Abstract
Four quaternary Zintl phase thermoelectric (TE) materials belonging to the Ba1-xEuxZn2Sb2 (x = 0.02(1), 0.04(1), 0.08(1), 0.15(1)) system were successfully synthesized using the molten Pb-flux or the conventional high-temperature reaction methods. Their crystal structures were characterized by both powder and single-crystal X-ray diffraction analyses, and all four isotypic title compounds adopted the orthorhombic BaCu2S2-type (Pnma, Z = 4, Pearson code oP20) structure. The radius ratio criterion, based on the cationic and anionic elements (i.e., r+/r−), was successfully verified in the title system, as in our previous reports, where r+/r− > 1 for the BaCu2S2-type structure. A series of density functional theory calculations were performed using a hypothetical model with the idealized compositions of Ba0.75Eu0.25Zn2Sb2, and the results were compared with the ternary parental compound BaZn2Sb2 to understand the influence of Eu substituents in the Ba1-xEuxZn2Sb2 system. A similar overall shape of the density of states (DOS) curves and the near-constant DOS values at EF before and after the cationic substitution suggest only marginal changes in the carrier concentration. Therefore, carrier mobility has a dominant role in rationalizing the observed variations in the electrical transport properties of the title system. Temperature-dependent TE property measurements proved that an increase in the Seebeck coefficient S and a decrease in electrical conductivity σ were observed as the Eu substituents gradually increased in the Ba1-xEuxZn2Sb2 system, although the overall S and σ values were lower than those in the parental compound BaZn2Sb2. The thermal conductivities of these title compounds were successfully lowered by phonon scattering, but due to the overall smaller electrical transport properties, the observed maximum ZT was 0.49 at 773 K for Ba0.98(1)Eu0.02Zn2Sb2.
1. Introduction
Thermoelectric (TE) devices can generate a significant amount of electricity from the wasted heat produced by vehicle exhausts, power plants, and other sources. To exploit this electricity for use in daily life, some issues including low energy conversion efficiencies in TE materials, thermal management problems within TE devices, TE device integration issues with heat sources should first be resolved [1,2,3,4]. The efficiency of a TE material is determined by the dimensionless figure of merit ZT = S2σT/κtot, where S, σ, T, and κtot are the Seebeck coefficient, electrical conductivity, absolute temperature, and total thermal conductivity, respectively [3]. In particular, the κtot comprises two main contributions: the electronic part κelec related to charge carriers and the lattice part κlatt caused by phonons. Except for κlatt, all of the TE material parameters that define ZT depend upon carrier concentration, n. Therefore, to achieve high ZT, the n of the TE material should be enhanced, and the κlatt should be lowered [2,5].
Quite interestingly, all of the aforementioned features, necessary for the development of efficient TE materials, are presented in the Zintl phase [6,7,8]. The Zintl phase is a small bandgap semiconductor with complex crystal structures. Their n can properly be tuned by vacancy formation, doping, or substitution [9,10,11,12], whereas their low κtot is largely attributed to their complex crystal structures, which impede phonon velocities and induce different phonon scattering mechanisms [13]. Among the trigonal CaAl2Si2-type AM2M’2 (A = Ca, Sr, Ba, Eu; M = Mg, Zn, Cd; M’ = P, As, Sb) system, many compounds have been reported as TE material of interest [12,14,15,16,17,18,19,20]. On the other hand, the orthorhombic BaCu2S2-type BaM2M’2 (M = Cu, Zn; M’ = P, S, As, Se, Sb, Te) system, displaying identical atomic stoichiometry, has rarely been investigated in the TE field due to its narrow phase width and poor metallic conductivity [21,22,23]. In our previous studies on the BaCu2S2-type Ba1-xSrxZn2-yCdySb2 system, we proved that the structural transformation between the BaCu2S2-type and the CaAl2Si2-type phase should be attributed to the radius ratio criterion of the cationic and anionic elements, i.e., r+/r−, with the BaCu2S2-phase preferring r+/r− > 1 [24]. In the same study, we also demonstrated a maximum ZT value of 0.64 for the quaternary Ba0.87(1)Sr0.13Zn2Sb2. In another study for the BaCu2S2-type Ba1-xSrxZn2-yCuySb2 system, we observed a small amount of σ improvement in TE power factor PF (PF = S2σ) upon some substitution on the parental BaZn2Sb2, which made further fine-tuning of the substituent amount necessary [25]. Lastly, we verified the aforementioned r+/r− ratio criterion by investigating the quaternary BaZn2-xCdxSb2 system, while simultaneously obtaining the ZT value of 0.54 in the Zn-rich compound BaZn1.62(2)Cd0.38Sb2 [26].
Encouraged by the aforementioned developments of the new BaCu2S2-type Ba1-xSrxM2Sb2 (M = Zn, Cd, Cu) systems for the TE material application, we have expanded our study to introduce the cationic Eu substituents in the parental BaZn2Sb2 system, since the Eu substitution was known to be successful for the improvement of TE properties [27]. Therefore, in this work, we conducted comprehensive experimental and theoretical investigations for the quaternary Ba1-xEuxZn2Sb2 system, including a series of powder and single-crystal X-ray diffraction (PXRD and SXRD) analyses for the crystal structural characterization, the density of states (DOS) curves and band structure analyses for the electronic structure analyses, and the electrical transport properties, as well as thermal conductivities measurements for the TE properties.
2. Results and Discussion
2.1. Crystal Structure Analysis
Four Zintl phase solid solutions in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system have been synthesized by the molten Pb-flux method or Nb-tube method, and their crystal structures were carefully analyzed by both PXRD and SXRD analyses. The phase purities of the four title compounds were initially evaluated by indexing peaks in the collected PXRD patterns with the simulated pattern generated by the SXRD refinement result of Ba0.85(1)Eu0.15Zn2Sb2. As displayed in Figure 1, all four title compounds were proven to be single-phase products. A more comprehensive crystal structure analysis was conducted by using the SXRD data presented in Table 1, Table 2 and Table 3. The refinement results proved that all four title compounds adopted the isotypic orthorhombic BaCu2S2-type structure (Pnma, Z = 4, Pearson code oP20) and contained five crystallographically independent atomic sites, i.e., one Ba/Eu-mixed site, two Zn sites, and two Sb sites. Energy dispersive X-ray spectroscopy (EDS) analysis was also performed for the nicely grown bar-shaped single-crystal samples (Figure 2). Further details regarding elemental analysis and distribution mapping of the title compounds are also provided in Supplementary Figures S1–S4.
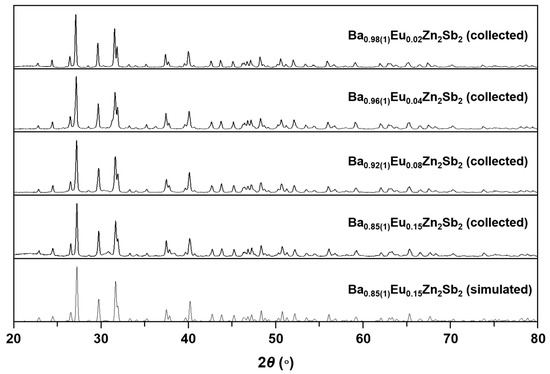
Figure 1.
PXRD patterns of four title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system. A simulated PXRD pattern of Ba0.85(1)Eu0.15Zn2Sb2 is also provided as a reference.

Table 1.
SXRD data and structure refinement results for the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system.
Table 2.
Atomic coordinates and equivalent isotropic displacement parameters (Ueq a) from the SXRD refinements for the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system.
Table 3.
Selected bond distances for the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system.

Figure 2.
Optical Microscope images of the single crystals of (a) Ba0.96(1)Eu0.04Zn2Sb2 and (b) Ba0.85(1)Eu0.15Zn2Sb2. Scale bars are also displayed.
As shown in Figure 3a, the overall crystal structure of Ba0.85(1)Eu0.15Zn2Sb2 displays the three-dimensional (3D) cage-like anionic frameworks, with the Ba/Eu-mixed cations filling the center of each 3D cage. In particular, these anionic frameworks consist of a one-dimensional (1D) infinite chain of by sharing two edges with their neighboring tetrahedra (Figure 3c). Moreover, each 1D chain is built by the tetrahedral [ZnSb4] moieties. Furthermore, the cage-shaped cationic site in the title can be described as a 16-coordinate site surrounded by nine Zn and seven Sb atoms, as displayed in Figure 3b [24].
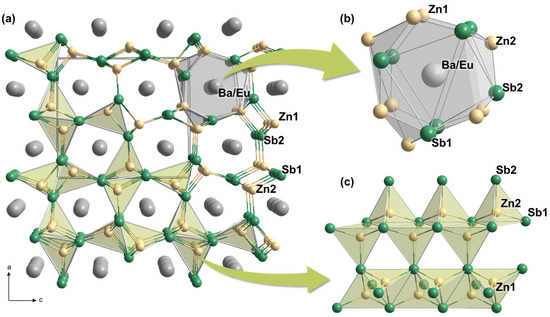
Figure 3.
(a) Crystal structures of Ba0.85(1)Eu0.15Zn2Sb2 illustrated by a combination of ball-and-stick and polyhedral representations. (b) A Eu/Ba mixed-cationic site and (c) the 3D anionic frameworks of are also displayed. The tetrahedral [ZnSb4] building blocks are highlighted in light green polyhedra, and a unit cell is outlined with a black line. Atomic labels are also provided. Color codes: Ba/Eu mixed-site, gray; Zn, light yellow; Sb, green.
In our recent studies of the quinary Ba1-xSrxZn2-yCdySb2 and Ba1-xSrxZn2-yCuySb2 systems, we rationalized, for the first time, the concept of phase selectivity between the CaAl2Si2-type and BaCu2S2-type structures in the AM2Sb2 (A = alkaline-earth metal, rare-earth metals; M = transition metal) system based on the overall radius ratio (herein referred to as r+/r−ratio, where r+ is the average radius of cationic elements, and r− is the average radius of anionic elements) criterion [24,25]. According to our comprehensive analysis, in the case of r+/r− > 1, the compounds adopted the BaCu2S2-type structure. On the other hand, if r+/r− < 1, the compounds crystallized in the CaAl2Si2-type structure. In addition, in our previous studies, the overall r+/r− ratio criterion depended upon both the cationic and anionic elements, with a certain amount of substituents [24,25]. However, in this work, our investigations based on the r+/r− ratio criterion are only limited to the radius difference of the cationic elements. As relatively smaller Eu substituted for the larger Ba (r(Eu2+) = 1.20 Å, r(Ba2+) = 1.38 Å) [28] in the title Ba1-xEuxZn2Sb2 system, the r+/r− ratio gradually decreased as follows: 1.035, 1.032, 1.028, and 1.017 for Ba0.98(1)Eu0.02Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, respectively. However, the r+/r− ratio of all four title compounds is still larger than 1, which proved that the r+/r− ratio criterion for the phase selectivity applies to the quaternary Ba1-xEuxZn2Sb2 system. Furthermore, based upon the r+/r− ratio criterion, we may “reverse predict” the maximum amount of Eu substituents that maintains the BaCu2S2-type phase. Theoretically, the Ba1-xEuxZn2Sb2 system can maintain the BaCu2S2-type phase until ca. 28% of the Eu substituents are included, where r+/r− = 1. Beyond this point, the size mismatch between the 3D cage-shaped framework and the cationic elements can escalate the repulsive interactions within the anionic frameworks in the BaCu2S2-type phase, which can eventually result in the phase transition into the more energetically favorable CaAl2Si2-type phase in the given r+/r− ratio.
2.2. Electronic Structure Analysis
A series of DFT calculations were performed using the TB-LMTO-ASA method [29,30,31,32] to understand the effects of Ba/Eu-mixing on the electronic structure of the title compounds. It should be noted that the use of the TB-LMTO-ASA method is known to provide an underestimated bandgap when compared to those obtained by actual experiments. For practical reasons, the hypothetical structural model was designed with the idealized composition of Ba0.75Eu0.25Zn2Sb2, and the symmetry was lowered from the experimentally refined Pnma to its subgroup Pm (No. 6). The lattice parameters and atomic positions were taken from the SXRD refinement results of Ba0.85(1)Eu0.15Zn2Sb2. An additional calculation for the ternary parental compound BaZn2Sb2 was also performed for comparison purposes. Further structural details about the two hypothetical models are provided in Supplementary Table S1.
The resultant TDOS and PDOS curves and band structures are shown and compared in Figure 4. The overall shapes of the DOS curves for the two structural models closely resemble each other, including the complex orbital mixing over the entire energy window (See Figure 4a,c). In particular, below the Fermi level (EF), the strong orbital contributions from the s-states of all components are mostly observed between −11.1 and −7.0 eV (the results not shown here in order to obtain better visualization of the overall orbital distribution and resonance peak near EF). On the other hand, the orbital contributions from the p-states of the anionic Zn and Sb are dominant in the energy window between ca. −6.2 and 0 eV (EF), with some contributions from the cationic elements as well. In addition, a small resonance peak was observed slightly above EF in the TDOS curve of the ternary BaZn2Sb2, and it became sharper and more noticeable for the idealized Eu-substituted quaternary Ba0.75Eu0.25Zn2Sb3. Furthermore, the DOS value at EF showed little change upon the introduction of Eu in the parental BaZn2Sb2 system. This suggests that the carrier mobility μ (not the n) is primarily dominant for the electrical conductivity of the title Ba1-xEuxZn2Sb2 system. This will be further discussed in Section 2.3, where we attempt to rationalize the observed variations in this section in terms of their chemical compositions, as well as their electronic structures.
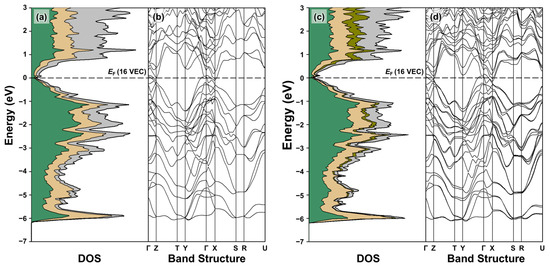
Figure 4.
TDOS, PDOS, and band structure of BaZn2Sb2 (a,b) and Ba0.75Eu0.25Zn2Sb2 (c,d). Color codes in the DOS curves are as follows: TDOS, bold black outline; Ba PDOS, gray region; Eu PDOS, dark yellow region; Zn PDOS, light yellow region; Sb PDOS, green region. EF (horizontal dashed line) is set as the energy reference at 0 eV, and the corresponding VEC is also displayed.
The density of states (DOS) analysis (Figure 4a,c) reveals that both BaZn2Sb2 and Ba0.75Eu0.25Zn2Sb2 possess a narrow pseudogap, indicating the absence of an actual bandgap. Previous DFT calculations for BaZn2Sb2 reported varying bandgap values (0.07, 0.2, and 0.35 eV), depending on the computational method being employed [23,33,34]. Since the TB-LMTO-ASA method tends to underestimate the bandgap size, BaZn2Sb2 likely behaves as a heavily doped semiconductor. Similarly, band structure analysis (Figure 4b,d) further reveals the lack of a clearly observable bandgap in both compounds. In the BaZn2Sb2, the valence band maxima appear near the Γ point along interval Γ-Y, while the conduction band minima are located near the X point along interval Γ-X. Moreover, the second conduction band minima exist along the interval Γ-Z. While BaZn2Sb2 and Ba0.75Eu0.25Zn2Sb2 show minimal differences in the band structure along the interval Γ-Y, the valence and conduction bands in Ba0.75Eu0.25Zn2Sb2 become closer along the Γ-Z and Γ-X intervals. Despite the narrowed energy gap, no overlap between the valence and conduction bands is observed, suggesting that Ba0.75Eu0.25Zn2Sb2 retains its heavily doped semiconducting characteristics.
2.3. TE Properties Measurements
To understand the influence of the Eu substitution for TE properties in the title Ba1-xEuxZn2Sb2 system, the temperature-dependent electrical transport properties and thermal conductivity measurements were performed for the four title compounds in the temperature range between 303 K and 797 K and between 323 K and 773 K, respectively.
First, the temperature-dependent electrical conductivities σ of four title compounds are illustrated in Figure 5a. All four title compounds show decreasing σ patterns as the temperature increases up to a “critical” temperature, which is a typical feature of the heavily doped semiconductor. Here, the increased phonon vibration obstructs the flow of free electrons, thus reducing the electrical conductivity. However, beyond this temperature, the electrical conductivities increase as the temperature increases, presenting the semiconducting characteristics, a phenomena which should be attributed to intrinsic carrier excitation across the bandgap, resulting in increased n. The overall temperature dependence of electrical conductivity was similar to those in previous reports on Ba1–xSrxZn2–yCdySb2 and Eu2ZnSb2 Zintl phase systems [24,35]. In addition, this critical temperature gradually increased as the amount of Eu substituents increased. In other words, as the Eu concentration increased, the metallic characteristics of each title compound also increased. However, with an increasing amount of Eu, the overall σ values decreased, which can be attributed to the reduced mobility in our Eu-substituted samples. Basically, as more Eu is added to the Ba1-xEuxZn2Sb2 system, an increased structural disorder at the cationic sites results in a high carrier scattering rate. Consequently, a gradual decrease in μ can occur. This observation is consistent with our previous results in Section 2.2, where we predicted a major role of μ (not n) in determining the electronic transport properties of the Eu-substituted title compounds. The observed maximum σ values are 131, 93.5, 93.1, and 82.8 S/cm for Ba0.98Eu0.02Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, respectively. Interestingly, in a previous study conducted on the Sr-substituted Ba1-xSrxZn2Sb2 system, we mentioned that the σ increase was explained in terms of the high electronegativity of Sr over Ba [24]. Due to its high electronegativity, Sr donated fewer electrons to the Zn–Sb anionic framework, resulting in an increased hole concentration. As a result, σ values in the investigated Ba1-xSrxZn2Sb2 system increased upon Sr introduction. However, in this current work, although Eu was even more electronegative than Sr (thus, more hole carriers are generated), the σ values decreased with the Eu substitution. This is presumably because the substantial decrease in μ due to the Eu-induced structural distortion was not fully compensated by the relatively smaller increase in hole concentration. This rationalization is in good agreement with the observed gradual decrease in σ values in our title Ba1-xEuxZn2Sb2 system upon the gradual introduction of Eu substituents.
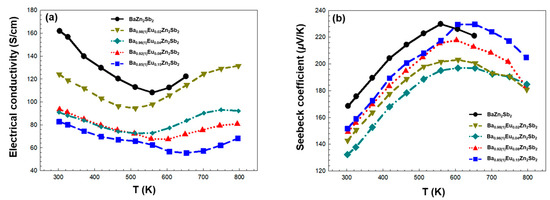
Figure 5.
Temperature-dependent (a) electrical conductivity σ and (b) Seebeck coefficients S of the four title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system measured between 303 and 793 K. The experimental data of the reference compound BaZn2Sb2 [24] is also plotted for comparison purposes.
Second, the temperature-dependent Seebeck coefficients S of the four title compounds are displayed in Figure 5b. The S values of all four compounds remained positive over the measured temperature range, confirming the p-type characteristics of all four samples. These S values of the four compounds increased as temperatures increased up to a certain critical temperature and then decreased. This decrease in S with temperature appears to be a consequence of thermally generated intrinsic carrier excitation across the bandgap and is consistent with the increase in electrical conductivity observed at the relatively higher temperature regions in Figure 5a. Except for Ba0.96(1)Eu0.04Zn2Sb2 composition, the S values gradually improved as the amount of Eu substituents increased. These results can be rationalized by considering that the S is proportional to the carrier effective mass, which in turn is related to μ. As the μ is reduced due to the Eu addition, the hole effective mass increases and eventually results in the following enhanced S values: (kB = the Boltzmann constant, e = the charge of electron, h = the Planck constant, and m* = the effective mass) [36]. The maximum S values are 230, 218, 203, and 197 μV/K for Ba0.85(1)Eu0.15Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, Ba0.98Eu0.02Zn2Sb2, and Ba0.96(1)Eu0.04Zn2Sb2, respectively. In particular, the S values for Ba0.85(1)Eu0.15Zn2Sb2 beyond 620 K are improved compared to those of the parental ternary compound BaZn2Sb2. The thermoelectric power factor PF, which can be calculated by PF = S2σ, was also evaluated, and the results are shown in Figure S5a. Due to the relatively smaller S and σ values of the title compounds, the overall PF is reduced considerably as the Eu substitution is introduced into the parental ternary phase BaZn2Sb2.
Thirdly, the temperature-dependent thermal conductivities κtot are provided in Figure 6a. Compared to the ternary BaZn2Sb2, the overall κtot of the four title compounds decreased upon the introduction of the Eu substituent in the Ba1-xEuxZn2Sb2 system, and this should be attributed to the enhanced cationic site disorder. This cationic site disorder caused by the Ba/Eu-mixture induces mass disorder scattering, enhances electron phonon scattering, and enables the manipulation of the phonon vibrational modes, thus causing stronger overall phonon scattering [37,38,39]. As a result, we see a significantly reduced κtot. In particular, the observed κtot in our system was considerably low and even comparable to those in the previously reported thermoelectric materials with the low κtot [36,40]. The κtot values of the four title compounds show a decreasing pattern up to ca. 550 K, then beyond this point, it slightly increases. The observed minimum κtot values are 0.54, 0.58, 0.61, and 0.69 W/mK for Ba0.92(1)Eu0.08Zn2Sb2, Ba0.85(1)Eu0.15Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, and Ba0.98Eu0.02Zn2Sb2, respectively. Like the previously discussed σ, κtot also tended to decrease as the amount of Eu substituent increased. In general, the κtot value is expressed as the summation of the electronic term κelec and the lattice term κlatt. Since the κelec value can be calculated by the Wiedemann–Franz law (κelec = LσT, L = Lorenz number), the lattice term κlatt was obtained by subtracting the κelec value from the κtot value (κlatt = κtot − κelec) [41]. By using this equation, we calculated the κelec values of 0.06, 0.05, 0.05, and 0.07 W/mK at 298 K for Ba0.85(1)Eu0.15Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, and Ba0.98Eu0.02Zn2Sb2, respectively. In addition, the κlatt values evaluated by subtracting κelec from κtot are plotted in Supplementary Figure S5b. In this work, the contribution of κelec seemed to be slight, so it was confirmed that κlatt is dominant for the overall κtot.
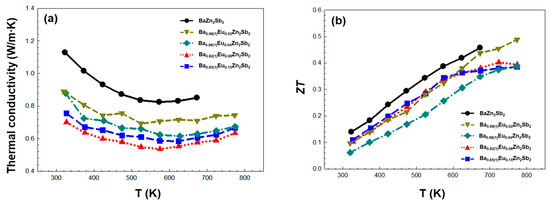
Figure 6.
Temperature-dependent (a) total thermal conductivity κtot and (b) figure-of-merit ZT of the four title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system measured between 323 and 773K. The experimental data of the reference compound BaZn2Sb2 [24] is also plotted for comparison purposes.
Finally, the temperature-dependent figure-of-merit ZT values of the title compounds are provided in Figure 6b. Although the κtot value significantly decreased in all title compounds, the PF values were also low. Therefore, the positive influence of decreasing the κtot value on ZT was nearly compensated by PF. Among the four title compounds, Ba0.98(1)Eu0.02Zn2Sb2 showed the highest ZT value of 0.49 at 773K. In order to further improve the ZT of the title system, some suitable anionic substitutions can be introduced into the anionic frameworks to possibly fine-tune the hole concentration, which eventually may promote the synergetic effect [42,43,44].
3. Materials and Methods
3.1. Synthesis
The quaternary title compound Ba0.96(1)Eu0.04Zn2Sb2 was synthesized by the molten Pb-metal flux method, while three other compounds, Ba0.98(1)Eu0.02Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, were synthesized by the conventional high-temperature method using a Nb ampoule (diameter = 1 cm, length = 4 cm) as a reaction container. The following reactant elements were purchased from Alfa Aesar (Ward Hill, MA, USA): Ba (rod, 99+ %), Eu (ingot, 99.9%), Zn (shot, 99.99%), Sb (shot, 99.9999%), and Pb (granules, 99.99%), and the slightly tanned surfaces of chunks of Ba and Eu elements were cleaned using a metal brush before loading. The reactants ratio used for the molten Pb-metal flux method was Ba:Eu:Zn:Sb:Pb = 0.7:0.3:2:2:10, and the mixture of reactants was loaded in an alumina crucible inside an Ar-filled glovebox (KOREAKIYON Ltd., Co., Seoul, Republic of Korea). To protect the reactants mixture from oxidation at the elevated temperature, the alumina crucible was sealed again with a fused-silica jacket under vacuum. The reactants ratio used for the conventional high-temperature reactions was Ba:Eu:Zn:Sb = (1–x):x:2:2 (x = 0.1, 0.3, 0.4). The reactants were cut into small pieces and loaded in the one-end-sealed Nb ampoule inside an Ar-filled glovebox, and the other end of the ampule was sealed by arc welding under the Ar atmosphere. Then, this Nb ampule was sealed again in a secondary container of a fused-silica jacket under vacuum to prevent the Nb ampoule from oxidation during the high-temperature reaction process. The reactants mixture used in the molten Pb-flux method was heated to 1223 K at the rate of 120 K/h, kept there for 24 h, and then cooled to 823 K for 92 h. At the last stage of the reaction, the reaction container was instantaneously removed from the furnace at 823 K and centrifuged for 2 min to remove the remaining molten metal flux. The reactant mixtures used for the high-temperature reaction method were heated to 1223 K at the rate of 60 K/h, kept there for 12 h, and then cooled to 893 K at the rate of 20 K/h, where the reactants were kept for 72 h. Four title compounds, Ba0.98Eu0.02Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, were also re-synthesized by ball-milling followed by the hot-pressing reaction method to create the samples for the TE property measurements. A mixture of reactants, based on the targeted stoichiometry, was loaded into a stainless-steel container with two 0.5-inch and two 0.25-inch stainless steel balls inside an Ar-filled glove box. After that, the reactant mixtures were ball-milled for 2 h 30 min using the SPEX 8000M machine (SPEX SamplePrep, Metuchen, NJ, USA). During the process, the machine was stopped every 30 min to scrape and re-mix the pulverized reactant mixtures to produce homogeneous samples. After the ball-milling process, the obtained powder products were placed in a 12.5 mm graphite mold and hot-pressed under 40 MPa at 873 K for 2 h.
3.2. X-Ray Diffraction Analysis
The phase purities and detailed crystal structures of the four isotypic title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system were determined by both PXRD and SXRD analyses. Four PXRD patterns were collected by the Miniflex 600 diffractometer (Rigaku Co., Tokyo, Japan) with Cu Kα1 radiation (λ = 1.54059 Å) and plotted in Figure 1. Data collection was performed for 30 min per sample, with a step size of 0.02° in the range of 20° ≤ 2θ ≤ 80°. The phase purities were verified by comparing the experimentally obtained PXRD patterns to the simulated pattern generated by using the SXRD result of Ba0.85(1)Eu0.15Zn2Sb2. All four collected patterns were in good agreement with the simulated pattern, indicating the BaCu2S2-type phase.
The SXRD data of Ba0.96(1)Eu0.04Zn2Sb2 was collected at room temperature using a SMART BREEZE CCD-based diffractometer (Bruker AXS Inc., Madison, WI, USA) with Mo Kα1 radiation (λ = 0.71073 Å). After the brief quality check for several well-grown single crystals, the best crystal was used for full data collection using Bruker’s APEX3 program (version 2019.1-0) [45]. Data reduction, integration, and unit cell parameter refinements were executed using the SAINT program (version 8.40A) [46]. The SADABS program (version 2016/2) [47] was also used to perform semi-empirical absorption corrections, based on equivalents. The SXRD data of Ba0.98(1)Eu0.02Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2 were collected using synchrotron radiation (λ = 0.65000 Å) on a MX225HS detector (Rayonix, Evanston, IL, USA) at BL2D SMC in the Pohang Accelerator Laboratory. Data collection was conducted using the PAL BL2D-SMDC program [48], and HKL3000sm (version 715) [49] was applied for cell refinement, reduction, and absorption correction. All SXRD data sets indicated the orthorhombic Pnma space group (No. 62). The detailed crystal structures, including the atomic positions accompanied by anisotropic displacement parameters (ADPs), the extinction coefficients, and the mixed ratio of Ba and Eu, were refined to convergences using full-matrix least-squares methods on F2. For the standardization of atomic positions, the STRUCTURE TIDY program (https://www.platonsoft.nl; accessed on 14 December 2024) [50] was used during the last step of structure refinement. More detailed crystallographic data, including atomic positions with ADP values and several selected bond distances, are presented in Table 1, Table 2 and Table 3. All these crystallographic data can be obtained free of charge from the Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif (accessed on 10 January 2025). The depository numbers are as follows: CCDC-2405340 for Ba0.98(1)Eu0.02Zn2Sb2, CCDC-2405341 for Ba0.96(1)Eu0.04Zn2Sb2, CCDC-2405342 for Ba0.92(1)Eu0.08Zn2Sb2, and CCDC-2405343 for Ba0.85(1)Eu0.15Zn2Sb2.
3.3. Electronic Structure Calculations
To understand the overall electronic structure of the title compounds, a series of DFT calculations were performed using the TB-LMTO47 method with the atomic sphere approximation (ASA) [29,30,31,32]. Two hypothetical structural models with the idealized compositions of BaZn2Sb2 and Ba0.75Eu0.25Zn2Sb2 were designed, and the DOS curves and band structures were thoroughly analyzed. The symmetry of these models was lowered from the experimentally obtained space group Pnma (No. 62) to its subgroup Pm (No. 6) to accommodate the idealized chemical compositions in each model. Further detailed crystallographic information, including the lattice parameters and the atomic positions, was extracted from the SXRD refinement results of BaZn2Sb2 and Ba0.85(1)Eu0.15Zn2Sb2, respectively. The structural details of these two models are provided in Supplementary Table S1. All the space in a unit cell is filled with overlapping Wigner−Seitz (WS) atomic spheres, and all relativistic effects (except spin−orbit coupling) were taken into account using a scalar relativistic approximation [51]. Each WS sphere is regarded to contain the symmetrically spherical potential, and the combined correction was applied for the overlapping regions. The radii of each WS sphere were calculated automatically to ensure that the overlapping potential exhibited the best approximation to the full potential [51]. The WS radii used for the two models are as follows: Ba, 2.462 Å; Zn, 1.489−1.572 Å; Sb, 1.734−1.787 Å for BaZn2Sb2; and Ba, 2.444−2.445 Å; Eu, 2.444 Å; Zn, 1.486−1.570 Å; Sb, 1.732−1.791 Å for Ba0.75Eu0.25Zn2Sb2. The basis sets included 6s, 6p, 5d, and 4f orbitals for Ba; 6s, 6p, and 5d orbitals for Eu; 4s, 4p, and 3d orbitals for Zn; and 5s, 5p, 5d, and 4f orbitals for Sb. The Löwdin downfolding technique was applied for the Ba 6p, Eu 6p, Sb 5d, and 4f orbitals [52]. The self-consistent charge density was obtained using 360 irreducible k-points in the Brillouin zone for both structure models [53].
3.4. Thermogravimetric (TGA) Analysis
The thermal stabilities of the four title compounds, Ba0.98Eu0.02Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, were checked by TGA using a TG 209 F1 Libra device (Netzsch-Gerätebau GmbH, Selb, Germany). A total of 20 mg of the pulverized sample was placed on an alumina pan and heated to 1173 K at a rate of 10 K/min under the continuous N2 flow condition. After that, the sample was naturally cooled down to room temperature. The TGA analysis proved that all four title compounds were thermally stable up to ca. 800 K (see Supplementary Figure S6).
3.5. EDS Analysis
Elemental analysis, including distribution mapping for the four title compounds, was performed by EDS using an ULTRA Plus field-emission (Carl Zeiss, Oberkochen, Germany) scanning electron microscope (SEM) system with an acceleration voltage of 30 kV. Several well-grown bar-shaped single crystals obtained using the molten Pb-flux method (See Figure 2) were carefully selected and placed on an aluminum puck with double-sided conductive carbon tapes under the Ar atmosphere. EDS analysis indicated comparable compositional results for Ba0.95Eu0.01Zn2.06Sb1.97, Ba0.98Eu0.06Zn1.97Sb2.00, Ba0.91Eu0.10Zn2.00Sb1.99, and Ba0.83Eu0.21Zn1.95Sb2.02 to the SXRD refinement results for Ba0.98(1)Eu0.02Zn2Sb2, Ba0.96(1)Eu0.04Zn2Sb2, Ba0.92(1)Eu0.08Zn2Sb2, and Ba0.85(1)Eu0.15Zn2Sb2, respectively. Detailed EDS analysis and distribution mapping results are provided in Supplementary Figures S1–S4.
3.6. Electrical Transport Property Measurement
Four disk-shaped title compounds, which were synthesized by ball-milling, followed by hot-pressing, were prepared into a bar shape (3 mm × 3 mm × 10 mm) for the electrical transport property measurements. The densities of these samples were proven to be higher than 90%, according to the geometric density measurement method. The longer direction of each bar-shaped sample coincided with the direction in which the properties were measured. The electrical conductivity σ and the Seebeck coefficient S were simultaneously measured between 303 and 793 K under the He atmosphere using a ZEM-3 instrument system (ULVAC-RIKO Inc., Yokohama, Japan).
3.7. Thermal Conductivity Measurement
Thermal diffusivity was measured for the four disk-shaped title compounds under an inert gas atmosphere from 323 to 773 K using a LFA 457 HyperFlash instrument (Netzsch-Gerätebau GmbH, Selb, Germany). The measurement was performed using a flash diffusion method in which the front surface of the disk was irradiated with a short laser burst, and the temperature change on the rear surface was recorded and analyzed using an IR detector. The thermal conductivity κtot was evaluated using the equation κtot = DCpρ (D = thermal diffusivity, Cp = heat capacity, and ρ = density) [54]. In this work, the Dulong−Petit value (3R/atom, R = gas constant) was exploited for Cp. The κtot was regarded as the sum of the lattice κlatt and the electronic κelec thermal conductivities [54]. κelec was calculated using the Wiedemann—Franz law (κelec = LσT, L = the temperature-dependent Lorenz number), and the L value was estimated using the single-parabolic band model from the temperature-dependent S [36]. Therefore, κlatt was obtained from the simple equation κlatt = κtot−κelec and is plotted in Supplementary Figure S5b.
4. Conclusions
Four new Zintl phase solid solutions in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system were obtained using the molten Pb metal-flux or the conventional high-temperature reaction methods. Their crystal structures were characterized by PXRD and SXRD, and all four title compounds adopted the BaCu2S2-type phase with the orthorhombic Pnma space group. The overall crystal structure of the isotypic title compounds can be described as an assembly of (1) the 3D anionic frameworks formed by the interconnection of four neighboring 1D chains and (2) the mixed cationic elements filling the cage-shaped voids within these frameworks. In addition, the applicability of the previously outlined radius ratio (r+/r−) criterion, which determines the structure-type selectivity between the CaAl2Si2-type and BaCu2S2-type phases, was strongly confirmed in the title system. A series of DFT calculations using hypothetical structure models revealed that the Ba1-xEuxZn2Sb2 system possesses heavily doped semiconducting characteristics, and the variations in transport properties upon Eu substitution should be explained in terms of changes in carrier mobilities. The temperature-dependent TE property measurements proved that despite the significantly lowered κtot, due to the relatively smaller S and σ values, the ZT values of the four Eu-substituted title compounds either decreased or slightly increased, with the maximum value of 0.49 at 773 K. We expect that the suitable anionic substitution may independently optimize the n, leading to overall enhanced TE properties.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules30020310/s1, Table S1: Structural details of the hypothetical model for BaZn2Sb2 and Ba0.75Eu0.25Zn2Sb2; Figure S1: EDS analysis and elemental mapping results for Ba0.98(1)Eu0.02Zn2Sb2. An evaluated EDS composition for this crystal is Ba0.95Eu0.01Zn2.06Sb1.97; Figure S2: EDS analysis and elemental mapping results for Ba0.96(1)Eu0.04Zn2Sb2. An evaluated EDS composition for this crystal is Ba0.98Eu0.06Zn1.97Sb2.00; Figure S3: EDS analysis and elemental mapping results for Ba0.92(1)Eu0.08Zn2Sb2. An evaluated EDS composition for this crystal is Ba0.91Eu0.10Zn2.00Sb1.99; Figure S4: EDS analysis and elemental mapping results for Ba0.85(1)Eu0.15Zn2Sb2. An evaluated EDS composition for this crystal is Ba0.83Eu0.21Zn1.95Sb2.02; Figure S5: Temperature-dependent (a) power factor PF of the four title compounds in Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system between 303 and 793 K and (b) lattice thermal conductivity κlatt of the four title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system measured between 323 and 773 K. The experimental data of the reference compound BaZn2Sb2 [24] is also plotted for comparison purposes.; Figure S6: TGA results for the four title compounds in the Ba1-xEuxZn2Sb2 (0.02(1) ≤ x ≤ 0.15(1)) system over the temperature range between 300 and 1173 K.
Author Contributions
Conceptualization, D.S. and T.-S.Y.; methodology, D.S. and T.-S.Y.; software, D.S. and J.L.; validation, D.S., J.L., A.A. and T.-S.Y.; formal analysis, D.S. and A.A.; investigation, D.S., J.L., A.A., J.H.P. and M.-H.C.; resources, K.M.O., K.H.L. and T.-S.Y.; data curation, D.S., J.L., M.-H.C., J.H.P. and A.A.; writing—original draft preparation, D.S. and A.A.; writing—review and editing, J.L., A.A. and T.-S.Y.; visualization, D.S., J.L. and T.-S.Y.; supervision, T.-S.Y.; project administration, T.-S.Y.; funding acquisition, T.-S.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (RS-2024-00337629 and NRF-2022M3H4A1A04076667).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original supplementary crystallographic data for this study is openly available in the Cambridge Crystallographic Data Center (12 Union Road, Cambridge CB2 1EZ, UK; fax: +44-1223-336033) at www.ccdc.cam.ac.uk/data_request/cif (accessed on 10 January 2025), or by emailing data_request@ccdc.cam.ac.uk. The reference/accession number to be used is CCDC 2405340-2405343. Furthermore, the raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tritt, T.M. Thermoelectric phenomena, materials, and applications. Annu. Rev. Mater. Res. 2011, 41, 433–448. [Google Scholar] [CrossRef]
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.M. Thermoelectrics Handbook—Macro to Nano; CRC-Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Fernández-Yáñez, P.; Romero, V.; Armas, O.; Cerretti, G. Thermal management of thermoelectric generators for waste energy recovery. Appl. Therm. Eng. 2021, 196, 117291. [Google Scholar] [CrossRef]
- Tritt, T.M.; Subramanian, M. Thermoelectric materials, phenomena, and applications: A bird’s eye view. MRS Bull. 2006, 31, 188–198. [Google Scholar] [CrossRef]
- Kauzlarich, S.M.; Brown, S.R.; Snyder, G.J. Zintl phases for thermoelectric devices. Dalton Trans. 2007, 21, 2099–2107. [Google Scholar] [CrossRef]
- Khatun, M.; Stoyko, S.S.; Mar, A. Quaternary Arsenides ACdGeAs2 (A = K, Rb) Built of Ethane-Like Ge2As6 Units. Inorg. Chem. 2014, 53, 7756–7762. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.M.; Mar, A. Layered Rare-Earth Gallium Antimonides RE GaSb2 (RE = La − Nd, Sm). J. Am. Chem. Soc. 2001, 123, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.I.; Zevalkink, A.; Snyder, G.J. Improved thermoelectric properties in Zn-doped Ca5Ga2Sb6. J. Mater. Chem. A 2013, 1, 4244–4249. [Google Scholar] [CrossRef]
- Aydemir, U.; Candolfi, C.; Borrmann, H.; Baitinger, M.; Ormeci, A.; Carrillo-Cabrera, W.; Chubilleau, C.; Lenoir, B.; Dauscher, A.; Oeschler, N. Crystal structure and transport properties of Ba8Ge43□ 3. Dalton Trans. 2010, 39, 1078–1088. [Google Scholar] [CrossRef]
- Aydemir, U.; Candolfi, C.; Ormeci, A.; Borrmann, H.; Burkhardt, U.; Oztan, Y.; Oeschler, N.; Baitinger, M.; Steglich, F.; Grin, Y. Synthesis, Crystal Structure, and Physical Properties of the Type-I Clathrate Ba8− δNix□ ySi46–x–y. Inorg. Chem. 2012, 51, 4730–4741. [Google Scholar] [CrossRef]
- Pomrehn, G.S.; Zevalkink, A.; Zeier, W.G.; Van De Walle, A.; Snyder, G.J. Defect-Controlled Electronic Properties in AZn2Sb2 Zintl Phases. Angew. Chem. Int. Ed. 2014, 53, 3422–3426. [Google Scholar] [CrossRef]
- Brown, S.R.; Kauzlarich, S.M.; Gascoin, F.; Snyder, G.J. Yb14MnSb11: New high efficiency thermoelectric material for power generation. Chem. Mater. 2006, 18, 1873–1877. [Google Scholar] [CrossRef]
- Gascoin, F.; Ottensmann, S.; Stark, D.; Haïle, S.M.; Snyder, G.J. Zintl phases as thermoelectric materials: Tuned transport properties of the compounds CaxYb1–xZn2Sb2. Adv. Funct. Mater. 2005, 15, 1860–1864. [Google Scholar] [CrossRef]
- Wartenberg, F.; Kranenberg, C.; Pocha, R.; Johrendt, D.; Mewis, A.; Hoffmann, R.-D.; Mosel, B.D.; Pöttgen, R. Neue Pnictide im CaAl2Si2-Typ und dessen Existenzgebiet. Z. Naturforsch. 2002, 57b, 1270–1276. [Google Scholar] [CrossRef]
- Pandey, T.; Singh, A.K. High thermopower and ultra low thermal conductivity in Cd-based Zintl phase compounds. Phys. Chem. Chem. Phys. 2015, 17, 16917–16926. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chanakian, S.; Zevalkink, A. Crystal chemistry and thermoelectric transport of layered AM2X 2 compounds. Inorg. Chem. Frontiers 2018, 5, 1744–1759. [Google Scholar] [CrossRef]
- Wang, Z.-C.; Been, E.; Gaudet, J.; Alqasseri, G.M.A.; Fruhling, K.; Yao, X.; Stuhr, U.; Zhu, Q.; Ren, Z.; Cui, Y. Anisotropy of the magnetic and transport properties of EuZn2As2. Phys. Rev. B 2022, 105, 165122. [Google Scholar] [CrossRef]
- Wang, X.-J.; Tang, M.-B.; Chen, H.-H.; Yang, X.-X.; Zhao, J.-T.; Burkhardt, U.; Grin, Y. Synthesis and high thermoelectric efficiency of Zintl phase YbCd2− xZnxSb2. Appl. Phys. Lett. 2009, 94, 092106. [Google Scholar] [CrossRef]
- Zhang, H.; Baitinger, M.; Tang, M.-B.; Man, Z.-Y.; Chen, H.-H.; Yang, X.-X.; Liu, Y.; Chen, L.; Grin, Y.; Zhao, J.-T. Thermoelectric properties of Eu(Zn1− xCdx)2Sb2. Dalton Trans. 2010, 39, 1101–1104. [Google Scholar] [CrossRef]
- Balvanz, A.; Baranets, S.; Ogunbunmi, M.O.; Bobev, S. Two polymorphs of BaZn2P2: Crystal structures, phase transition, and transport properties. Inorg. Chem. 2021, 60, 14426–14435. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Guo, K.; Yang, X.; Xing, J.; Wang, K.; Luo, J.; Zhao, J.-T. Realizing high thermoelectric performance in BaCu2–xAgxTe2 through enhanced carrier effective mass and point-defect scattering. ACS Appl. Energy Mater. 2018, 2, 889–895. [Google Scholar] [CrossRef]
- Wang, X.-J.; Tang, M.-B.; Zhao, J.-T.; Chen, H.-H.; Yang, X.-X. Thermoelectric properties and electronic structure of Zintl compound BaZn2Sb2. Appl. Phys. Lett. 2007, 90, 232107. [Google Scholar] [CrossRef]
- Jeong, J.; Shim, D.; Yox, P.; Choi, M.-H.; Ok, K.M.; Kovnir, K.; Miller, G.J.; You, T.-S. Tuning the Radius Ratio to Enhance Thermoelectric Properties in the Zintl Compounds AM2Sb2 (A = Ba, Sr; M = Zn, Cd). Chem. Mater. 2023, 35, 3985–3997. [Google Scholar] [CrossRef]
- Jeong, J.; Shim, D.; Choi, M.H.; Ok, K.M.; You, T.S. Effect of co-substitution on complex thermoelectric compounds: The Zintl phase Ba1-xSrxZn2-yCuySb2 system. Bull. Korean Chem. Soc. 2024, 45, 165–170. [Google Scholar] [CrossRef]
- Jeong, J.; Shim, D.; Choi, M.-H.; Yunxiu, Z.; Kim, D.-H.; Ok, K.M.; You, T.-S. Golden ratio of the r+/r- for the structure-selectivity in the thermoelectric BaZn2-xCdxSb2 system. J. Alloys Compd. 2024, 1002, 175272. [Google Scholar] [CrossRef]
- Hong, Y.; Yeon, S.; Yox, P.; Yunxiu, Z.; Choi, M.-H.; Moon, D.; Ok, K.M.; Kim, D.-H.; Kovnir, K.; Miller, G.J.; et al. Role of Eu-Doping in the Electron Transport Behavior in the Zintl Thermoelectric Ca5–x–yYbxEuyAl2Sb6 System. Chem. Mater. 2022, 34, 9903–9914. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Andersen, O.K. Linear methods in band theory. Phys. Rev. B 1975, 12, 3060. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O. Explicit, first-principles tight-binding theory. Phys. Rev. Lett. 1984, 53, 2571. [Google Scholar] [CrossRef]
- Lambrecht, W.R.; Andersen, O.K. Minimal basis sets in the linear muffin-tin orbital method: Application to the diamond-structure crystals C, Si, and Ge. Phys. Rev. B 1986, 34, 2439. [Google Scholar] [CrossRef]
- Jepsen, O.; Burkhardt, A. The TB-LMTO-ASA Program, version 4.7; Max-Plank-Institut fur Festkorperforschung: Shuttgart, Germany, 1999. [Google Scholar]
- Madsen, G.K.H. Automated Search for New Thermoelectric Materials: The Case of LiZnSb. J. Am. Chem. Soc. 2006, 128, 12140–12146. [Google Scholar] [CrossRef] [PubMed]
- Hamidani, A.; Bennecer, B.; Zanat, K. Effect of Sr substitution on the structural, electronic and thermoelectric properties of the Zintl-phase compound BaZn2Sb2. Phys. Scr. 2023, 98, 065910. [Google Scholar] [CrossRef]
- Chen, C.; Xue, W.; Li, S.; Zhang, Z.; Li, X.; Wang, X.; Liu, Y.; Sui, J.; Liu, X.; Cao, F. Zintl-phase Eu2ZnSb2: A promising thermoelectric material with ultralow thermal conductivity. Proc. Nat. Acad. Sci. 2019, 116, 2831–2836. [Google Scholar] [CrossRef]
- Chen, C.; Xue, W.; Li, X.; Lan, Y.; Zhang, Z.; Wang, X.; Zhang, F.; Yao, H.; Li, S.; Sui, J. Enhanced Thermoelectric Performance of Zintl Phase Ca9Zn4+xSb9 by Beneficial Disorder on the Selective Cationic Site. ACS Appl. Mater. Interfaces. 2019, 11, 37741–37747. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, P.; Balasubramanian, G. Engineering phonon transport through cation disorder in dimensionally constricted high entropy MXen. Carbon 2024, 223, 119015. [Google Scholar] [CrossRef]
- Toberer, E.S.; May, A.F.; Snyder, G.J. Zintl Chemistry for Designing High Efficiency Thermoelectric Materials. Chem. Mater. 2010, 22, 624–634. [Google Scholar] [CrossRef]
- Yang, S.; Lin, C.; He, X.; Huang, J.; Snyder, G.J.; Lin, Y.; Luo, M. Unlocking Ultralow Thermal Conductivity in α-CuTeI via Specific Symmetry Breaking in Cu Sublattice. Adv. Funct. Mater. 2024, 2419776. [Google Scholar] [CrossRef]
- Borgsmiller, L.; Snyder, G.J. Thermoelectric properties and low thermal conductivity of Zintl compound Yb10MnSb9. J. Mater. Chem. A 2022, 10, 15127–15135. [Google Scholar] [CrossRef]
- Kim, H.-S.; Gibbs, Z.M.; Tang, Y.; Wang, H.; Snyder, G.J. Characterization of Lorenz number with Seebeck coefficient measurement. APL Mater. 2015, 3, 041506. [Google Scholar] [CrossRef]
- Lim, S.-J.; Nam, G.; Shin, S.; Ahn, K.; Lee, Y.; You, T.-S. Anionic Doping and Cationic Site Preference in CaYb4Al2Sb6–xGex (x = 0.2, 0.5, 0.7): Origin of the Enhanced Seebeck Coefficient and the Structural Transformation. Inorg. Chem. 2019, 58, 5827–5836. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Misra, D. Enhancing thermoelectric properties of a p-type Mg3Sb2-based Zintl phase compound by Pb substitution in the anionic framework. RSC Adv. 2014, 4, 34552–34560. [Google Scholar] [CrossRef]
- Zhu, T.; Yu, C.; He, J.; Zhang, S.; Zhao, X.; Tritt, T.M. Thermoelectric properties of Zintl compound YbZn2Sb2 with Mn substitution in anionic framework. J. Electron. Mater. 2009, 38, 1068–1071. [Google Scholar] [CrossRef]
- Bruker AXS Inc. APEX3, version 2019.1-0; Bruker AXS Inc.: Madison, WI, USA, 2006. [Google Scholar]
- Bruker AXS Inc. SAINT Program, version 8.40A; Bruker AXS Inc.: Madison, WI, USA, 2002. [Google Scholar]
- Sheldrick, G.M. SADABS, version 2016/2; University of Göttingen: Göttingen, Germany, 2003. [Google Scholar]
- Shin, J.W.; Eom, K.; Moon, D. BL2D-SMC, the supramolecular crystallography beamline at the Pohang Light Source II, Korea. J. Synchrotron Radiat. 2016, 23, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Gelato, L.; Parthé, E. STRUCTURE TIDY—A computer program to standardize crystal structure data. J. Appl. Crystallogr. 1987, 20, 139–143. [Google Scholar] [CrossRef]
- Andersen, O.K.; Jepsen, O.; Glötzel, D. Canonical Description of the Band Structures of Metals. In Highlights of Condensed Matter Theory; Bassani, F., Fumi, F., Tosi, M., Eds.; Elsevier North Holland: New York, NY, USA, 1985; pp. 65–72. [Google Scholar]
- Jepsen, O.; Andersen, O. Calculated electronic structure of the sandwich d1 metals LaI2 and CeI2: Application of new LMTO techniques. Z. Phys. B Condens. Matter. 1995, 97, 35–47. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef]
- Borup, K.A.; De Boor, J.; Wang, H.; Drymiotis, F.; Gascoin, F.; Shi, X.; Chen, L.; Fedorov, M.I.; Müller, E.; Iversen, B.B. Measuring thermoelectric transport properties of materials. Energy Environ. Sci. 2015, 8, 423–435. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).