
Determination of 14 Benzodiazepine Multiresidues in Aquaculture Environment by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Instruments and Reagents
2.2. Preparation of Stock, Working, and Calibration Curve
2.3. Sample Pretreatment
2.3.1. Sample Collection and Preparation
2.3.2. Extraction
2.4. UHPLC–MS/MS Analysis
2.4.1. Chromatographic Conditions
2.4.2. Mass Spectrometry Conditions
2.5. Validation of Analytical Method
3. Results and Discussion
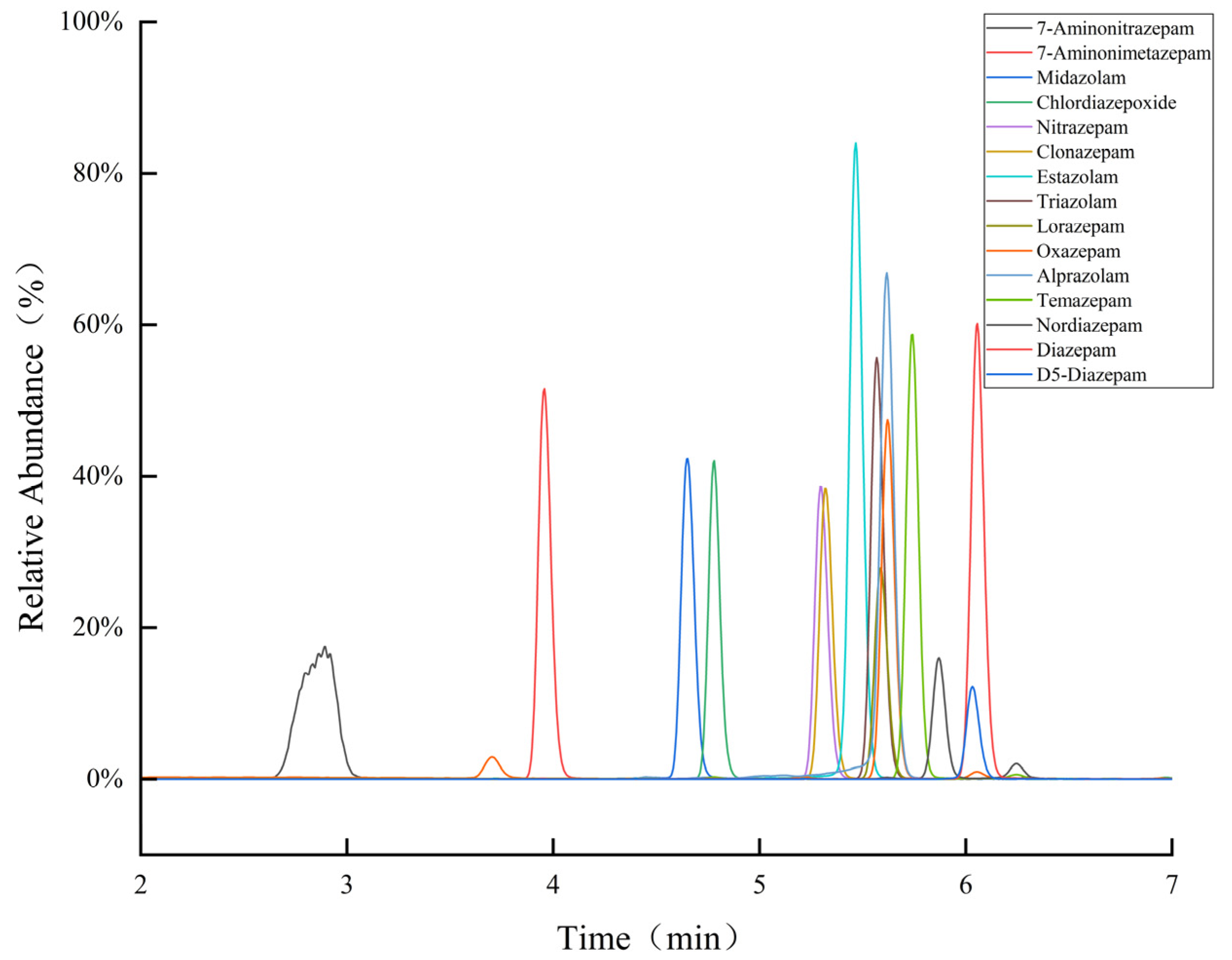
3.1. Optimization of Chromatographic Conditions
3.2. Optimization of Mass Spectrometry Parameters
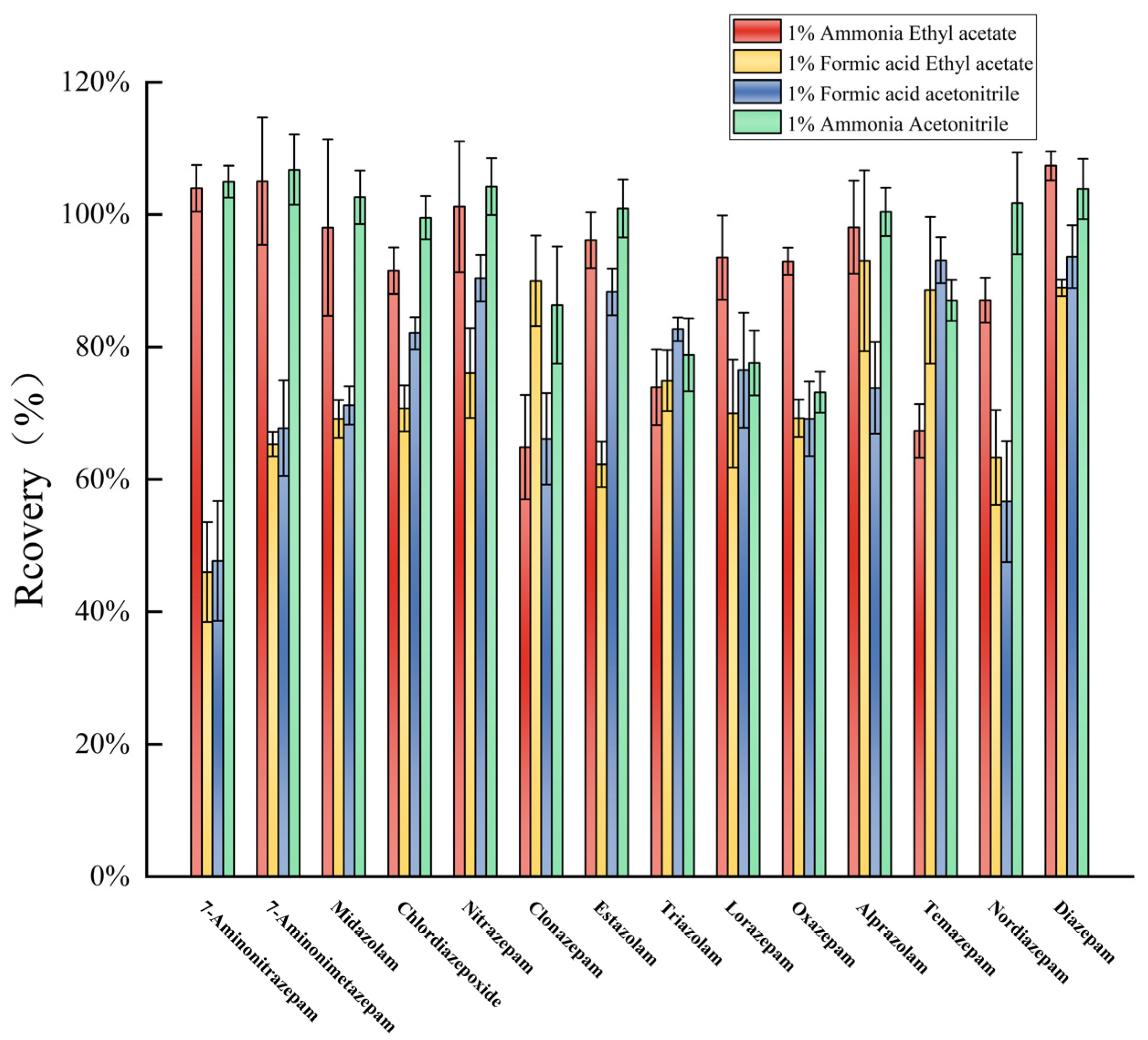
3.3. Optimization of Water Sample Pretreatment Conditions
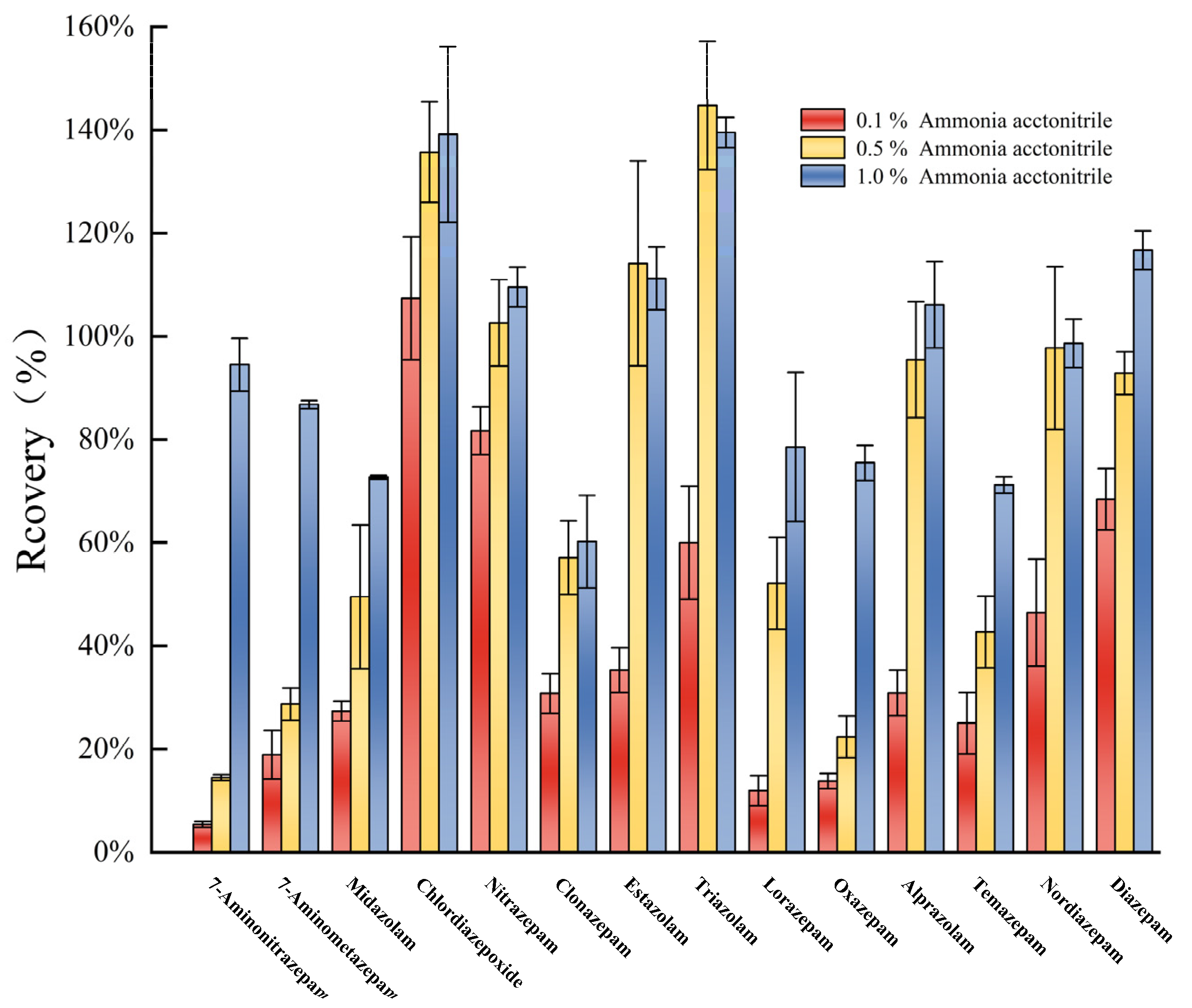
3.4. Optimization of Sediment Pretreatment Conditions
3.5. Method Validation
3.5.1. Linear Range and Sensitivity
3.5.2. Matrix Effect
3.5.3. Accuracy and Precision
3.6. Comparison with Other Methods
4. Conclusions
- The recoveries of most BDZs ranged from 70% to 120% at three different concentration levels for both water and sediment matrices, and the intra- and inter-day RSDs were <15% at the three different concentration levels. The LOD and LOQ ranges were 0.002–0.01 μg/L and 0.01–0.03 μg/L and 0.01–0.50 μg/kg and 0.04–1.00 μg/kg for the water samples and sediment matrix, respectively.
- The proposed method is highly sensitive and has good recovery. It is simple, efficient, rapid, and low-cost and could simultaneously analyze and detect 14 BDZs.
- With potential applications such as locating contamination hotspots, monitoring compliance with permissible limits, and evaluating how various technologies reduce the presence of these contaminants, this method provides technical support to detect BDZ residues in aquaculture environments.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hofe, I.v.; Stricker, B.H.; Vernooij, M.W.; Ikram, M.K.; Ikram, M.A.; Wolters, F.J. Benzodiazepine use in relation to long-term dementia risk and imaging markers of neurodegeneration: A population-based study. BMC Med. 2024, 22, 266. [Google Scholar] [CrossRef] [PubMed]
- Wachełko, O.; Szpot, P.; Tusiewicz, K.; Nowak, K.; Chłopaś-Konowałek, A.; Zawadzki, M. An ultra-sensitive UHPLC-QqQ-MS/MS method for determination of 54 benzodiazepines (pharmaceutical drugs, NPS and metabolites) and z-drugs in biological samples. Talanta 2023, 251, 123816. [Google Scholar] [CrossRef] [PubMed]
- Maust, D.T.; Petzold, K.; Strominger, J.; Kim, H.M.; Bohnert, A.S.B. Benzodiazepine Discontinuation and Mortality Among Patients Receiving Long-Term Benzodiazepine Therapy. JAMA Netw. Open 2023, 6, e2348557. [Google Scholar] [CrossRef]
- Aldaz, P.; Garjón, J.; Beitia, G.; Beltrán, I.; Librero, J.; Ibáñez, B.; Arroyo, P.; Ariz, M.J. Association between benzodiazepine use and development of dementia. Med. Clin. 2021, 156, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Zetsen, S.P.G.; Schellekens, A.F.A.; Paling, E.P.; Kan, C.C.; Kessels, R.P.C. Cognitive Functioning in Long-Term Benzodiazepine Users. Eur. Addict. Res. 2022, 28, 377–381. [Google Scholar] [CrossRef]
- Kawano, T.; Oshibuchi, H.; Kawano, M.; Muraoka, H.; Tsutsumi, T.; Yamada, M.; Ishigooka, J.; Nishimura, K.; Inada, K. Diazepam suppresses the stress-induced dopaminergic release in the amygdala of methamphetamine-sensitized rat. Eur. J. Pharmacol. 2018, 833, 247–254. [Google Scholar] [CrossRef]
- Mathieu, C.; Joly, P.; Jacqmin-Gadda, H.; Wanneveich, M.; Bégaud, B.; Pariente, A. Patterns of Benzodiazepine Use and Excess Risk of All-Cause Mortality in the Elderly: A Nationwide Cohort Study. Drug Saf. 2020, 44, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, S.; Kumar, R.; Driver, E.M.; Bowes, D.A.; Ng, K.T.; Sosa-Hernandez, J.E.; Oyervides-Munoz, M.A.; Melchor-Martinez, E.M.; Martinez-Ruiz, M.; Coronado-Apodaca, K.G.; et al. Occurrence of Z-drugs, benzodiazepines, and ketamine in wastewater in the United States and Mexico during the COVID-19 pandemic. Sci. Total Environ. 2023, 857, 159351. [Google Scholar] [CrossRef]
- Nunes, C.N.; dos Anjos, V.E.; Quináia, S.P. Are there pharmaceutical compounds in sediments or in water? Determination of the distribution coefficient of benzodiazepine drugs in aquatic environment. Environ. Pollut. 2019, 251, 522–529. [Google Scholar] [CrossRef]
- Carter, L.J.; Williams, M.; Martin, S.; Kamaludeen, S.P.B.; Kookana, R.S. Sorption, plant uptake and metabolism of benzodiazepines. Sci. Total Environ. 2018, 628–629, 18–25. [Google Scholar] [CrossRef]
- Cerveny, D.; Brodin, T.; Cisar, P.; McCallum, E.S.; Fick, J. Bioconcentration and behavioral effects of four benzodiazepines and their environmentally relevant mixture in wild fish. Sci. Total Environ. 2020, 702, 134780. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, A.; Daryasari, A.P.; Soleimani, M. A Molecularly Imprinted Polymer as the Adsorbent for the Selective Determination of Oxazepam in Urine and Plasma Samples by High-Performance Liquid Chromatography with Diode Array Detection. J. Anal. Chem. 2021, 76, 1414–1421. [Google Scholar] [CrossRef]
- Laura, D.; Ana, M.F.P.; Robert, K.; Elena, L.; Ana, d.C.; Marta, C. Fast and Sensitive Method for the Determination of 17 Designer Benzodiazepines in Hair by Liquid Chromatography–Tandem Mass Spectrometry. J. Anal. Toxicol. 2022, 46, 807. [Google Scholar] [CrossRef]
- Liu, J.; Wurita, A.; Wei, X.; Yang, H.; Gu, J.; Chen, L. Supramolecular solvent (SUPRASs) extraction method for detecting benzodiazepines and zolpidem in human urine and blood using gas chromatography tandem mass spectrometry. Leg. Med. 2021, 48, 101822. [Google Scholar] [CrossRef]
- Sorribes-Soriano, A.; Albert Esteve-Turrillas, F.; Armenta, S.; Manuel Herrero-Martínez, J. Molecularly imprinted polymer–stir bar sorptive extraction of diazepam from natural water. Microchem. J. 2023, 186, 108354. [Google Scholar] [CrossRef]
- Sheikh, M.; Hadjmohammadi, M.R.; Fatemi, M.H. Simultaneous extraction and analysis of clozapine and lorazepam from human plasma using dual solvent-stir bar microextraction with different acceptor phases followed by high-performance liquid chromatography ultra-violet detection. Anal. Methods 2021, 13, 110–116. [Google Scholar] [CrossRef]
- Rossi, B.; Freni, F.; Vignali, C.; Stramesi, C.; Collo, G.; Carelli, C.; Moretti, M.; Galatone, D.; Morini, L. Comparison of Two Immunoassay Screening Methods and a LC-MS/MS in Detecting Traditional and Designer Benzodiazepines in Urine. Molecules 2021, 27, 112. [Google Scholar] [CrossRef]
- Huang, W.; Moody, D.E.; Andrenyak, D.M.; Rollins, D.E. Immunoassay detection of nordiazepam, triazolam, lorazepam, and alprazolam in blood. J. Anal. Toxicol. 1993, 17, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, K.C. Review of Electroanalytical-Based Approaches for the Determination of Benzodiazepines. Biosensors 2019, 9, 130. [Google Scholar] [CrossRef]
- Schram, J.; Parrilla, M.; Sleegers, N.; Slosse, A.; Van Durme, F.; van Nuijs, A.L.N.; De Wael, K. Electrochemical classification of benzodiazepines: A comprehensive approach combining insights from voltammetry and liquid chromatography−mass spectrometry. Talanta 2024, 279, 126623. [Google Scholar] [CrossRef]
- Cirimele, V.; Kintz, P. Identification of Benzodiazepines in Human Hair. In Benzodiazepines and GHB: Detection and Pharmacology; Salamone, S.J., Ed.; Humana Press: Totowa, NJ, USA, 2001; pp. 77–93. [Google Scholar]
- Banaszkiewicz, L.; Woźniak, M.K.; Kaliszan, M.; Kot-Wasik, A. Determination of benzodiazepines and Z-hypnotic drugs in whole blood samples by GC–MS/MS: Method development, validation and application. Microchem. J. 2023, 193, 109115. [Google Scholar] [CrossRef]
- Vincenti, F.; Montesano, C.; Babino, P.; Carboni, S.; Napoletano, S.; De Sangro, G.; Di Rosa, F.; Gregori, A.; Curini, R.; Sergi, M. Finding evidence at a crime scene: Sensitive determination of benzodiazepine residues in drink and food paraphernalia by HPLC-HRMS/MS. Forensic Chem. 2021, 23, 100327. [Google Scholar] [CrossRef]
- Abad, M.O.K.; Masrournia, M.; Javid, A. Synthesis of novel MOF-on-MOF composite as a magnetic sorbent to dispersive micro solid phase extraction of benzodiazepine drugs prior to determination with HPLC-UV. Microchem. J. 2024, 197, 109797. [Google Scholar] [CrossRef]
- Brian, R.; Francesca, F.; Claudia, C.; Matteo, M.; Dario, G.; Claudia, V.; Luca, M. Determination of Traditional and Designer Benzodiazepines in Urine through. Curr. Pharm. Des. 2022, 28, 2622–2638. [Google Scholar] [CrossRef]
- Bajaj, A.O.; Ly, D.; Johnson-Davis, K.L. Retrospective analysis of metabolite patterns of clobazam and N-desmethylclobazam in human plasma by LC-MS/MS. J. Mass Spectrom. Adv. Clin. Lab 2022, 24, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Guideline, I.H.T.J.Q. Validation of analytical procedures: Text and methodology. Q2 (R1) 2005, 1, 05. [Google Scholar]
- Khaled, O.; Ryad, L.; Eissa, F. Determination of tetracycline residues in potatoes and soil by LC-MS/MS: Method development, validation, and risk assessment. Food Chem. 2024, 461, 140841. [Google Scholar] [CrossRef]
- Hou, Q.; Wang, Y.; Hu, J.; Zhang, J.; Zhang, C.; Song, W.; Wang, X.; Zheng, B.; Zhou, X. Simultaneous determination of phenothiazine drugs and their metabolites residues in animal derived foods by high performance liquid chromatography tandem mass spectrometry. Food Control 2025, 167, 110799. [Google Scholar] [CrossRef]
- Santini, S.; Fortini, M.; Martellini, T.; Marinelli, C.; Cincinelli, A. Multi-residue method validation of a LC-MS/MS method for quantitative determination of 349 pesticides in tomato and their health risk assessment through monitoring studies. J. Food Compos. Anal. 2024, 128, 106049. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Lin, J.; Bao, R. A modified QuEChERS-based UPLC-MS/MS method for rapid determination of multiple antibiotics and sedative residues in freshwater fish. Food Chem. X 2024, 22, 101268. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Zhang, Y.; Bian, Y.; Liu, Y.-J.; Ren, A.; Zhou, Y.; Shi, D.; Feng, X.-S. Benzodiazepines in complex biological matrices: Recent updates on pretreatment and detection methods. J. Pharm. Anal. 2023, 13, 442–462. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, Z.; Shi, Q.; Zhang, R.; Sun, A.; Zhao, J.; Wu, Y.; Shi, X. Rapid determination and risk evaluation of multi-class antibiotics in aquatic products by one-step purification process coupled with ultra-high performance liquid chromatography–tandem mass spectrometry. Talanta 2024, 277, 126421. [Google Scholar] [CrossRef]
- Albishri, H.M.; Aldawsari, N.A.; Abd El-Hady, D. A Simple and Reliable Liquid Chromatographic Method for Simultaneous Determination of Five Benzodiazepine Drugs in Human Plasma. Analytica 2022, 3, 251–265. [Google Scholar] [CrossRef]
- Bazmi, E.; Behnoush, B.; Akhgari, M.; Bahmanabadi, L. Quantitative analysis of benzodiazepines in vitreous humor by high-performance liquid chromatography. SAGE Open Med. 2016, 4, 2050312116666243. [Google Scholar] [CrossRef] [PubMed]
- Furugen, A.; Nishimura, A.; Kobayashi, M.; Umazume, T.; Narumi, K.; Iseki, K. Quantification of eight benzodiazepines in human breastmilk and plasma by liquid-liquid extraction and liquid-chromatography tandem mass spectrometry: Application to evaluation of alprazolam transfer into breastmilk. J. Pharm. Biomed. Anal. 2019, 168, 83–93. [Google Scholar] [CrossRef]
- He, H.; Xu, L.; Zhang, C.; Fang, N.; Jiang, J.; Wang, X.; Yu, J.; Zhao, X. Determination of three new herbicide residues in soil, sediment and water by liquid chromatography-tandem mass spectrometry. Chin. J. Chromatogr. 2024, 42, 256–263. [Google Scholar] [CrossRef]
- Rabea, D.; Ryad, L.; Shehata, M.R.; Khalaf-Alla, P.A. Determination of 7 nitroimidazoles compounds in meat and natural casing using modified QuEChERS combined with HPLC Orbitrap MS: Impact of meat processing approaches on the analytes residue. Int. J. Environ. Anal. Chem. 2024, 1–16. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; García-Campaña, A.M.; Cruces-Blanco, C. Development and validation of a QuEChERS method for the analysis of 5-nitroimidazole traces in infant milk-based samples by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1562, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Mitrowska, K.; Posyniak, A.; Zmudzki, J. Multiresidue method for the determination of nitroimidazoles and their hydroxy-metabolites in poultry muscle, plasma and egg by isotope dilution liquid chromatography–mass spectrometry. Talanta 2010, 81, 1273–1280. [Google Scholar] [CrossRef]
- González-Mariño, I.; Castro, V.; Montes, R.; Rodil, R.; Lores, A.; Cela, R.; Quintana, J.B. Multi-residue determination of psychoactive pharmaceuticals, illicit drugs and related metabolites in wastewater by ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1569, 91–100. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, S.; Chen, Q.; Cao, L. Salting-out assisted liquid-liquid extraction for the simple and rapid determination of veterinary antibiotic residues in aquatic products by HPLC-MS/MS. Food Chem. 2024, 460, 140775. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.L.; Sunyer-Caldú, A.; Barbosa, S.C.; Primel, E.G.; Fillmann, G.; Diaz Cruz, M.S. Rapid and cost-effective multiresidue analysis of pharmaceuticals, personal care products, and antifouling booster biocides in marine sediments using matrix solid phase dispersion. Chemosphere 2021, 267, 129085. [Google Scholar] [CrossRef] [PubMed]
- Mravcová, L.; Amrichová, A.; Navrkalová, J.; Hamplová, M.; Sedlář, M.; Gargošová, H.Z.; Fučík, J. Optimization and validation of multiresidual extraction methods for pharmaceuticals in Soil, Lettuce, and Earthworms. Environ. Sci. Pollut. Res. 2024, 31, 33120–33140. [Google Scholar] [CrossRef]
- Antignac, J.-P.; de Wasch, K.; Monteau, F.; De Brabander, H.; Andre, F.; Le Bizec, B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal. Chim. Acta 2005, 529, 129–136. [Google Scholar] [CrossRef]
- Zhao, L.; Lucas, D.; Long, D.; Richter, B.; Stevens, J. Multi-class multi-residue analysis of veterinary drugs in meat using enhanced matrix removal lipid cleanup and liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2018, 1549, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.B.; Rübensam, G.; Jank, L.; Barreto, F.; Ruaro Peralba, M.d.C.; Pizzolato, T.M.; Silvia Díaz-Cruz, M.; Barceló, D. Analytical quality assurance in veterinary drug residue analysis methods: Matrix effects determination and monitoring for sulfonamides analysis. Talanta 2015, 132, 443–450. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Bonfiglio, R.; Fernandez-Metzler, C.; Miller-Stein, C.; Olah, T. Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 2000, 11, 942–950. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, L.; Peng, Y.; Luo, M.; Wang, W.; Liu, X. Multiresidue analysis of over 200 pesticides in cereals using a QuEChERS and gas chromatography–tandem mass spectrometry-based method. Food Chem. 2015, 169, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Łozowicka, B.; Rutkowska, E.; Jankowska, M. Influence of QuEChERS modifications on recovery and matrix effect during the multi-residue pesticide analysis in soil by GC/MS/MS and GC/ECD/NPD. Environ. Sci. Pollut. Res. 2017, 24, 7124–7138. [Google Scholar] [CrossRef]
- Robin, J.; Binson, G.; Albouy, M.; Sauvaget, A.; Pierre-Eugène, P.; Migeot, V.; Dupuis, A.; Venisse, N. Analytical method for the biomonitoring of bisphenols and parabens by liquid chromatography coupled to tandem mass spectrometry in human hair. Ecotoxicol. Environ. Saf. 2022, 243, 113986. [Google Scholar] [CrossRef]
- Gupta, N.; Thakur, R.S.; Patel, D.K. Detection, quantification and degradation kinetic for five benzodiazepines using VAUS-ME-SFO/LC-MS/MS method for water, alcoholic and non-alcoholic beverages. Talanta 2023, 260, 124572. [Google Scholar] [CrossRef]
- Ghadi, M.; Hadjmohammadi, M.R. Extraction and determination of three benzodiazepines in aqueous and biological samples by air-assisted liquid–liquid microextraction and high-performance liquid chromatography. J. Iran. Chem. Soc. 2019, 16, 1147–1155. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | B (%) | C (%) |
|---|---|---|
| 0–0.5 | 10 | 90 |
| 0.5–2.0 | 40 | 60 |
| 2.0–2.5 | 60 | 40 |
| 2.5–8.0 | 90 | 10 |
| 8.0–9.1 | 10 | 90 |
| 9.1–11 | 10 | 90 |
| Compound | Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy (V) | Retention Time (min) |
|---|---|---|---|---|
| 7-Aminonitrazepam | 252.2 | 121.1 * | 25.35 | 2.89 |
| 224.1 | 19.49 | |||
| 7-Aminonimetazepam | 266.2 | 135.1 * | 24.17 | 3.96 |
| 209.1 | 23.49 | |||
| Midazolam | 326.2 | 291.1 * | 24.55 | 4.65 |
| 249.1 | 34.19 | |||
| Chlordiazepoxide | 300.2 | 227.1 * | 23.37 | 4.78 |
| 283.1 | 12.16 | |||
| Nitrazepam | 282.2 | 236.1 * | 22.61 | 5.29 |
| 180.1 | 34.99 | |||
| Clonazepam | 316.1 | 270.0 * | 23.41 | 5.32 |
| 214.1 | 35.92 | |||
| Estazolam | 295.2 | 267.1 * | 22.57 | 5.47 |
| 205.1 | 38.36 | |||
| Triazolam | 343.1 | 308.1 * | 25.05 | 5.57 |
| 239.0 | 38.70 | |||
| Lorazepam | 321.1 | 275.1 * | 20.37 | 5.59 |
| 229.1 | 28.59 | |||
| Oxazepam | 287.2 | 241.1 * | 20.80 | 5.62 |
| 269.0 | 13.21 | |||
| Alprazolam | 309.1 | 281.0 * | 24.92 | 5.63 |
| 205.1 | 39.92 | |||
| Temazepam | 301.1 | 255.0 * | 20.92 | 5.74 |
| 177.1 | 37.10 | |||
| Nordiazepam | 271.1 | 140.1 * | 25.98 | 5.87 |
| 165.1 | 26.65 | |||
| Diazepam | 285.2 | 193.1 * | 30.06 | 6.06 |
| 154.1 | 25.05 | |||
| D5-Diazepam | 290.2 | 198.2 * | 31.33 | 6.03 |
| Compound | Linear Range (μg/L) | Standard Curve | R2 | LOD (μg/L) | LOQ (μg/L) |
|---|---|---|---|---|---|
| 7-Aminonitrazepam | 1~20 | y = 0.6632x − 0.2578 | 0.9939 | 0.010 | 0.030 |
| 7-Aminometazepam | 1~20 | y = 0.6396x − 0.1917 | 0.9910 | 0.008 | 0.030 |
| Midazolam | 1~20 | y = 0.7182x − 0.1904 | 0.9968 | 0.004 | 0.010 |
| Chlordiazepoxide | 1~20 | y = 0.6944x − 0.1643 | 0.9934 | 0.004 | 0.010 |
| Nitrazepam | 1~20 | y = 0.7837x − 0.2544 | 0.9924 | 0.005 | 0.020 |
| Clonazepam | 1~20 | y = 0.6022x − 0.1123 | 0.9928 | 0.003 | 0.010 |
| Estazolam | 1~20 | y = 1.2087x − 0.2424 | 0.9959 | 0.002 | 0.010 |
| Triazolam | 1~20 | y = 0.6004x − 0.1367 | 0.9959 | 0.003 | 0.010 |
| Lorazepam | 1~20 | y = 0.7173x − 0.2568 | 0.9919 | 0.006 | 0.020 |
| Oxazepam | 1~20 | y = 1.0835x − 0.3744 | 0.9927 | 0.005 | 0.020 |
| Alprazolam | 1~20 | y = 0.7672x − 0.2215 | 0.9945 | 0.003 | 0.010 |
| Temazepam | 1~20 | y = 1.8753x + 0.621 | 0.9971 | 0.004 | 0.010 |
| Nordiazepam | 1~20 | y = 0.2429x + 0.0558 | 0.9965 | 0.004 | 0.010 |
| Diazepam | 1~20 | y = 0.8432x − 0.0293 | 0.9991 | 0.004 | 0.010 |
| Compound | Linear Range (μg/L) | Standard Curve | R2 | LOD (μg/kg) | LOQ (μg/kg) |
|---|---|---|---|---|---|
| 7-Aminonitrazepam | 1~20 | y = 0.5021x − 0.3507 | 0.9971 | 0.50 | 1.00 |
| 7-Aminometazepam | 1~20 | y = 0.4486x − 0.4404 | 0.9908 | 0.03 | 0.10 |
| Midazolam | 1~20 | y = 0.5562x − 0.8013 | 0.9946 | 0.02 | 0.05 |
| Chlordiazepoxide | 1~20 | y = 0.3724x − 0.1371 | 0.9974 | 0.03 | 0.10 |
| Nitrazepam | 1~20 | y = 0.4965x − 0.3090 | 0.9955 | 0.03 | 0.08 |
| Clonazepam | 1~20 | y = 0.5298x − 0.0741 | 0.9992 | 0.50 | 1.00 |
| Estazolam | 1~20 | y = 0.865x − 0.3691 | 0.9973 | 0.02 | 0.05 |
| Triazolam | 1~20 | y = 0.6614x − 0.3945 | 0.9949 | 0.02 | 0.06 |
| Lorazepam | 1~20 | y = 0.0664x − 0.0305 | 0.9949 | 0.20 | 0.70 |
| Oxazepam | 1~20 | y = 0.1094x − 0.0619 | 0.9934 | 0.10 | 0.40 |
| Alprazolam | 1~20 | y = 0.9659x − 0.6602 | 0.9917 | 0.01 | 0.04 |
| Temazepam | 1~20 | y = 1.1089x − 0.5865 | 0.9929 | 0.02 | 0.07 |
| Nordiazepam | 1~20 | y = 0.3578x − 0.2268 | 0.9915 | 0.04 | 0.10 |
| Diazepam | 1~20 | y = 0.3075x − 0.099 | 0.9990 | 0.03 | 0.10 |
| Compound | Spiked (μg/L) | Matrix Effect (%) | RSD (%) |
|---|---|---|---|
| 7-Aminonitrazepam | 0.5 | 108.33 | 12.02 |
| 1.0 | 84.56 | 8.80 | |
| 5.0 | 109.57 | 3.18 | |
| 7-Aminometazepam | 0.5 | 111.45 | 14.13 |
| 1.0 | 102.60 | 3.94 | |
| 5.0 | 105.48 | 6.69 | |
| Midazolam | 0.5 | 118.26 | 4.46 |
| 1.0 | 114.77 | 5.73 | |
| 5.0 | 107.28 | 1.50 | |
| Chlordiazepoxide | 0.5 | 119.57 | 11.10 |
| 1.0 | 114.77 | 5.96 | |
| 5.0 | 87.10 | 8.23 | |
| Nitrazepam | 0.5 | 92.59 | 4.47 |
| 1.0 | 95.72 | 1.72 | |
| 5.0 | 97.68 | 5.18 | |
| Clonazepam | 0.5 | 117.53 | 8.63 |
| 1.0 | 91.01 | 2.43 | |
| 5.0 | 95.53 | 14.25 | |
| Estazolam | 0.5 | 108.16 | 7.49 |
| 1.0 | 84.55 | 6.08 | |
| 5.0 | 90.90 | 7.08 | |
| Triazolam | 0.5 | 119.09 | 2.24 |
| 1.0 | 117.70 | 3.07 | |
| 5.0 | 91.84 | 5.65 | |
| Lorazepam | 0.5 | 118.92 | 1.58 |
| 1.0 | 117.37 | 10.34 | |
| 5.0 | 119.63 | 9.92 | |
| Oxazepam | 0.5 | 119.61 | 5.20 |
| 1.0 | 115.15 | 3.99 | |
| 5.0 | 93.38 | 9.06 | |
| Alprazolam | 0.5 | 119.62 | 6.43 |
| 1.0 | 98.36 | 10.22 | |
| 5.0 | 92.70 | 6.18 | |
| Temazepam | 0.5 | 118.15 | 4.66 |
| 1.0 | 115.31 | 13.5 | |
| 5.0 | 88.03 | 5.47 | |
| Nordiazepam | 0.5 | 81.39 | 7.68 |
| 1.0 | 103.76 | 14.36 | |
| 5.0 | 106.11 | 4.60 | |
| Diazepam | 0.5 | 118.07 | 6.72 |
| 1.0 | 94.49 | 14.19 | |
| 5.0 | 98.16 | 3.26 |
| Compound | Spiked (μg/kg) | Matrix Effect (%) | RSD (%) |
|---|---|---|---|
| 7-Aminonitrazepam | 0.5 | 84.56 | 7.29 |
| 2.5 | 108.33 | 11.4 | |
| 5.0 | 87.10 | 4.64 | |
| 7-Aminometazepam | 0.5 | 80.15 | 5.67 |
| 2.5 | 95.72 | 8.81 | |
| 5.0 | 97.68 | 9.19 | |
| Midazolam | 0.5 | 118.26 | 6.19 |
| 2.5 | 115.31 | 3.56 | |
| 5.0 | 101.49 | 10.42 | |
| Chlordiazepoxide | 0.5 | 114.77 | 9.55 |
| 2.5 | 103.76 | 5.3 | |
| 5.0 | 96.11 | 6.4 | |
| Nitrazepam | 0.5 | 91.84 | 10.97 |
| 2.5 | 98.16 | 7.99 | |
| 5.0 | 94.49 | 3.32 | |
| Clonazepam | 0.5 | 88.03 | 2.5 |
| 2.5 | 81.20 | 12.59 | |
| 5.0 | 91.01 | 3.21 | |
| Estazolam | 0.5 | 80.06 | 9.31 |
| 2.5 | 83.55 | 3.3 | |
| 5.0 | 90.90 | 3.25 | |
| Triazolam | 0.5 | 118.07 | 14.68 |
| 2.5 | 120.09 | 12.37 | |
| 5.0 | 107.70 | 2.01 | |
| Lorazepam | 0.5 | 150.25 | 8.66 |
| 2.5 | 130.70 | 10.76 | |
| 5.0 | 126.30 | 7.51 | |
| Oxazepam | 0.5 | 93.38 | 11.48 |
| 2.5 | 115.15 | 4.65 | |
| 5.0 | 121.28 | 3.87 | |
| Alprazolam | 0.5 | 163.29 | 11.47 |
| 2.5 | 138.77 | 14.7 | |
| 5.0 | 82.56 | 2.16 | |
| Temazepam | 0.5 | 95.92 | 11.49 |
| 2.5 | 102.60 | 6.74 | |
| 5.0 | 109.57 | 13.8 | |
| Nordiazepam | 0.5 | 117.53 | 7.25 |
| 2.5 | 90.61 | 8.03 | |
| 5.0 | 111.45 | 9.63 | |
| Diazepam | 0.5 | 108.16 | 3.55 |
| 2.5 | 118.90 | 11.19 | |
| 5.0 | 118.11 | 10.78 |
| Compound | Spiked (μg/L) | Precision (Intra-Day) | Precision (Inter-Day) | ||
|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| 7-Aminonitrazepam | 0.5 | 132.0 | 5.5 | 122.4 | 4.9 |
| 1.0 | 116.2 | 4.1 | 108.5 | 5.4 | |
| 5.0 | 99.3 | 8.2 | 97.8 | 6.2 | |
| 7-Aminometazepam | 0.5 | 124.7 | 4.8 | 119.2 | 4.3 |
| 1.0 | 118.5 | 3.3 | 110.5 | 3.8 | |
| 5.0 | 110.7 | 5.2 | 104.7 | 6.9 | |
| Midazolam | 0.5 | 113.7 | 5.3 | 108.8 | 7.3 |
| 1.0 | 113.5 | 8.8 | 106.7 | 5.3 | |
| 5.0 | 103.7 | 4.2 | 109.8 | 4.2 | |
| Chlordiazepoxide | 0.5 | 115.7 | 2.7 | 112.7 | 3.1 |
| 1.0 | 114.3 | 5.8 | 111.2 | 4.6 | |
| 5.0 | 107.2 | 5.1 | 103.9 | 8.9 | |
| Nitrazepam | 0.5 | 146.7 | 4.7 | 137.5 | 4.9 |
| 1.0 | 119.3 | 4.8 | 113.4 | 5.7 | |
| 5.0 | 94.0 | 6.0 | 102.3 | 6.8 | |
| Clonazepam | 0.5 | 123.7 | 5.8 | 125.8 | 7.3 |
| 1.0 | 112.2 | 5.3 | 108.7 | 3.1 | |
| 5.0 | 104.5 | 3.3 | 106.9 | 4.6 | |
| Estazolam | 0.5 | 112.8 | 2.6 | 109.9 | 3.7 |
| 1.0 | 118.9 | 5.1 | 115.8 | 3.4 | |
| 5.0 | 101.6 | 7.0 | 111.9 | 5.9 | |
| Triazolam | 0.5 | 113.1 | 3.2 | 107.7 | 8.1 |
| 1.0 | 114.4 | 2.3 | 115.8 | 3.2 | |
| 5.0 | 102.2 | 9.5 | 104.9 | 4.3 | |
| Lorazepam | 0.5 | 116.0 | 5.6 | 117.1 | 6.7 |
| 1.0 | 113.4 | 4.2 | 109.3 | 4.5 | |
| 5.0 | 103.0 | 13.6 | 107.6 | 5.2 | |
| Oxazepam | 0.5 | 119.8 | 6.8 | 111.9 | 5.9 |
| 1.0 | 114.5 | 3.5 | 115.4 | 6.9 | |
| 5.0 | 95.8 | 4.2 | 99.7 | 3.2 | |
| Alprazolam | 0.5 | 121.8 | 4.9 | 117.9 | 4.5 |
| 1.0 | 117.9 | 3.1 | 109.7 | 3.6 | |
| 5.0 | 101.9 | 3.2 | 107.3 | 4.9 | |
| Temazepam | 0.5 | 117.1 | 7.1 | 115.9 | 3.2 |
| 1.0 | 114.1 | 5.3 | 108.7 | 7.4 | |
| 5.0 | 95.7 | 3.0 | 98.5 | 6.1 | |
| Nordiazepam | 0.5 | 128.6 | 11.0 | 122.9 | 6.3 |
| 1.0 | 116.7 | 6.8 | 117.8 | 4.5 | |
| 5.0 | 94.9 | 3.8 | 98.4 | 6.8 | |
| Diazepam | 0.5 | 90.3 | 6.7 | 103.8 | 7.2 |
| 1.0 | 114.0 | 4.4 | 109.4 | 6.4 | |
| 5.0 | 96.2 | 6.5 | 101.5 | 5.3 | |
| Compound | Spiked (μg/L) | Precision (Intra-Day) | Precision (Inter-Day) | ||
|---|---|---|---|---|---|
| Recovery (%) | RSD (%) | Recovery (%) | RSD (%) | ||
| 7-Aminonitrazepam | 0.5 | 131.9 | 3.1 | 127.9 | 4.6 |
| 1.0 | 94.0 | 6.2 | 98.2 | 5.1 | |
| 5.0 | 93.7 | 9.1 | 89.3 | 7.9 | |
| 7-Aminometazepam | 0.5 | 117.7 | 2.2 | 107.4 | 3.9 |
| 1.0 | 99.7 | 9.2 | 97.3 | 4.7 | |
| 5.0 | 98.1 | 5.4 | 101.8 | 6.4 | |
| Midazolam | 0.5 | 113.9 | 2.4 | 109.9 | 4.5 |
| 1.0 | 95.3 | 3.7 | 105.8 | 3.3 | |
| 5.0 | 92.8 | 9.0 | 95.7 | 8.2 | |
| Chlordiazepoxide | 0.5 | 112.2 | 3.1 | 115.9 | 4.2 |
| 1.0 | 89.9 | 10.3 | 93.2 | 7.6 | |
| 5.0 | 82.5 | 11.0 | 88.7 | 3.4 | |
| Nitrazepam | 0.5 | 109.8 | 5.1 | 113.2 | 3.2 |
| 1.0 | 93.4 | 3.7 | 103.7 | 4.5 | |
| 5.0 | 77.7 | 7.7 | 94.4 | 6.6 | |
| Clonazepam | 0.5 | 102.2 | 6.0 | 104.8 | 5.7 |
| 1.0 | 86.8 | 4.7 | 96.7 | 5.2 | |
| 5.0 | 82.5 | 6.0 | 92.2 | 6.7 | |
| Estazolam | 0.5 | 116.8 | 0.7 | 113.6 | 4.5 |
| 1.0 | 84.2 | 5.0 | 89.7 | 4.9 | |
| 5.0 | 84.7 | 7.0 | 93.5 | 5.7 | |
| Triazolam | 0.5 | 115.6 | 6.3 | 109.7 | 5.4 |
| 1.0 | 95.5 | 5.3 | 103.5 | 6.1 | |
| 5.0 | 91.0 | 7.7 | 96.8 | 4.7 | |
| Lorazepam | 0.5 | 130.5 | 4.1 | 133.7 | 6.4 |
| 1.0 | 77.1 | 3.8 | 89.4 | 3.2 | |
| 5.0 | 80.1 | 10.5 | 91.1 | 2.7 | |
| Oxazepam | 0.5 | 134.7 | 3.3 | 129.5 | 4.2 |
| 1.0 | 76.6 | 8.2 | 83.3 | 5.9 | |
| 5.0 | 82.2 | 9.6 | 84.9 | 6.8 | |
| Alprazolam | 0.5 | 148.0 | 4.4 | 131.1 | 7.1 |
| 1.0 | 94.0 | 5.6 | 99.4 | 2.4 | |
| 5.0 | 88.6 | 7.7 | 92.3 | 4.5 | |
| Temazepam | 0.5 | 126.1 | 3.7 | 118.9 | 5.6 |
| 1.0 | 84.2 | 6.4 | 88.9 | 8.3 | |
| 5.0 | 77.9 | 6.4 | 87.8 | 4.1 | |
| Nordiazepam | 0.5 | 117.3 | 2.5 | 113.7 | 5.2 |
| 1.0 | 90.9 | 5.9 | 104.5 | 7.7 | |
| 5.0 | 86.0 | 12.2 | 98.0 | 4.3 | |
| Diazepam | 0.5 | 114.2 | 3.2 | 117.2 | 7.2 |
| 1.0 | 96.4 | 6.7 | 105.6 | 3.5 | |
| 5.0 | 90.7 | 8.7 | 103.3 | 6.2 | |
| Method | Matrix | Linear Range (μg/L) | LOD (μg/L) | Ref. |
|---|---|---|---|---|
| VAUS-ME-SFO/LC-MS/MS | Water, alcoholic, and non-alcoholic | 0.124–7.810 | 0.316–0.968 | [52] |
| AALLME–HPLC–UV | Water, plasma, and urine | 2.3–800 | 0.7–0.9 | [53] |
| GC-MS | Urine and blood | 0.2–100 | 0.06–1.5 | [14] |
| HPLC-DAD | Human plasma | 3–500 | 1.26–2.09 | [34] |
| UHPLC-MS/MS | Water and sediment | 1–20 | 0.002–0.01 μg/L (water) 0.01–0.5 μg/kg (sediment) | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Chen, J.; Jiang, G.; Mei, Y.; Gong, Z.; Liu, M.; Li, J.; Gan, J. Determination of 14 Benzodiazepine Multiresidues in Aquaculture Environment by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Molecules 2025, 30, 775. https://doi.org/10.3390/molecules30040775
Guo H, Chen J, Jiang G, Mei Y, Gong Z, Liu M, Li J, Gan J. Determination of 14 Benzodiazepine Multiresidues in Aquaculture Environment by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Molecules. 2025; 30(4):775. https://doi.org/10.3390/molecules30040775
Chicago/Turabian StyleGuo, Hongyang, Jianwu Chen, Guangjun Jiang, Yuqing Mei, Zhiqiang Gong, Mingdian Liu, Jinping Li, and Jinhua Gan. 2025. "Determination of 14 Benzodiazepine Multiresidues in Aquaculture Environment by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry" Molecules 30, no. 4: 775. https://doi.org/10.3390/molecules30040775
APA StyleGuo, H., Chen, J., Jiang, G., Mei, Y., Gong, Z., Liu, M., Li, J., & Gan, J. (2025). Determination of 14 Benzodiazepine Multiresidues in Aquaculture Environment by Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry. Molecules, 30(4), 775. https://doi.org/10.3390/molecules30040775