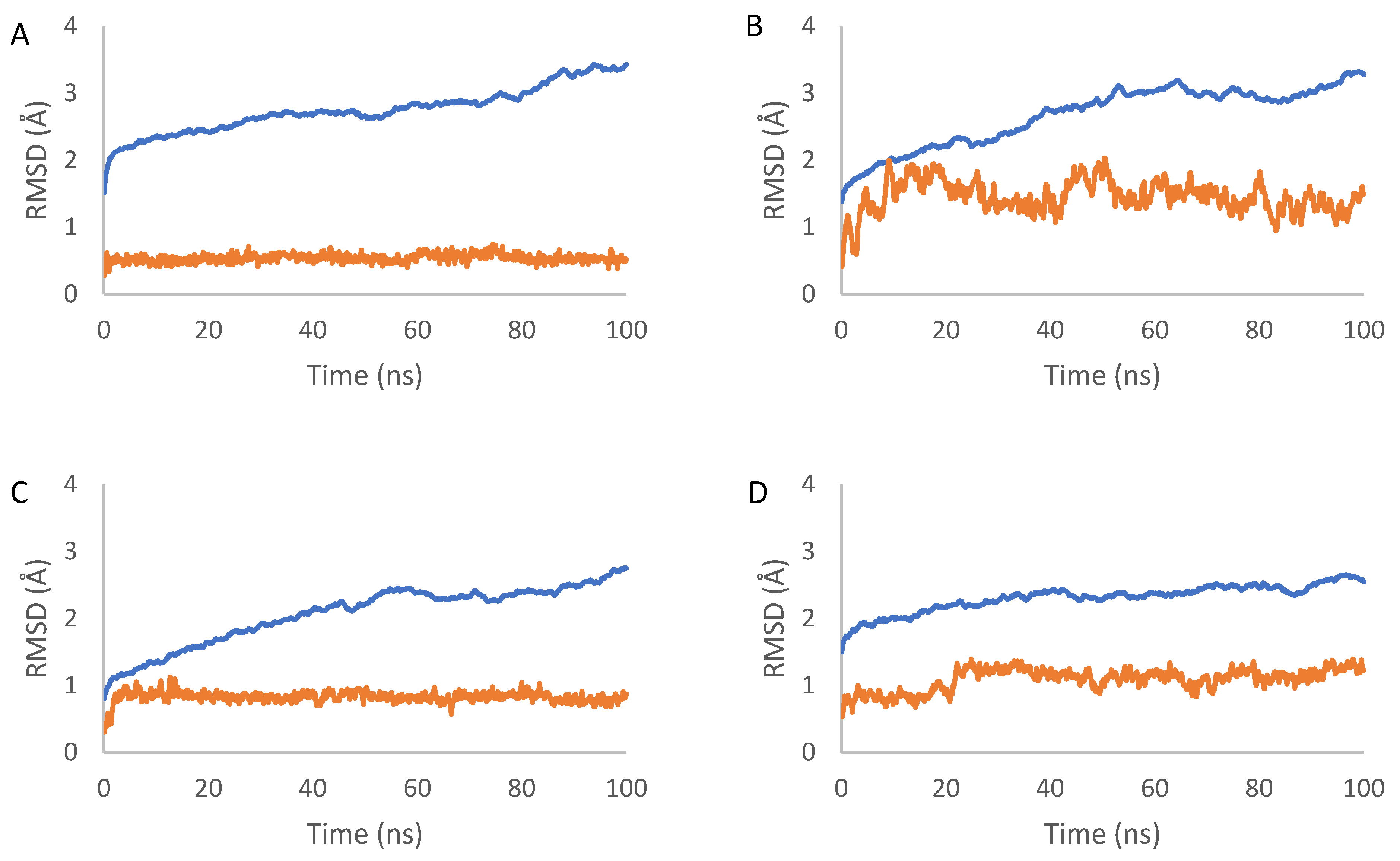
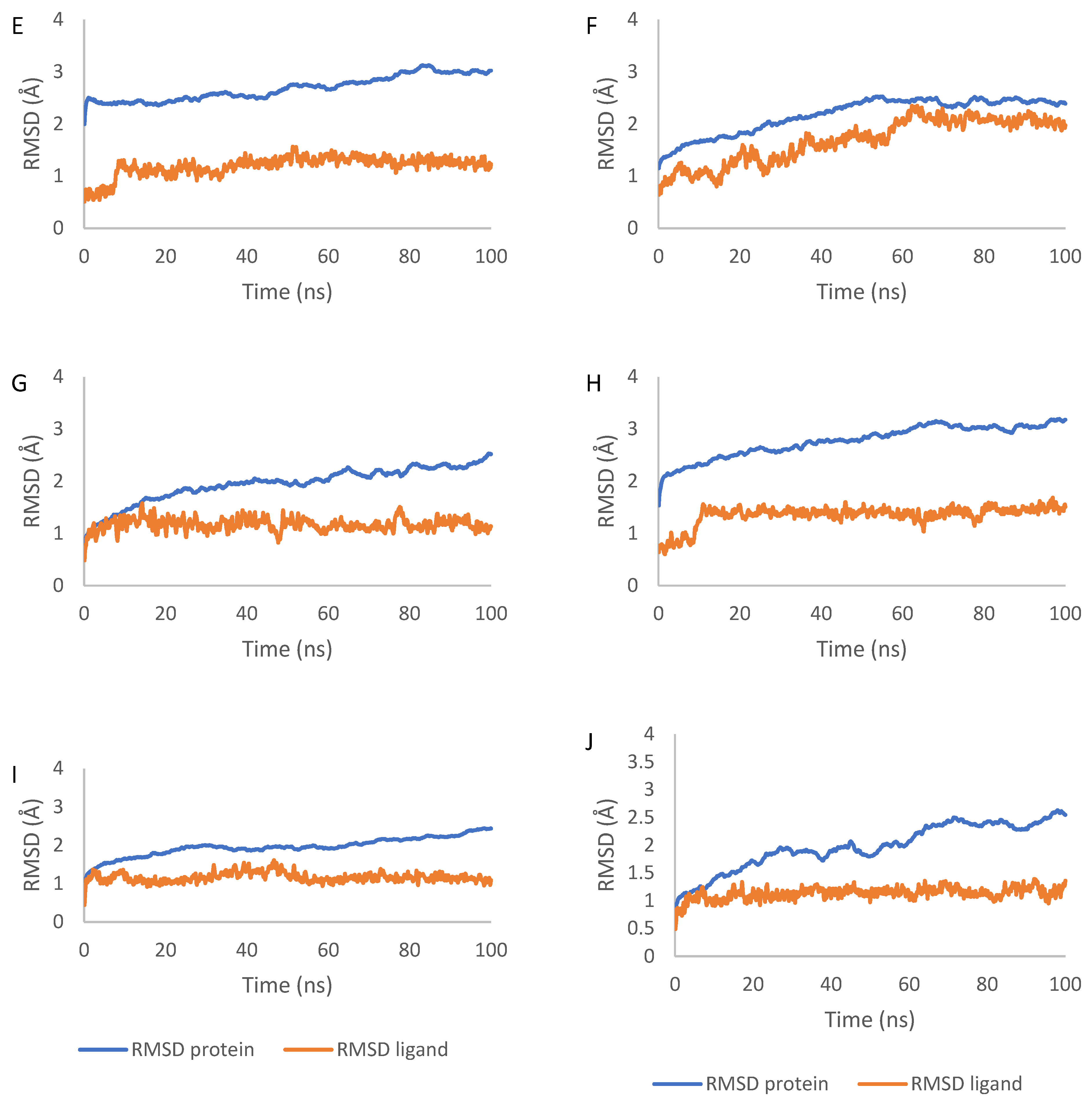
2.7. Molecular Dynamics Simulation Results
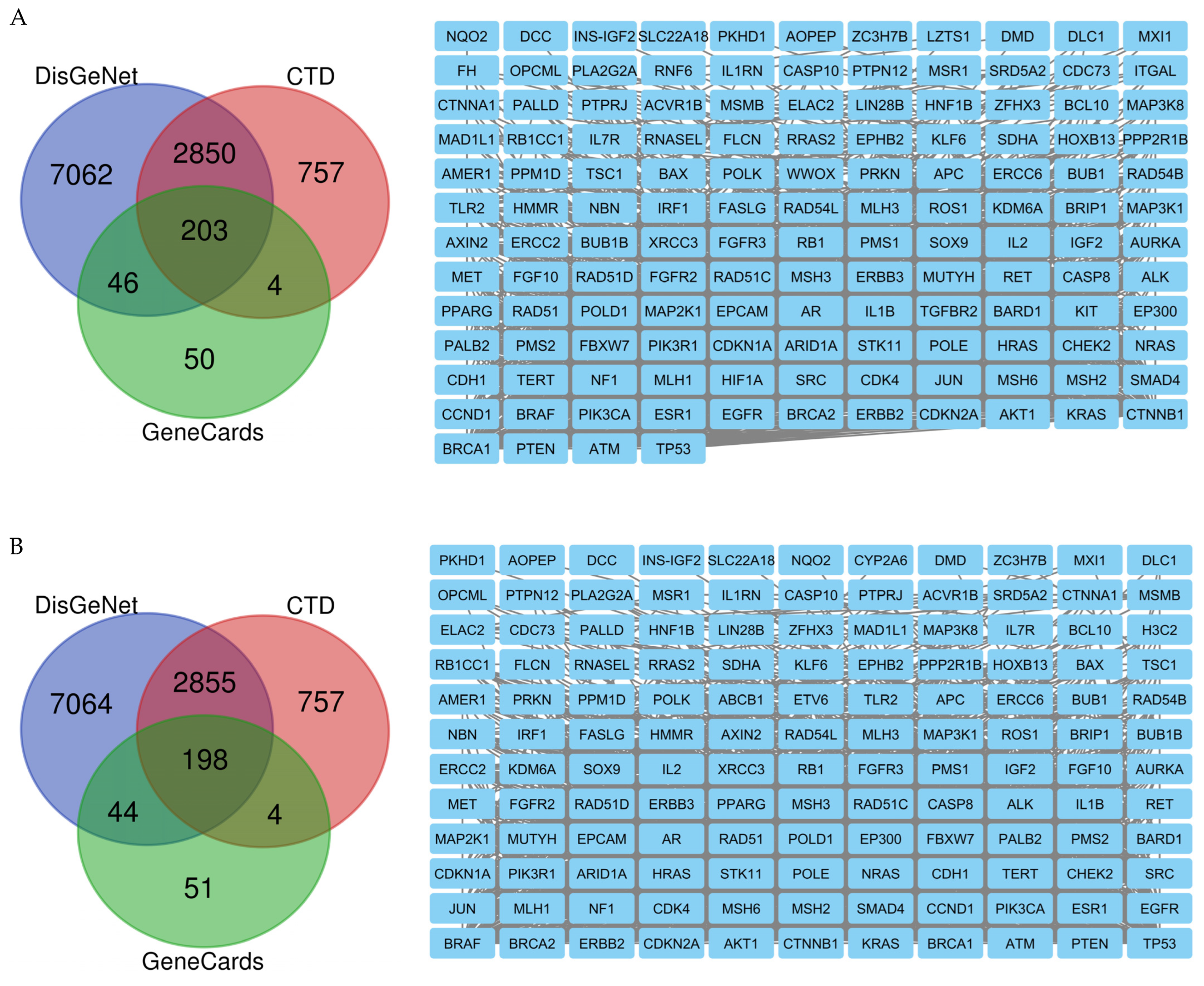
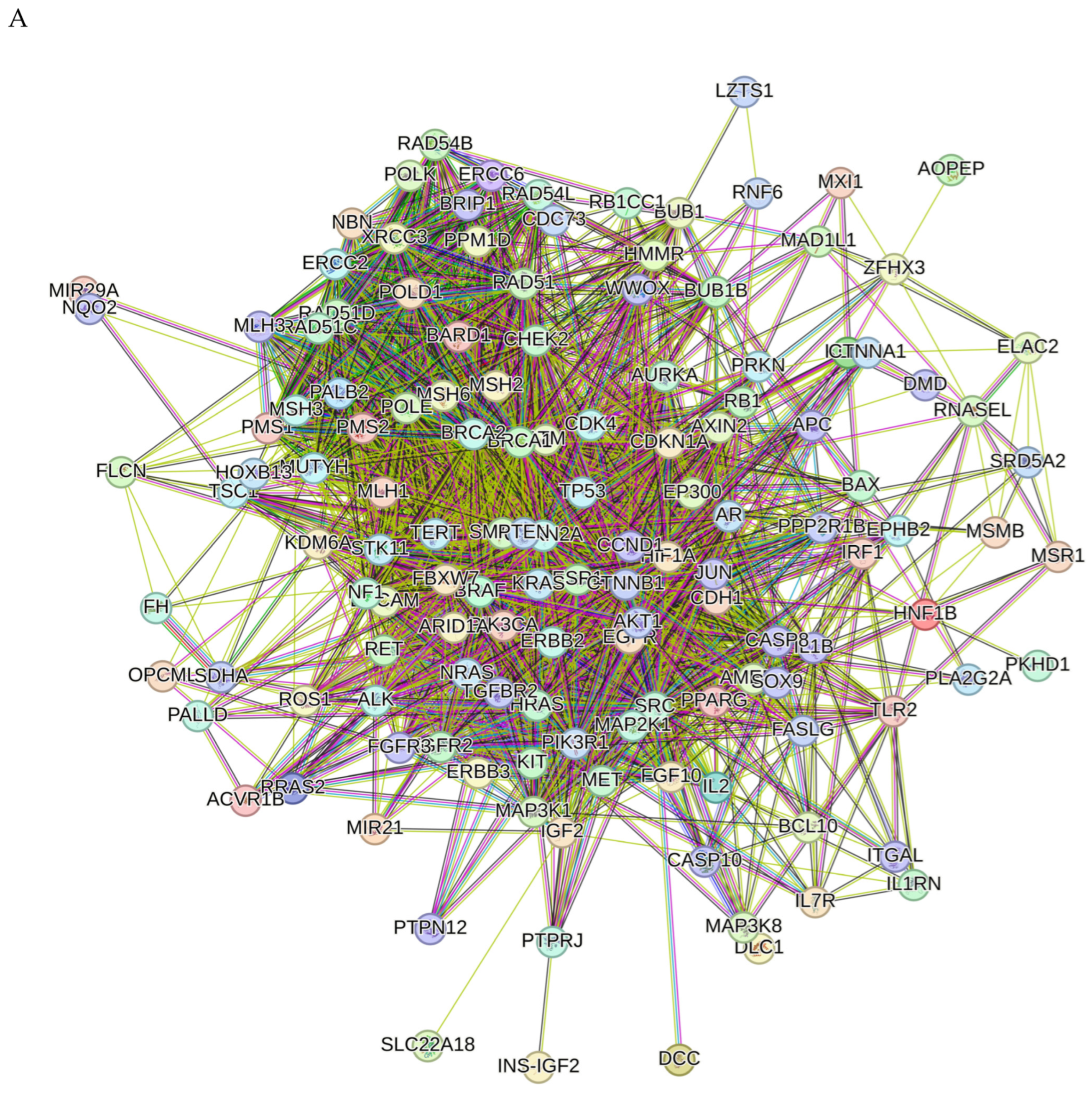
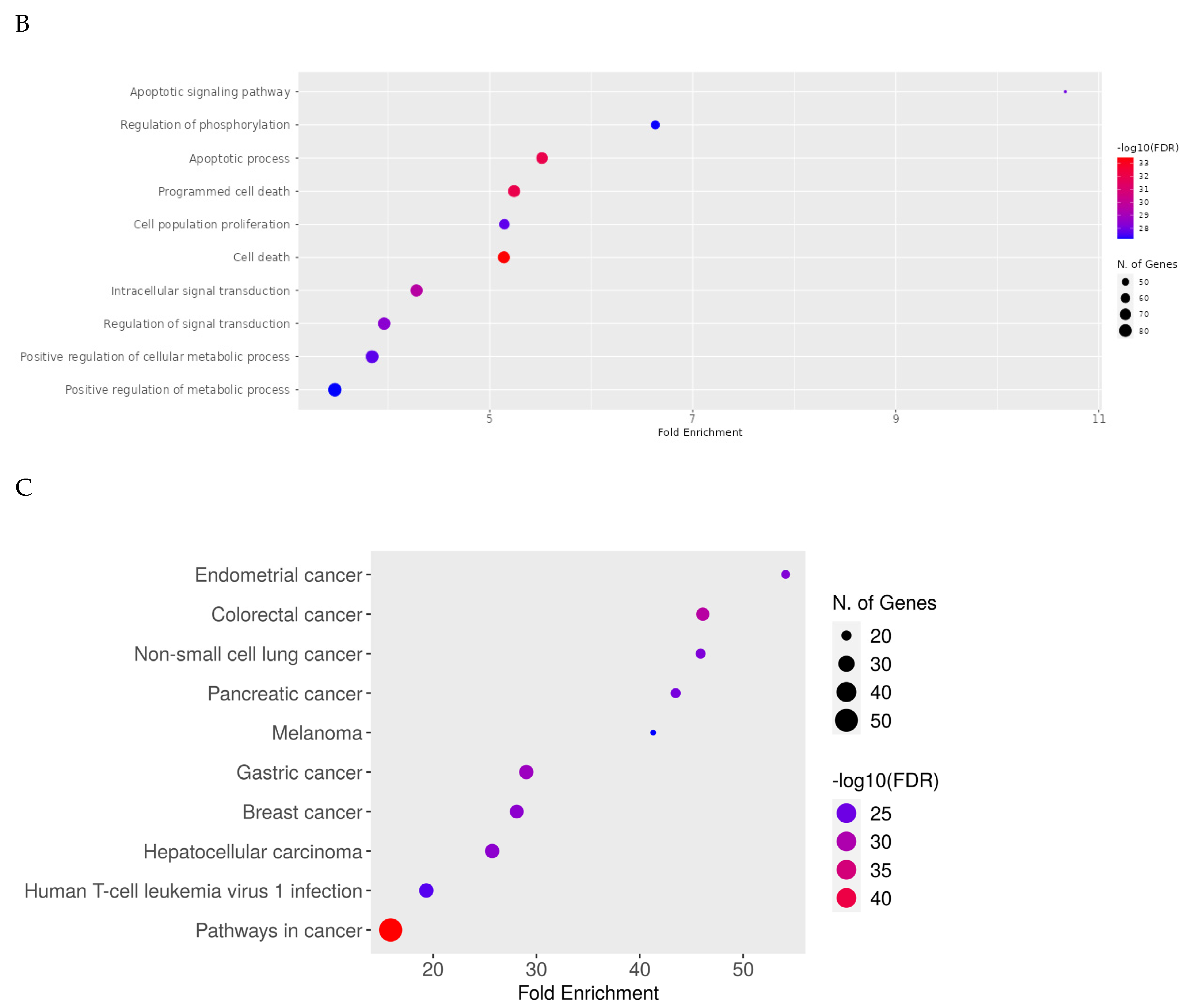
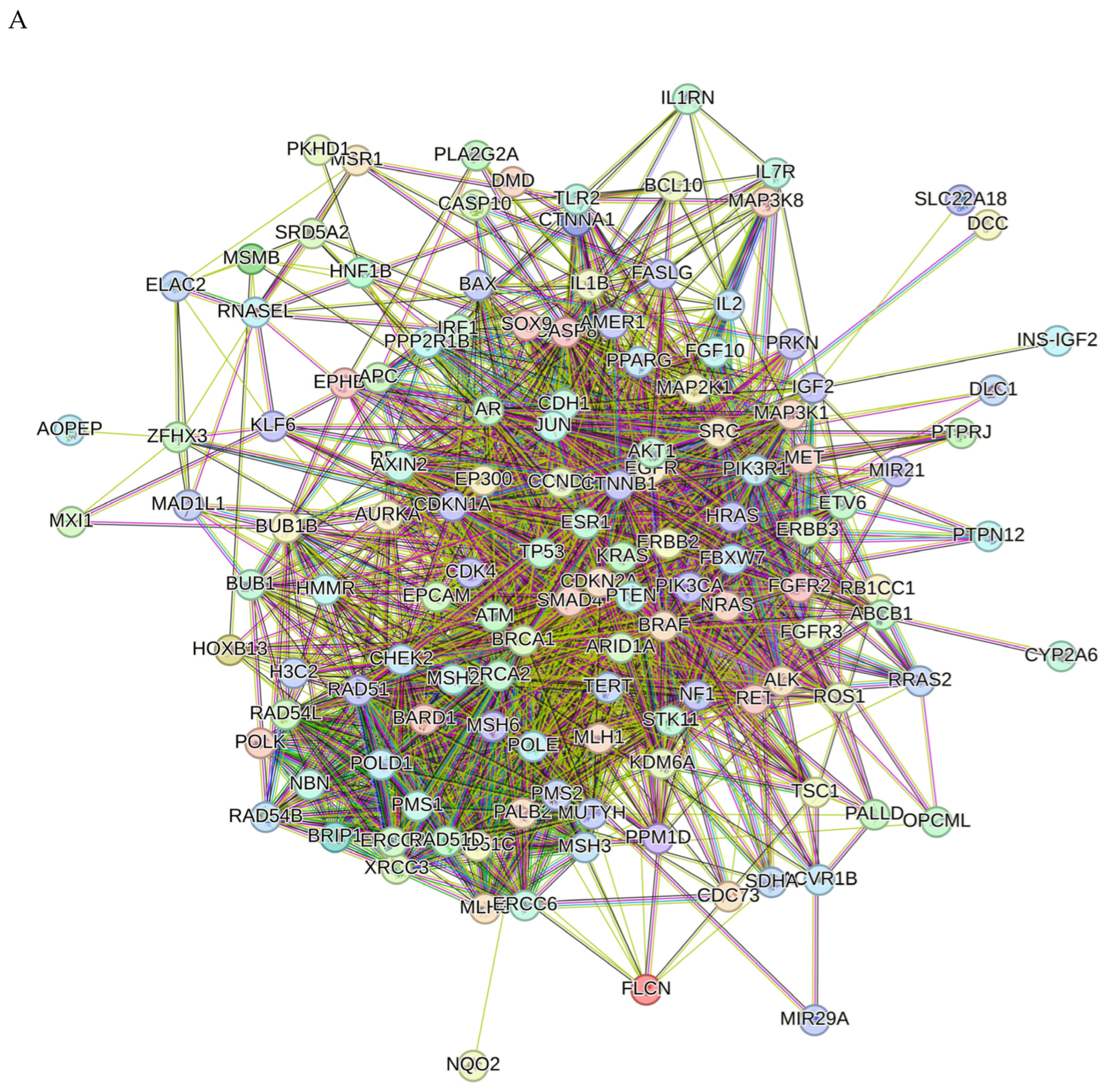
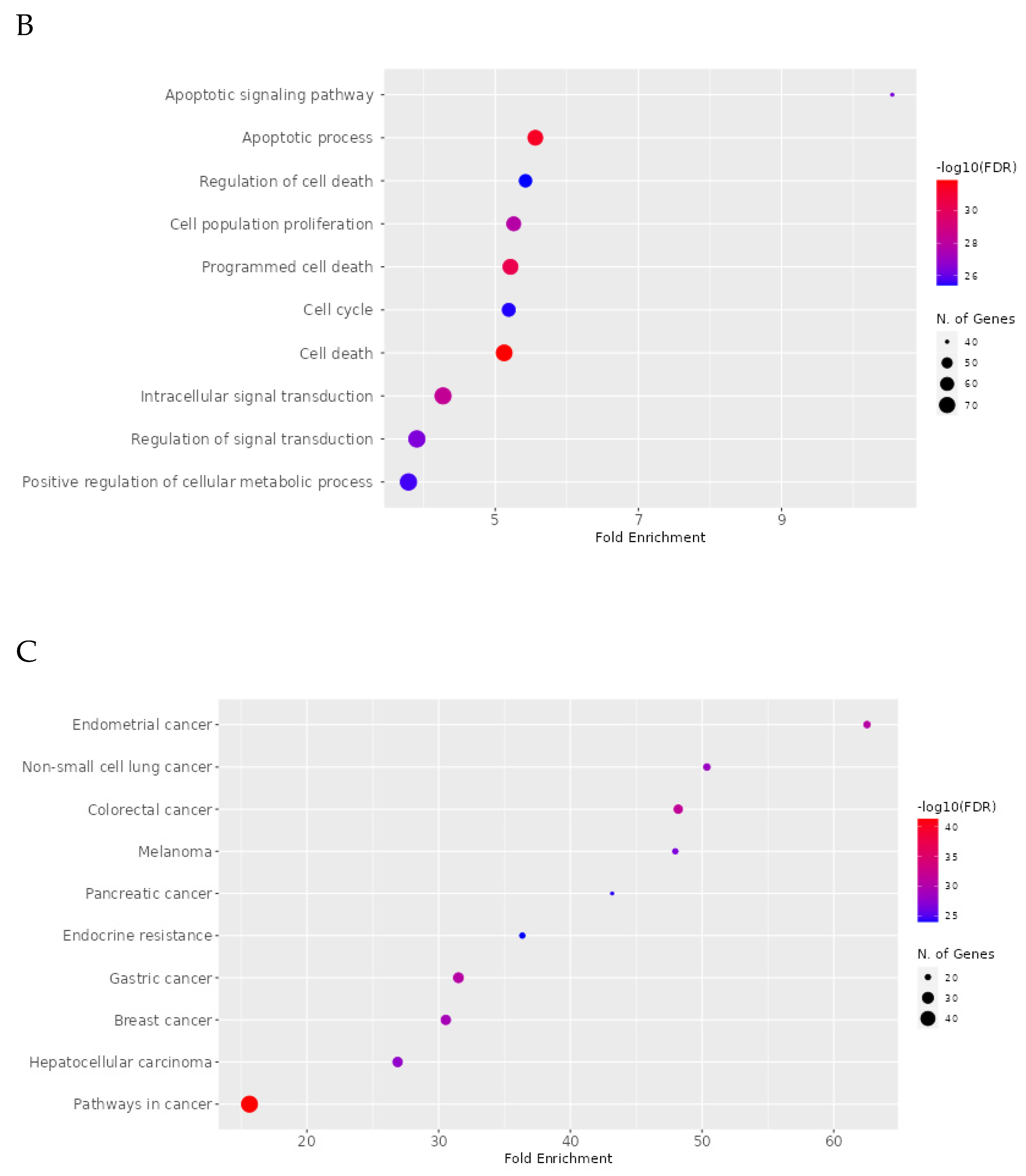
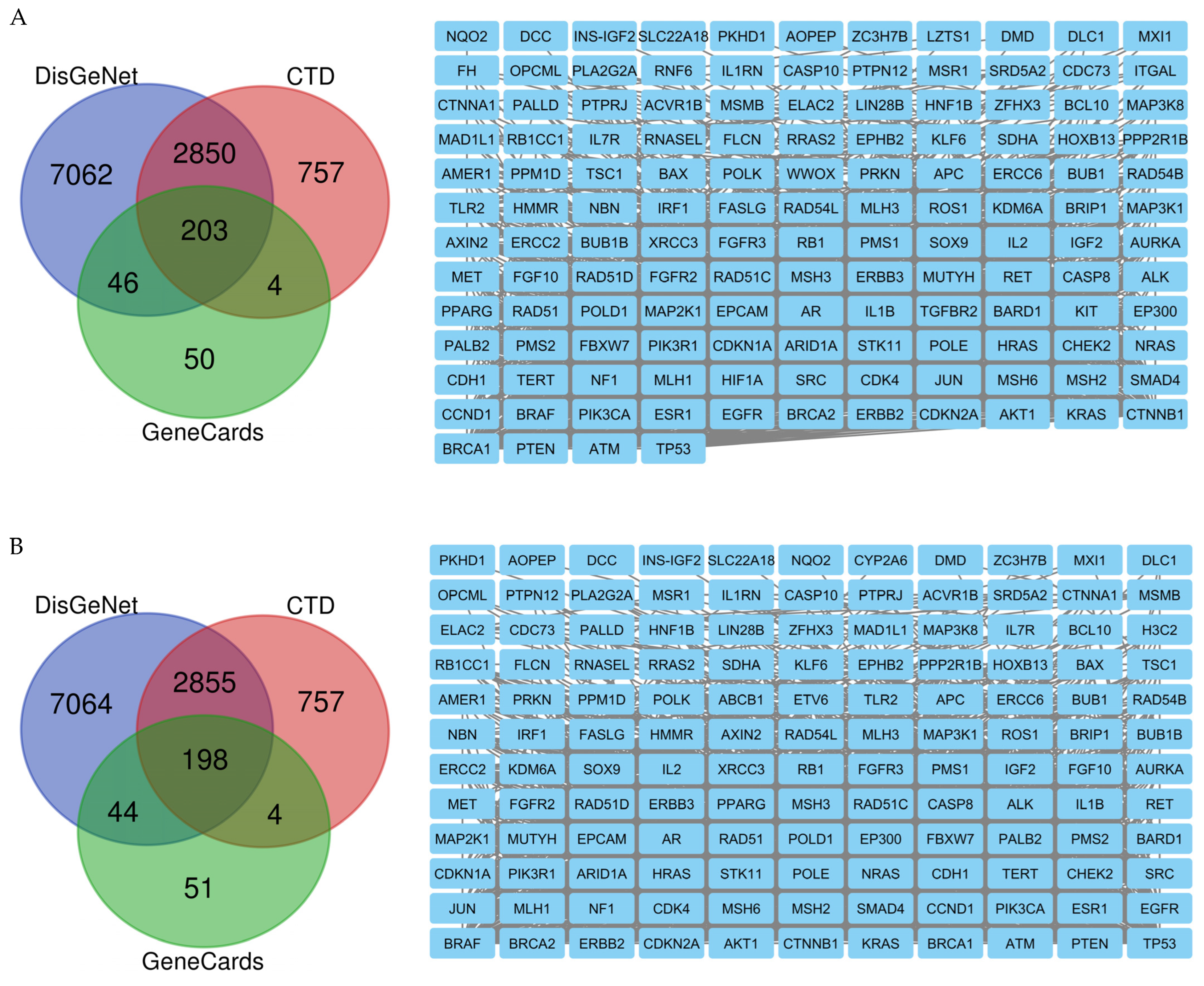
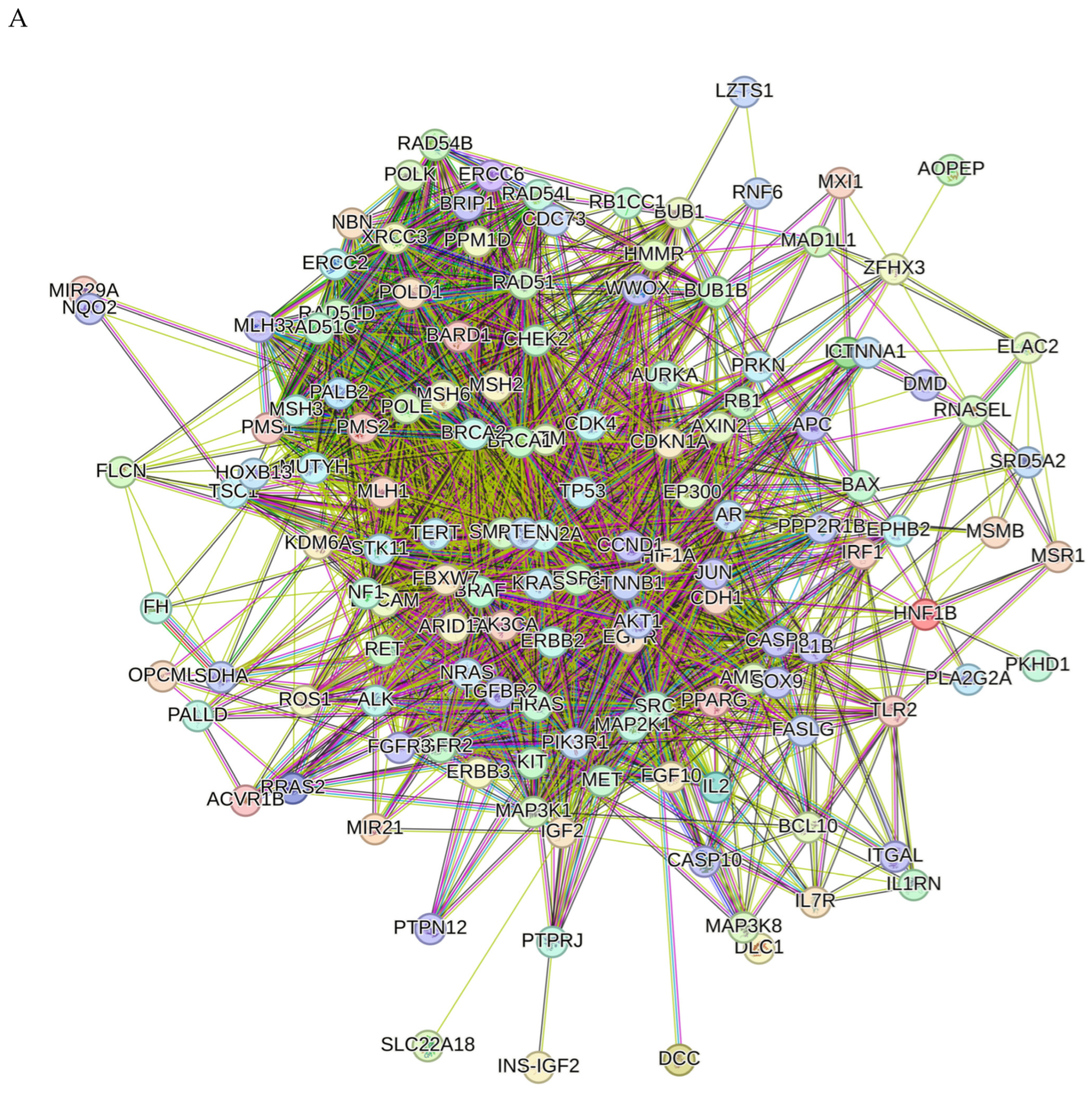
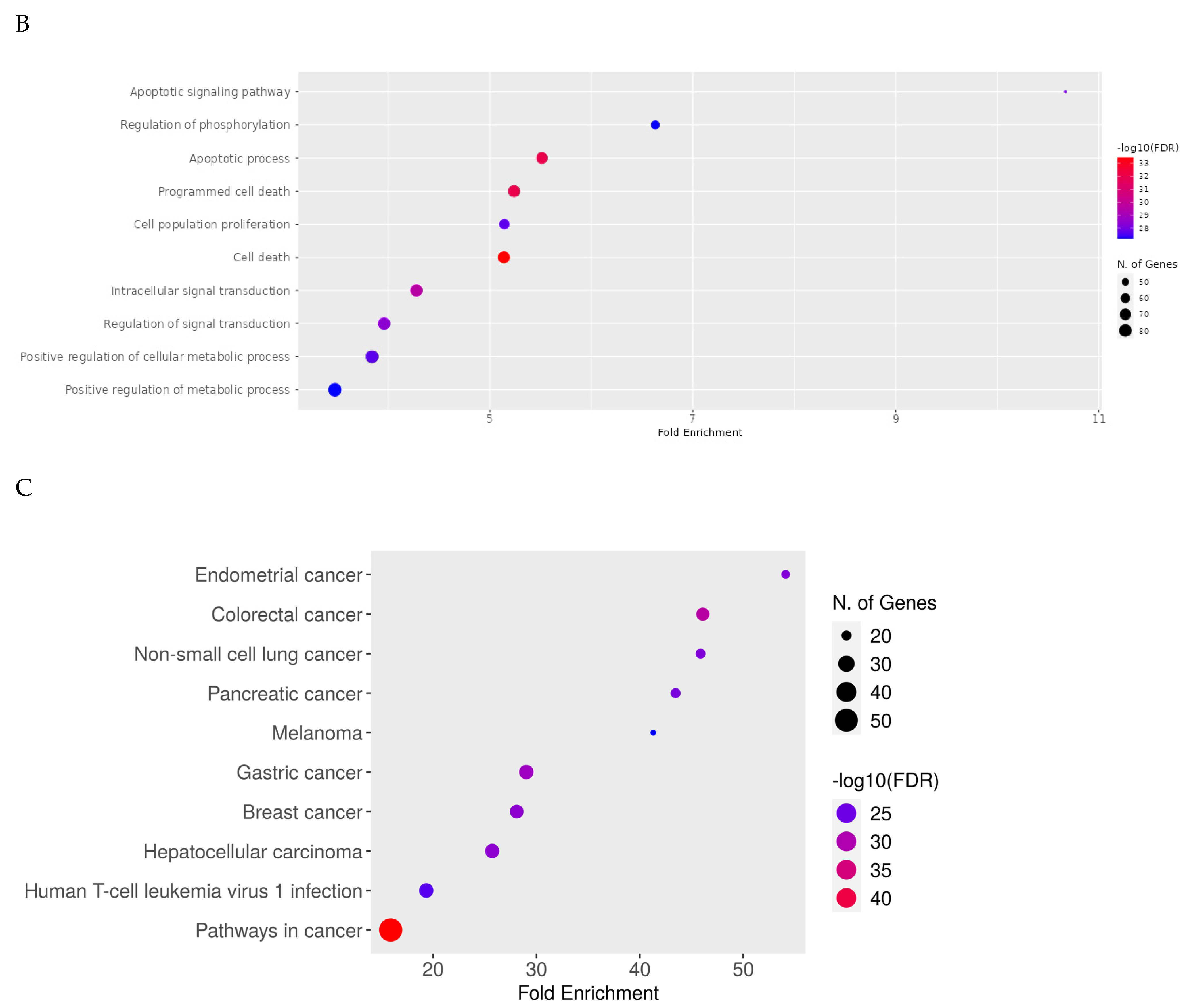
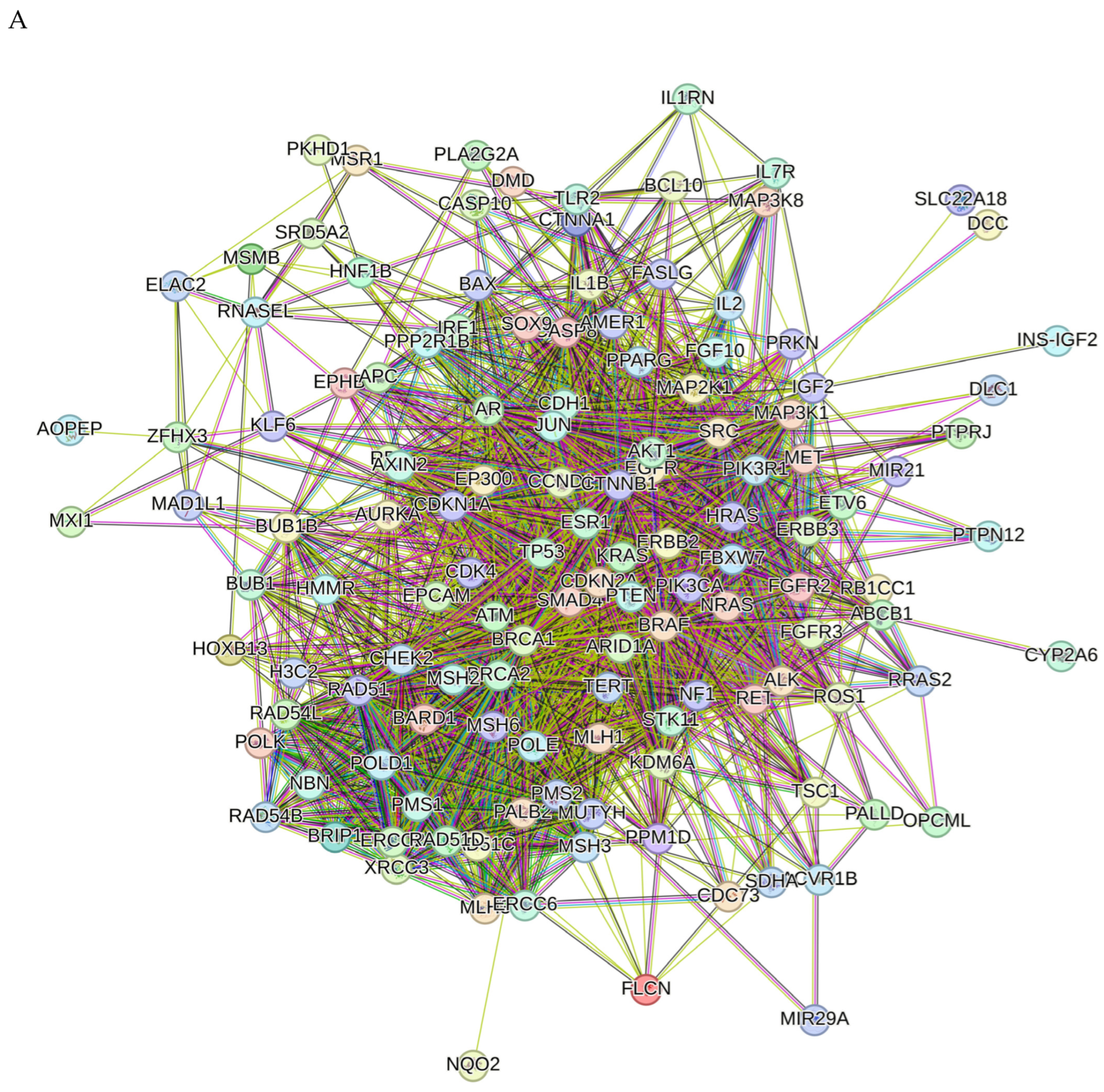
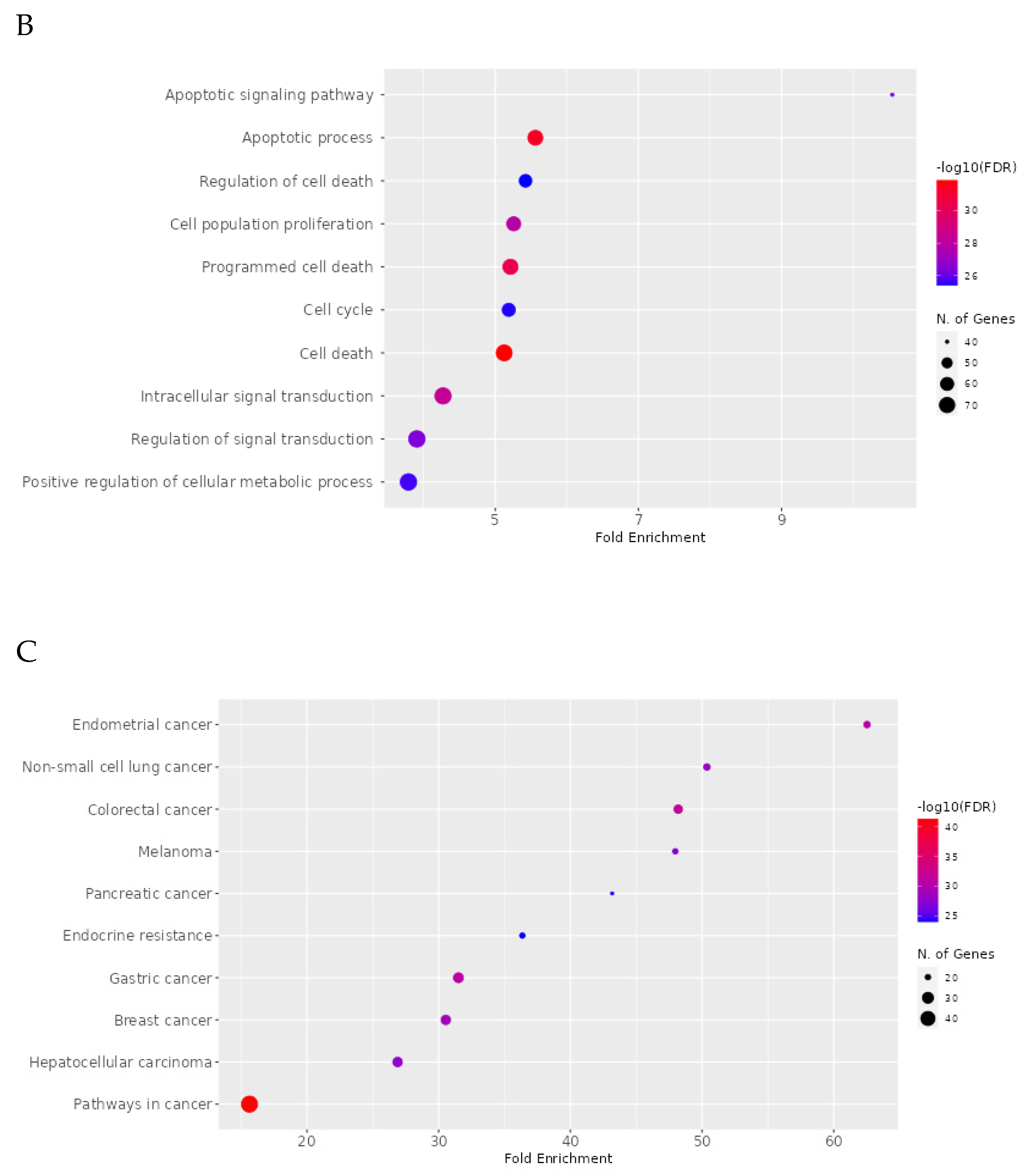
2.8. Network Pharmacology Results
3. Discussion
4. Materials and Methods
4.1. Compounds
4.1.1. General Experimental Procedures
4.1.2. Plant Material
4.1.3. Extraction and Isolation
- 7α-acetoxy-6β-hydroxyroyleanone (Roy) (1): 1H-NMR (300 MHz, Chloroform-d, ppm): δ 7.22 (s, 1H, 12-OH), 5.66 (dd, J = 2.2, 0.7 Hz, 1H, H-7β), 4.31 (s, 1H, H-6α), 3.16 (sept, J = 7.1 Hz, 1H, H-15), 2.63 (d, J = 12.8 Hz, 1H, H-1β), 2.04 (s, 3H, 7α-OAc), 1.89–1.78 (m, 1H, H-2β), 1.61 (s, 3H, Me-20), 1.55–1.46* (m, 2H, H-2α and H-3β), 1.33 (s, 1H, H-5α), 1.23* (s, 3H, Me-19), 1.22* (d, J = 7.1 Hz, 3H, Me-17), 1.21* (s, 1H, H-3α +), 1.20* (d, J = 7.1 Hz, 3H Me-16), 1.18* (s, 1H H-1α +), 0.94 (s, 3H, Me-18). *Overlapped signals, +Can be changed.
- 7α-acetoxy-6β-hydroxy-12-O-phenylacetyl-royleanone (2): The compound was prepared according to the general procedure, with phenylacetyl chloride (422.6 μmol, 10 equiv.) and then let to react for 60 min. The crude mixture was purified by preparative chromatography with a mixture of dichloromethane/acetone (99:1). The pure product (16%) was obtained as a dark yellow oil. (c 0.200, CHCl3). IR : 3516.3, 2965.8, 2937.5, 2872.7, 1771.8, 1727.3, 1670.6, 1456.0, 1225.4, 1136.3, 1104.0, 1027.0, 751.8, 723.5, 703.2 cm−1. 1H NMR (400 MHz, Chloroform-d, ppm): δ 7.37 (d, J = 4.2 Hz, 4H, H-2′, H-3′), 7.31 (dt, J = 8.5, 4.2 Hz, 1H, H-4′), 5.63 (s, 1H, H-7β), 4.31 (s, 1H, H-6α), 3.93 (s, 2H, 12-COCH2), 2.99 (qui, J = 7.1 Hz, 1H, H-15), 2.51 (br d, J = 12.6 Hz, 1H, H-1β), 2.04 (s, 3H, 7α-OAc), 1.91–1.75 (m, 1H, H-2β), 1.63 (s, 3H, Me-20), 1.58–1.54 (m, 1H, H-2α), 1.46 (d, J = 13.1 Hz, 1H, H-3β), 1.32 (s, 1H, H-5α), 1.22* (s, 5H, Me-19, H-3α, H-1α), 1.04* (d, J = 7.1 Hz, 6H, Me-16, Me-17), 0.93 (s, 3H, Me-18). *Overlapped signal. 13C NMR (101 MHz, Chloroform-d, ppm): δ 185.86 (C-14), 179.69 (C-11), 169.82 (7α-COCH3), 168.94 (12-COCH2), 153.05 (C-9), 149.59 (C-12), 135.67 (C-8), 132.65 (C-1′), 129.68 (C-2′ +), 128.88 (C-3′ +), 127.69 (C-4′), 68.98 (C-7), 67.36 (C-6), 49.89 (C-5), 42.40 (C-3), 40.89 (12-COCH2), 39.00 (C-10), 38.45 (C-1), 33.86 (C-4), 33.65 (C-18), 25.05 (C-15), 23.99 (C-19), 21.85 (C-20), 21.04 (7α-COCH3), 20.18 (C-17), 20.04 (C-16), 19.03 (C-2). +Can be changed. HRMS (ESI-MS): m/z calculated for C30H37ClO7 [2M + Na]+ 1039.4814, found 1039.48210.
- 7α-acetoxy-6β-hydroxy-12-O-(4-methyl)benzoylroyleanone (3): The compound was prepared according to the general procedure, with 4-toluoylbenzoyl chloride (88.4 μmol, 3 equiv.) and then let to react for 30 min. The crude mixture was purified by preparative chromatography with a mixture of dichloromethane/ethyl acetate (97:3). The pure product (53%) was obtained as a yellow amorphous powder. mp: 226–228 °C. (c 0.280, CHCl3). IR : 3551.8, 2961.7, 2929.1, 2870.5, 1739.3, 1726.3, 1667.6, 1648.0, 1609.0, 1609.0, 1465.5, 1374.2, 1250.3, 1224.3, 1178.6, 1139.5, 1100.4, 1067.8, 1009.1, 963.5, 898.3, 836.3, 823.3, 741.8, 686.4 cm−1. 1H NMR (400 MHz, Chloroform-d, ppm): δ 8.04 (d, J = 8.3 Hz, 2H, H-2′) +, 7.32 (d, J = 8.0 Hz, 2H, H-3′) +, 5.69 (d, J = 2.0 Hz, 1H, H-7β), 4.34 (s, 1H, H-6α), 3.19 (hept, J = 7.1 Hz, 1H, H-15), 2.52–2.50 (m, 1H, H-1β), 2.45 (s, 3H, Me-7′), 2.07 (s, 3H, 7α-OAc), 1.80 (dt, J = 13.7, 3.6 Hz, 1H, H-2β), 1.63 (s, 3H, Me-20), 1.55 (dt, J = 14.1, 3.7 Hz, 1H, H-2α), 1.46 (dt, J = 12.8, 3.4 Hz, 1H, H-3β), 1.37 (s, 1H, H-5α), 1.25–1.19* (m, 11H, Me-19, Me-17, H-3α, Me-16, H-1α), 0.95 (s, 3H, Me-18). *Overlapped signal. 13C NMR (101 MHz, Chloroform-d, ppm): δ 186.00 (C-14), 179.94 (C-11), 169.87 (7α-COCH3), 164.19 (12-COO), 153.20 (C-9), 149.98 (C-12), 145.37 (C-4′), 139.61 (C-13), 135.71 (C-8), 130.72 (C-3′ +), 129.63 (C-2′ +), 125.39 (C-1′), 69.07 (C-7), 67.41 (C-6), 49.93 (C-5), 42.45 (C-3), 39.04 (C-10), 38.47 (C-1), 33.87 (C-4), 33.67 (C-18), 25.26 (C-15), 24.00 (C-19), 21.98 (C-5′), 21.91 (C-20), 21.07 (7α-COCH3), 20.58 (C-16), 20.35 (C-17), 19.03 (C-2). +Can be changed. HRMS (ESI-MS): m/z calculated for C30H36O7 [M + H]+ 509.2534, found 509.25375.
- 7α-acetoxy-6β-hydroxy-12-O-(2-naphtoate)benzoylroyleanone (4): The compound was prepared according to the general procedure, with 2-Naphthoyl chloride (384.2 μmol, 10 equiv.) and then let to react overnight. The crude mixture was purified by preparative chromatography with a mixture of dichloromethane/acetone (99:1). The pure product (68%) was obtained as a yellow amorphous powder. mp: 230–232 °C. (c 0.167, CHCl3). IR : 3467.0, 2965.0, 2922.6, 2854.2, 1745.8, 1736.0, 1657.8, 1631.7, 1609.0, 1576.3, 1459.0, 1367.7, 1276.4, 1250.3, 1217.7, 1185.1, 1142.8, 1123.2, 1097.1, 1061.3, 1022.1, 1009.1, 829.8, 777.7, 761.4, 732.0 cm−1. 1H NMR (400 MHz, Chloroform-d): δ 8.75 (d, J = 1.7 Hz, 1H, H-1′ +), 8.13 (dd, J = 8.5, 1.7 Hz, 1H, H-3′ +), 8.00 (d, J = 8.1 Hz, 1H, H-4′), 7.96 (d, J = 8.7 Hz, 1H, H-8′ ++), 7.93 (d, J = 8.0 Hz, 1H, H-5′ ++), 7.65 (t, J = 7.4 Hz, 1H H-6′ +++), 7.59 (t, J = 7.5 Hz, 1H, H-7′ +++), 5.70 (d, J = 2.0 Hz, 1H, H-7β), 4.35 (s, 1H, H-6α), 3.23 (qui, J = 7.1 Hz, 1H, H-15), 2.52 (br s, 1H, H-1β), 2.09 (s, 3H, 7α-OAc), 1.89–1.74 (m, 1H, H-2β), 1.65 (s, 3H, Me-20), 1.55 (dt, J = 14.1, 3.6 Hz, 1H, H-2α), 1.47 (d, J = 13.2 Hz, 1H, H-3β), 1.39 (s, 1H, H-5α), 1.24* (d, J = 7.1 Hz, 11H, Me-19, Me-17, H-3α, Me-16, H-1α), 0.96 (s, 3H, Me-18). *Overlapped signal; +, ++, and +++Can be changed. 13C NMR (101 MHz, Chloroform-d, ppm): δ 185.98 (C-14), 179.90 (C-11), 169.87 (7α-COCH3), 164.44 (12-COO), 153.18 (C-9), 150.00 (C-12), 136.22 (C-4a), 132.76 (C-8a), 132.58 (C-1′ +), 129.73 (C-4′ ++), 129.15 (C-6′ +++), 128.81 (C-8′ ++), 128.02 (C-5′ ++), 127.15 (C-7′ +++), 125.55 (C-3′ +), 125.29 (C-2′), 69.06 (C-7), 67.42 (C-6), 49.93 (C-5), 42.44 (C-3), 39.07 (C-10), 38.49 (C-1), 33.88 (C-4), 25.34 (C-15), 24.00 (C-19), 21.92 (C-20), 21.09 (7α-COCH3), 20.65 (C-16), 20.39 (C-17), 19.03 (C-2). +, ++, and +++Can be changed. HRMS (ESI-MS): m/z calculated for C33H36O7 [M + H]+ 545.2534, found 545.25428.
- 7α-acetoxy-6β-hydroxy-12-O-butanoylroyleanone (5): The compound was prepared according to the general procedure, with butyryl chloride (181.3 μmol, 6 equiv.) and then let to react for 5 min. The crude mixture was purified by preparative chromatography with dichloromethane. The pure product (94%) was obtained as an amber amorphous powder. mp: 148–150 °C. (c 0.241, CHCl3). IR : 3516.0, 2965.0, 2932.4, 2877.0, 1768.6, 1742.6, 1732.8, 1670.9, 1657.8, 1612.2, 1465.5, 1367.7, 1276.4, 1224.3, 741.8 cm−1. 1H NMR (400 MHz, Chloroform-d, ppm): δ 5.65 (d, J = 2.0 Hz, 1H, H-7β), 4.31 (s, 1H, H-6α), 3.10 (p, J = 7.1 Hz, 1H, H-15), 2.59 (q, J = 7.4 Hz, 2H, H-1′), 2.49 (d, J = 12.9 Hz, 1H, H-1β), 2.05 (s, 3H, 7α-OAc), 1.79* (q, J = 7.4 Hz, 3H, H-2β, H-2′), 1.62 (s, 1H, Me-20), 1.55 (dq, J = 14.3, 3.7 Hz, 1H, H-2α), 1.45 (dt, J = 13.5, 3.3 Hz, 1H, H-3β), 1.33 (s, 1H, H-5α), 1.23 (d, J = 11.4 Hz, 4H, Me-19, H-3α +), 1.18* (dd, J = 7.1, 3.1 Hz, 7H, Me-16, Me-17, H-1α +), 1.05 (t, J = 7.4 Hz, 1H, Me-3′), 0.93 (s, 3H, Me-18). *Overlapped signal; +Can be changed. 13C NMR (101 MHz, Chloroform-d, ppm): δ 185.98 (C-14), 179.93 (C-11), 171.10 (12-COO), 169.88 (7α-COCH3), 153.06 (C-9), 149.65 (C-12), 139.30 (C-13), 135.69 (C-8), 69.03 (C-7), 67.32 (C-6), 49.90 (C-5), 42.43 (C-3), 38.98 (C-10), 38.43 (C-1), 35.81 (C-1′), 33.85 (C-4), 33.65 (C-18), 25.25 (C-15), 23.97 (C-19), 21.84 (C-20), 21.05 (7α-COCH3), 20.41 (C-16), 20.33 (C-17), 19.04 (C-2), 18.36 (C-2′), 13.77 (C-3′). HRMS (ESI-MS): m/z calculated for C26H36O7 [M + H]+ 461.2534, found 461.25346.
4.2. ADMET and Drug-Likeness Analysis
4.3. Toxicity Prediction and Molecular Properties
4.4. Anticarcinogenic Activity
4.5. DFT Calculations
4.6. Molecular Docking
4.6.1. Protein and Ligand Preparation
4.6.2. Active Site Prediction
4.6.3. Receptor–Ligand Docking
4.7. Molecular Dynamics Simulations
4.8. Network Pharmacology
4.8.1. Identification of Potential Targets of Analyzed Compounds
4.8.2. Associated Targets of Cancer Diseases
4.8.3. Visualization and Analysis of the Network of the Protein–Protein Interactions
4.8.4. Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dar, R.A.; Shahnawaz, M.; Ahanger, M.A.; ul Majid, I. Exploring the Diverse Bioactive Compounds from Medicinal Plants: A Review. J. Phytopharm. 2023, 12, 189–195. [Google Scholar] [CrossRef]
- Fotsing Yannick Stéphane, F.; Kezetas Jean Jules, B.; El-Saber Batiha, G.; Ali, I.; Ndjakou Bruno, L. Extraction of Bioactive Compounds from Medicinal Plants and Herbs. In Natural Medicinal Plants; IntechOpen Limited: London, UK, 2022. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.H.; Ruoff, H.J.; Volm, M. Intrinsic and acquired drug resistance in malignant tumors: The main reason for therapeutic failure. Arzneim.-Forsch./Drug Res. 2008, 58, 261–264. [Google Scholar] [CrossRef]
- Debela, D.T.; Muzazu, S.G.Y.; Heraro, K.D.; Ndalama, M.T.; Mesele, B.W.; Haile, D.C.; Kitui, S.K.; Manyazewal, T. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021, 9, 20503121211034366. [Google Scholar] [CrossRef] [PubMed]
- Hong, J. Natural product diversity and its role in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 2012, 15, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Moshari-Nasirkandi, A.; Alirezalu, A.; Alipour, H.; Amato, J. Screening of 20 species from Lamiaceae family based on phytochemical analysis, antioxidant activity and HPLC profiling. Sci. Rep. 2023, 13, 16987. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mogib, M.; Albar, H.A.; Batterjee, S.M. Chemistry of the Genus Plectranthus. Molecules 2002, 7, 271–301. [Google Scholar] [CrossRef]
- Lambrechts, I.A.; Lall, N. Traditional usage and biological activity of Plectranthus madagascariensis and its varieties: A review. J. Ethnopharmacol. 2021, 269, 113663. [Google Scholar] [CrossRef]
- Barbosa, M.d.O.; Wilairatana, P.; Leite, G.M.d.L.; Delmondes, G.d.A.; Silva, L.Y.S.d.; Júnior, S.C.A.; Dantas, L.B.R.; Bezerra, D.S.; Beltrão, I.C.S.L.d.; Dias, D.d.Q.; et al. Plectranthus Species with Anti-Inflammatory and Analgesic Potential: A Systematic Review on Ethnobotanical and Pharmacological Findings. Molecules 2023, 28, 5653. [Google Scholar] [CrossRef]
- Nizar Ahamed, A.; Mohamed Yaser, S.; Mohammad Idhris, S.; Syed Ali Padusha, M.; Ahamed Sherif, N. Phytochemical and pharmacological potential of the genus Plectranthus—A review. S. Afr. J. Bot. 2023, 154, 159–189. [Google Scholar] [CrossRef]
- Matias, D.; Nicolai, M.; Fernandes, A.S.; Saraiva, N.; Almeida, J.; Saraiva, L.; Faustino, C.; Díaz-Lanza, A.M.; Reis, C.P.; Rijo, P. Comparison Study of Different Extracts of Plectranthus madagascariensis, P. neochilus and the Rare P. porcatus (Lamiaceae): Chemical Characterization, Antioxidant, Antimicrobial and Cytotoxic Activities. Biomolecules 2019, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Gáborová, M.; Šmejkal, K.; Kubínová, R. Abietane diterpenes of the genus plectranthus sensu lato. Molecules 2022, 27, 166. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Teodósio, C.; Oliveira, C.; Oliveira, C.; Díaz-Lanza, A.; Reis, C.; Duarte, N.; Rijo, P. Naturally Occurring Plectranthus-derived Diterpenes with Antitumoral Activities. Curr. Pharm. Des. 2019, 24, 4207–4236. [Google Scholar] [CrossRef]
- Śliwiński, T.; Sitarek, P.; Skała, E.; Isca, V.M.S.; Synowiec, E.; Kowalczyk, T.; Bijak, M.; Rijo, P. Diterpenoids from plectranthus spp. As potential chemotherapeutic agents via apoptosis. Pharmaceuticals 2020, 13, 123. [Google Scholar] [CrossRef]
- Sitarek, P.; Synowiec, E.; Kowalczyk, T.; Bangay, G.; Śliwiński, T.; Picot, L.; Princiotto, S.; Rijo, P. Anticancer Properties of Plectranthus ornatus-Derived Phytochemicals Inducing Apoptosis via Mitochondrial Pathway. Int. J. Mol. Sci. 2022, 23, 11653. [Google Scholar] [CrossRef]
- Yu, J.; Su, N.Q.; Yang, W. Describing Chemical Reactivity with Frontier Molecular Orbitalets. JACS Au 2022, 2, 1383–1394. [Google Scholar] [CrossRef]
- Sindhu, M.S.; Poonkothai, M.; Thirumalaisamy, R. Phenolic and terpene compounds from Plectranthus amboinicus (Lour.) Spreng. Act as promising hepatic anticancer agents screened through in silico and in vitro approaches. S. Afr. J. Bot. 2022, 149, 145–159. [Google Scholar] [CrossRef]
- Van Mourik, T.; Bühl, M.; Gaigeot, M.P. Density functional theory across chemistry, physics and biology. Philos. Trans. A Math. Phys. Eng. Sci. 2014, 372, 20120488. [Google Scholar] [CrossRef]
- Thanikaivelan, P.; Subramanian, V.; Raghava Rao, J.; Nair, B.U. Application of quantum chemical descriptor in quantitative structure activity and structure property relationship. Chem. Phys. Lett. 2000, 323, 59–70. [Google Scholar] [CrossRef]
- Kaya, S.; Putz, M.V. Atoms-In-Molecules’ Faces of Chemical Hardness by Conceptual Density Functional Theory. Molecules 2022, 27, 8825. [Google Scholar] [CrossRef]
- Pal, R.; Chattaraj, P.K. Chemical reactivity from a conceptual density functional theory perspective. J. Indian Chem. Soc. 2021, 98, 100008. [Google Scholar] [CrossRef]
- Geerlings, P. From Density Functional Theory to Conceptual Density Functional Theory and Biosystems. Pharmaceuticals 2022, 15, 1112. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Rong, C.; Zhang, R.; Liu, S. Evaluating frontier orbital energy and HOMO/LUMO gap with descriptors from density functional reactivity theory. J. Mol. Model. 2017, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Pegu, D.; Deb, J.; Van Alsenoy, C.; Sarkar, U. Theoretical investigation of electronic, vibrational, and nonlinear optical properties of 4-fluoro-4-hydroxybenzophenone. Spectrosc. Lett. 2017, 50, 232–243. [Google Scholar] [CrossRef]
- Landeros-Martínez, L.-L.; Campos-Almazán, M.I.; Sánchez-Bojorge, N.-A.; Flores, R.; Palomares-Báez, J.P.; Rodríguez-Valdez, L.M. Theoretical Studies for the Discovery of Potential Sucrase-Isomaltase Inhibitors from Maize Silk Phytochemicals: An Approach to Treatment of Type 2 Diabetes. Molecules 2023, 28, 6778. [Google Scholar] [CrossRef]
- Landeros-Martínez, L.-L.; Gutiérrez-Méndez, N.; Palomares-Báez, J.P.; Sánchez-Bojorge, N.-A.; Flores-De los Ríos, J.P.; Piñón-Castillo, H.A.; Chávez-Rojo, M.A.; Rodríguez-Valdez, L.-M. The Oxidative Process of Acarbose, Maysin, and Luteolin with Maltase-Glucoamylase: Molecular Docking and Molecular Dynamics Study. Appl. Sci. 2021, 11, 4067. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Rashmi, R.; Toppo, V.; Chole, P.B.; Banadka, A.; Sudheer, W.N.; Nagella, P.; Shehata, W.F.; Al-Mssallem, M.Q.; Alessa, F.M.; et al. Plant Secondary Metabolites: The Weapons for Biotic Stress Management. Metabolites 2023, 13, 716. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Fareed, S.; Ansari, S.; Rahman, M.A.; Ahmad, I.Z.; Saeed, M. Current approaches toward production of secondary plant metabolites. J. Pharm. Bioallied Sci. 2012, 4, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381. [Google Scholar] [CrossRef]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern Approaches in the Discovery and Development of Plant-Based Natural Products and Their Analogues as Potential Therapeutic Agents. Molecules 2022, 27, 349. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Shaker, B.; Ahmad, S.; Lee, J.; Jung, C.; Na, D. In silico methods and tools for drug discovery. Comput. Biol. Med. 2021, 137, 104851. [Google Scholar] [CrossRef] [PubMed]
- Rijo, P.; Esteves, M.; Simões, M.; Silva, A.; Duarte, A.; Rodriguez, B. Antimicrobial activity of 7α-acetoxy-6β-hydroxyroyleanone 12-O-benzoyl esters. Planta Med. 2008, 74, PB81. [Google Scholar] [CrossRef]
- Gaspar-Marques, C.; Rijo, P.; Simões, M.F.; Duarte, M.A.; Rodriguez, B. Abietanes from Plectranthus grandidentatus and P. hereroensis against methicillin- and vancomycin-resistant bacteria. Phytomedicine 2006, 13, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Ndjoubi, K.O.; Sharma, R.; Badmus, J.A.; Jacobs, A.; Jordaan, A.; Marnewick, J.; Warner, D.F.; Hussein, A.A. Antimycobacterial, cytotoxic, and antioxidant activities of abietane diterpenoids isolated from plectranthus madagascariensis. Plants 2021, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Napagoda, M.; Gerstmeier, J.; Butschek, H.; Lorenz, S.; De Soyza, S.; Qader, M.; Nagahawatte, A.; Wijayaratne, G.B.; Schneider, B.; Svatoš, A.; et al. Plectranthus zeylanicus: A Rich Source of Secondary Metabolites with Antimicrobial, Disinfectant and Anti-Inflammatory Activities. Pharmaceuticals 2022, 15, 436. [Google Scholar] [CrossRef]
- Cerqueira, F.; Cordeiro-Da-Silva, A.; Gaspar-Marques, C.; Simões, F.; Pinto, M.M.M.; Nascimento, M.S.J. Effect of abietane diterpenes from Plectranthus grandidentatus on T- and B-lymphocyte proliferation. Bioorg. Med. Chem. 2004, 12, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Nicolai, M.; Saraiva, L.; Pinheiro, R.; Faustino, C.; Diaz Lanza, A.; Pinto Reis, C.; Stankovic, T.; Dinic, J.; Pesic, M.; et al. Cytotoxic Activity of Royleanone Diterpenes from Plectranthus madagascariensis Benth. ACS Omega 2019, 4, 8094–8103. [Google Scholar] [CrossRef]
- Pereira, F.; Figueiredo, T.; de Almeida, R.F.M.; Antunes, C.A.C.; Garcia, C.; Reis, C.P.; Ascensão, L.; Sobral, R.G.; Rijo, P. Unveiling the mechanism of action of 7α-acetoxy-6β-hydroxyroyleanone on an mrsa/visa strain: Membrane and cell wall interactions. Biomolecules 2020, 10, 983. [Google Scholar] [CrossRef]
- Domínguez-Martín, E.M.; Magalhães, M.; Díaz-Lanza, A.M.; Marques, M.P.; Princiotto, S.; Gómez, A.M.; Efferth, T.; Cabral, C.; Rijo, P. Phytochemical Study and Antiglioblastoma Activity Assessment of Plectranthus hadiensis (Forssk.) Schweinf. ex Sprenger var. hadiensis Stems. Molecules 2022, 27, 3813. [Google Scholar] [CrossRef] [PubMed]
- Isca, V.M.S.; Sencanski, M.; Filipovic, N.; Dos Santos, D.J.V.A.; Gašparović, A.Č.; Saraíva, L.; Afonso, C.A.M.; Rijo, P.; García-Sosa, A.T. Activity to breast cancer cell lines of different malignancy and predicted interaction with protein kinase C isoforms of royleanones. Int. J. Mol. Sci. 2020, 21, 3671. [Google Scholar] [CrossRef] [PubMed]
- Ntungwe, E.; Domínguez-Martín, E.M.; Teodósio, C.; Teixidó-Trujillo, S.; Armas Capote, N.; Saraiva, L.; Díaz-Lanza, A.M.; Duarte, N.; Rijo, P. Preliminary biological activity screening of Plectranthus spp. Extracts for the search of anticancer lead molecules. Pharmaceuticals 2021, 14, 402. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, C.E.S.; Garcia, C.; Pereira, F.; Mota, J.; Pereira, P.; Cebola, M.J.; Reis, C.P.; Correia, I.; Piedade, M.F.M.; Minas Da Piedade, M.E.; et al. Extraction Optimization and Structural and Thermal Characterization of the Antimicrobial Abietane 7α-Acetoxy-6β-hydroxyroyleanone. Mol. Pharm. 2018, 15, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.F.; Rijo, P.; Duarte, A.; Barbosa, D.; Matias, D.; Delgado, J.; Cirilo, N.; Rodríguez, B. Two new diterpenoids from Plectranthus species. Phytochem. Lett. 2010, 3, 221–225. [Google Scholar] [CrossRef]
- Hensch, M.; Rüedi, P.; Eugster, C.H. Horminon, Taxochinon und weitere Royleanone aus 2 abessinischen Plectranthus-Spezies (Labiatae). Helv. Chim. Acta 1975, 58, 1921–1934. [Google Scholar] [CrossRef]
- Isca, V.; Bangay, G.; Princiotto, S.; Saraíva, L.; Santos, D.; García-Sosa, A.; RIJO, P. Reactivity of 7α-acetoxy-6β-hydroxyroyleanone and ability of its derivatives to modulate PKC isoforms. Res. Sq. 2023, preprint. [Google Scholar] [CrossRef]
- Garcia, C.; Isca, V.M.S.; Pereira, F.; Monteiro, C.M.; Ntungwe, E.; Sousa, F.; Dinic, J.; Holmstedt, S.; Roberto, A.; Díaz-Lanza, A.; et al. Royleanone Derivatives From Plectranthus spp. as a Novel Class of P-Glycoprotein Inhibitors. Front. Pharmacol. 2020, 11, 557789. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Bakchi, B.; Krishna, A.D.; Sreecharan, E.; Ganesh, V.B.J.; Niharika, M.; Maharshi, S.; Puttagunta, S.B.; Sigalapalli, D.K.; Bhandare, R.R.; Shaik, A.B. An overview on applications of SwissADME web tool in the design and development of anticancer, antitubercular and antimicrobial agents: A medicinal chemist’s perspective. J. Mol. Struct. 2022, 1259, 132712. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed]
- Borba, J.V.B.; Alves, V.M.; Braga, R.C.; Korn, D.R.; Overdahl, K.; Silva, A.C.; Hall, S.U.S.; Overdahl, E.; Kleinstreuer, N.; Strickland, J.; et al. STopTox: An in Silico Alternative to Animal Testing for Acute Systemic and Topical Toxicity. Environ. Health Perspect. 2022, 130, 27012. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Reich, J.R.; Rufener, C. OSIRIS, an entirely in-house developed drug discovery informatics system. J. Chem. Inf. Model. 2009, 49, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8.14.1–8.14.40. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Jejurikar, B.L.; Rohane, S.H. Drug Designing in Discovery Studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Mukhtar, S. Protein–Protein Interaction Network Exploration Using Cytoscape. In Methods in Molecular Biology; Humana: New York, NY, USA, 2023; Volume 2690, pp. 419–427. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Predicted LD50 (mg/kg) | Predicted Toxicity Class | Prediction Accuracy (%) |
|---|---|---|---|
| 1 | 1000 | 4 | 69.26 |
| 2 | 75 | 3 | 67.38 |
| 3 | 100 | 3 | 67.38 |
| 4 | 100 | 3 | 67.38 |
| 5 | 75 | 3 | 68.07 |
| 6 | 100 | 3 | 67.38 |
| Compounds | Antineoplastic Activity | Anticarcinogenic Activity | ||
|---|---|---|---|---|
| Pa Value | Pi Value | Pa Value | Pi Value | |
| 1 | 0.879 | 0.005 | 0.419 | 0.028 |
| 2 | 0.819 | 0.010 | 0.332 | 0.047 |
| 3 | 0.834 | 0.008 | 0.398 | 0.031 |
| 4 | 0.822 | 0.009 | 0.312 | 0.054 |
| 5 | 0.858 | 0.006 | 0.378 | 0.035 |
| 6 | 0.851 | 0.007 | 0.347 | 0.042 |
| Compound | EHOMO (eV) | ELUMO (eV) | ΔE (eV) | I (eV) | A (eV) | χ (eV) | μ (eV) | η (eV) | S (eV−1) | ω (eV) | ΔNmax |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −6.885 | −3.433 | 3.45 | 6.89 | 3.43 | 5.16 | −5.16 | 1.73 | 0.29 | 7.71 | 2.99 |
| 2 | −6.971 | −3.579 | 3.39 | 6.97 | 3.58 | 5.28 | −5.28 | 1.70 | 0.29 | 8.20 | 3.11 |
| 3 | −7.167 | −3.462 | 3.71 | 7.17 | 3.46 | 5.31 | −5.31 | 1.85 | 0.27 | 7.62 | 2.87 |
| 4 | −6.365 | −2.862 | 3.50 | 6.37 | 2.86 | 4.61 | −4.61 | 1.75 | 0.29 | 6.08 | 2.63 |
| 5 | −7.252 | −3.537 | 3.72 | 7.25 | 3.54 | 5.39 | −5.39 | 1.86 | 0.27 | 7.83 | 2.90 |
| 6 | −7.197 | −3.412 | 3.79 | 7.20 | 3.41 | 5.30 | −5.30 | 1.89 | 0.26 | 7.43 | 2.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isca, V.M.S.; Sitarek, P.; Merecz-Sadowska, A.; Małecka, M.; Owczarek, M.; Wieczfińska, J.; Zajdel, R.; Nowak, P.; Rijo, P.; Kowalczyk, T. Anticancer Effects of Abietane Diterpene 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus grandidentatus and Its Semi-Synthetic Analogs: An In Silico Computational Approach. Molecules 2024, 29, 1807. https://doi.org/10.3390/molecules29081807
Isca VMS, Sitarek P, Merecz-Sadowska A, Małecka M, Owczarek M, Wieczfińska J, Zajdel R, Nowak P, Rijo P, Kowalczyk T. Anticancer Effects of Abietane Diterpene 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus grandidentatus and Its Semi-Synthetic Analogs: An In Silico Computational Approach. Molecules. 2024; 29(8):1807. https://doi.org/10.3390/molecules29081807
Chicago/Turabian StyleIsca, Vera M. S., Przemysław Sitarek, Anna Merecz-Sadowska, Magdalena Małecka, Monika Owczarek, Joanna Wieczfińska, Radosław Zajdel, Paweł Nowak, Patricia Rijo, and Tomasz Kowalczyk. 2024. "Anticancer Effects of Abietane Diterpene 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus grandidentatus and Its Semi-Synthetic Analogs: An In Silico Computational Approach" Molecules 29, no. 8: 1807. https://doi.org/10.3390/molecules29081807
APA StyleIsca, V. M. S., Sitarek, P., Merecz-Sadowska, A., Małecka, M., Owczarek, M., Wieczfińska, J., Zajdel, R., Nowak, P., Rijo, P., & Kowalczyk, T. (2024). Anticancer Effects of Abietane Diterpene 7α-Acetoxy-6β-hydroxyroyleanone from Plectranthus grandidentatus and Its Semi-Synthetic Analogs: An In Silico Computational Approach. Molecules, 29(8), 1807. https://doi.org/10.3390/molecules29081807