


The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
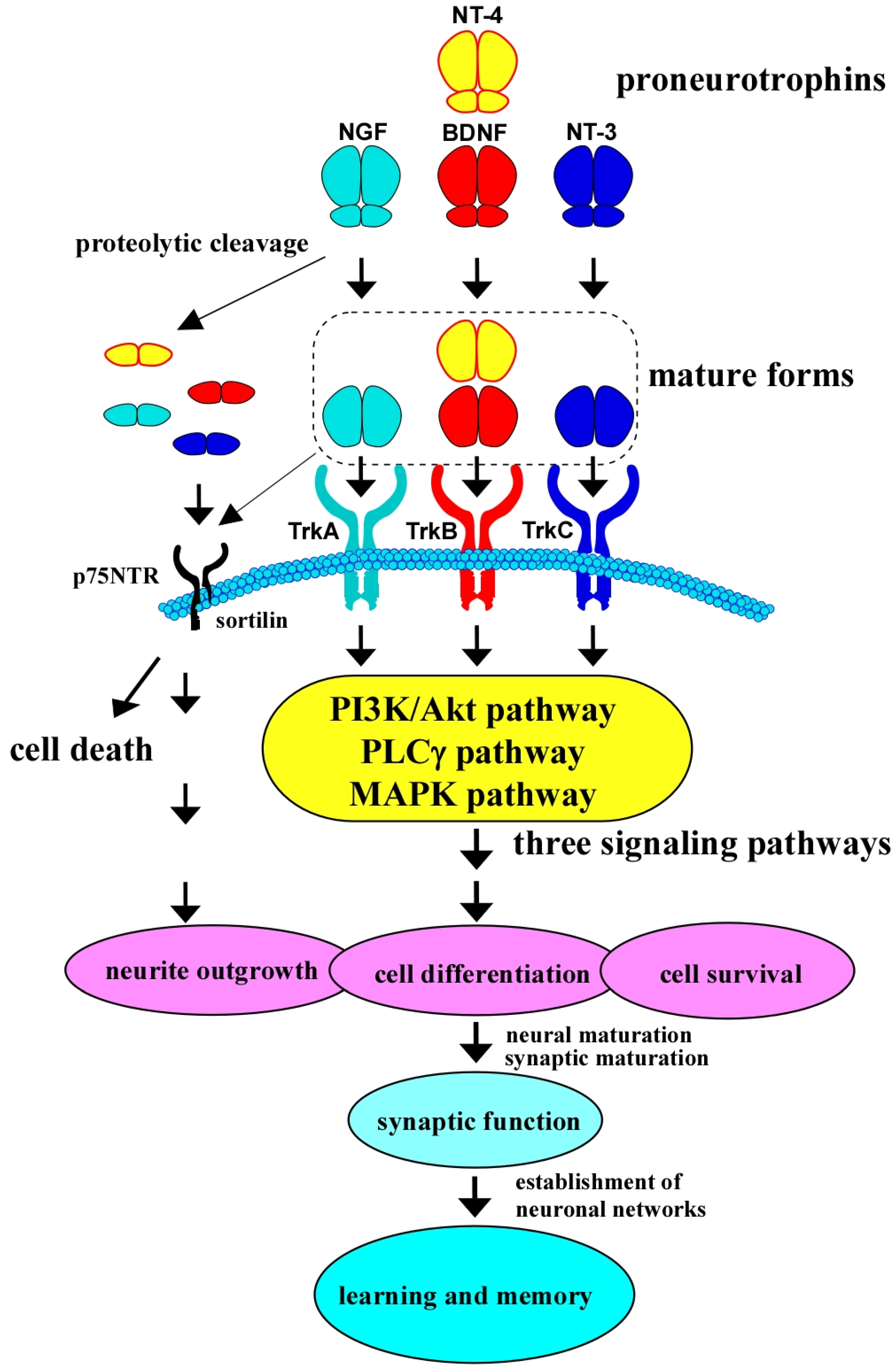
2. Biological Roles of BDNF/TrkB System and Its Downstream Intracellular Signaling
3. BDNF/TrkB System and Antidepressant Effects of Natural Compounds in Depression Models
4. BDNF Mimetics and Their Antidepressant Effects in Depression Models
5. A Variety of Mechanisms Under the Influence of BDNF in Depression Models
6. Relationship Between BDNF/TrkB System and Schizophrenia
7. Contribution of BDNF/TrkB System in Antipsychotic Effects of Natural Compounds in Schizophrenia Models
8. The Therapeutic Potential of BDNF Mimetics in Alzheimer’s Disease (AD)
8.1. The Role of BDNF in AD
8.2. Challenges in Direct BDNF Therapy
8.3. Small-Molecule BDNF Mimetics: Early Efforts
8.4. Next-Generation BDNF Mimetics: Improved Selectivity and Potency
8.5. Preclinical and Clinical Development
9. The Therapeutic Potential of BDNF Mimetics in Parkinson’s Disease (PD) and Huntington’s Disease (HD)
9.1. The Role of BDNF in PD
9.2. BDNF Mimetics in PD
9.3. The Role of BDNF in HD
9.4. BDNF Mimetics in HD
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skaper, S.D. Neurotrophic Factors: An Overview. Methods Mol. Biol. 2018, 1727, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Alfonsetti, M.; d’Angelo, M.; Castelli, V. Neurotrophic factor-based pharmacological approaches in neurological disorders. Neural Regen. Res. 2023, 18, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Kasemeier-Kulesa, J.C.; Morrison, J.A.; Lefcort, F.; Kulesa, P.M. TrkB/BDNF signalling patterns the sympathetic nervous system. Nat. Commun. 2015, 6, 8281. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Xu, B.; Lu, B.; Hempstead, B.L. New insights in the biology of BDNF synthesis and release: Implications in CNS function. J. Neurosci. 2009, 29, 12764–12767. [Google Scholar] [CrossRef]
- Numakawa, T.; Kajihara, R. Involvement of brain-derived neurotrophic factor signaling in the pathogenesis of stress-related brain diseases. Front. Mol. Neurosci. 2023, 16, 1247422. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF signaling in context: From synaptic regulation to psychiatric disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alnaaim, S.A.; Saad, H.M.; Batiha, G.E. The Molecular Pathway of p75 Neurotrophin Receptor (p75NTR) in Parkinson’s Disease: The Way of New Inroads. Mol. Neurobiol. 2024, 61, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.K.; Teng, K.K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R.D.; Kermani, P.; Torkin, R.; Chen, Z.Y.; Lee, F.S.; et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005, 25, 5455–5463. [Google Scholar] [CrossRef]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Meeker, R.B.; Williams, K.S. The p75 neurotrophin receptor: At the crossroad of neural repair and death. Neural Regen. Res. 2015, 10, 721–725. [Google Scholar] [CrossRef]
- Autry, A.E. Function of brain-derived neurotrophic factor in the hypothalamus: Implications for depression pathology. Front. Mol. Neurosci. 2022, 15, 1028223. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Luan, X.; Wang, X.; Li, H.; Zhao, H.; Li, S.; Li, X.; Qiu, Z. Exploring the Association between BDNF related Signaling Pathways and Depression: A Literature Review. Brain Res. Bull. 2024, 220, 111143. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Richards, M.; Nakajima, S.; Adachi, N.; Furuta, M.; Odaka, H.; Kunugi, H. The role of brain-derived neurotrophic factor in comorbid depression: Possible linkage with steroid hormones, cytokines, and nutrition. Front. Psychiatry 2014, 5, 136. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Huang, T.; Zhang, M.; Zhang, Y.; Zeng, Y.; Chen, X. Glucocorticoid receptor signaling in the brain and its involvement in cognitive function. Neural Regen. Res. 2025, 20, 2520–2537. [Google Scholar] [CrossRef]
- Farcas, A.; Hindmarch, C.; Iftene, F. BDNF gene Val66Met polymorphisms as a predictor for clinical presentation in schizophrenia—Recent findings. Front. Psychiatry 2023, 14, 1234220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, S.; Zhang, T.; Yang, F.; Lu, B. Corticosterone antagonist or TrkB agonist attenuates schizophrenia-like behavior in a mouse model combining Bdnf-e6 deficiency and developmental stress. iScience 2022, 25, 104609. [Google Scholar] [CrossRef] [PubMed]
- Tigaret, C.M.; Lin, T.E.; Morrell, E.R.; Sykes, L.; Moon, A.L.; O’Donovan, M.C.; Owen, M.J.; Wilkinson, L.S.; Jones, M.W.; Thomas, K.L.; et al. Neurotrophin receptor activation rescues cognitive and synaptic abnormalities caused by hemizygosity of the psychiatric risk gene Cacna1c. Mol. Psychiatry 2021, 26, 1748–1760. [Google Scholar] [CrossRef]
- Boxer, A.L.; Sperling, R. Accelerating Alzheimer’s therapeutic development: The past and future of clinical trials. Cell 2023, 186, 4757–4772. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Dhaliwal, J.; Sah, S.P. 7,8-Dihydroxyflavone improves cognitive functions in ICV-STZ rat model of sporadic Alzheimer’s disease by reversing oxidative stress, mitochondrial dysfunction, and insulin resistance. Psychopharmacology 2021, 238, 1991–2009. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jeong, Y.J.; Kang, E.J.; Kang, B.S.; Lee, S.H.; Kim, Y.J.; Kang, S.S.; Suh, S.W.; Ahn, E.H. GAP-43 closely interacts with BDNF in hippocampal neurons and is associated with Alzheimer’s disease progression. Front. Mol. Neurosci. 2023, 16, 1150399. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kajihara, R. An Interaction between Brain-Derived Neurotrophic Factor and Stress-Related Glucocorticoids in the Pathophysiology of Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 1596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, X.; Schroeder, J.P.; Chan, C.B.; Song, M.; Yu, S.P.; Weinshenker, D.; Ye, K. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New Avenues for the Treatment of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Moya-Alvarado, G.; Gonzalez-Billaut, C.; Bronfman, F.C. Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton 2016, 73, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. USA 2009, 106, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Matsumoto, T.; Ooshima, Y.; Chiba, S.; Furuta, M.; Izumi, A.; Ninomiya-Baba, M.; Odaka, H.; Hashido, K.; Adachi, N.; et al. Impairments in brain-derived neurotrophic factor-induced glutamate release in cultured cortical neurons derived from rats with intrauterine growth retardation: Possible involvement of suppression of TrkB/phospholipase C-γ activation. Neurochem. Res. 2014, 39, 785–792. [Google Scholar] [CrossRef]
- Alcántara, S.; Frisén, J.; del Río, J.A.; Soriano, E.; Barbacid, M.; Silos-Santiago, I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J. Neurosci. 1997, 17, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [CrossRef] [PubMed]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Kunugi, H. Glucocorticoid suppresses BDNF-stimulated MAPK/ERK pathway via inhibiting interaction of Shp2 with TrkB. FEBS Lett. 2011, 585, 3224–3228. [Google Scholar] [CrossRef] [PubMed]
- Puranik, N.; Jung, H.; Song, M. SPROUTY2, a Negative Feedback Regulator of Receptor Tyrosine Kinase Signaling, Associated with Neurodevelopmental Disorders: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2024, 25, 11043. [Google Scholar] [CrossRef] [PubMed]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Datta, S.R.; Greenberg, M.E. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr. Opin. Neurobiol. 2001, 11, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Nishi, T.; Chan, P.H. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow. Metab. 2006, 26, 1479–1489. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Zarneshan, S.N.; Fakhri, S.; Khan, H. Targeting Akt/CREB/BDNF signaling pathway by ginsenosides in neurodegenerative diseases: A mechanistic approach. Pharmacol. Res. 2022, 177, 106099. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Reddy, D.H. Apoptosis in Alzheimer’s disease: Insight into the signaling pathways and therapeutic avenues. Apoptosis 2023, 28, 943–957. [Google Scholar] [CrossRef]
- Airaksinen, E.; Larsson, M.; Lundberg, I.; Forsell, Y. Cognitive functions in depressive disorders: Evidence from a population-based study. Psychol. Med. 2004, 34, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Peng, R.; Guo, Q.; Cui, J.; Chen, G.; Wang, Z. Bupleurum in Treatment of Depression Disorder: A Comprehensive Review. Pharmaceuticals 2024, 17, 512. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Mo, X.; He, L.; Ma, Q.; Cai, L.; Zheng, Y.; Huang, L.; Lin, X.; Wu, M.; Ding, W.; et al. The role of BDNF transcription in the antidepressant-like effects of 18β-glycyrrhetinic acid in a chronic social defeat stress model. Phytomedicine 2024, 132, 155332. [Google Scholar] [CrossRef] [PubMed]
- Langlois, C.; Potvin, S.; Khullar, A.; Tourjman, S.V. Down and High: Reflections Regarding Depression and Cannabis. Front. Psychiatry 2021, 12, 625158. [Google Scholar] [CrossRef]
- Anand, R.; Anand, L.K.; Rashid, N.; Painuli, R.; Malik, F.; Singh, P.P. Synthesis and Evaluation of Natural and Unnatural Tetrahydrocannabiorcol for Its Potential Use in Neuropathologies. J. Nat. Prod. 2024, 87, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.A.; Yadav, D.; Khan, F.; Song, M. Indole-3-Carbinol and Its Derivatives as Neuroprotective Modulators. Brain Sci. 2024, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gu, H.; Ye, D.; Li, Y.; Chen, Y.; Qiao, H.; Huang, Y.; Tao, R.; Yu, S.; Zhang, J.; et al. NMC-4 Ameliorates Depression-Like Behavior and Neuroinflammation Caused by Chronic Unpredictable Mild Stress. Chem. Biol. Drug Des. 2024, 104, e14626. [Google Scholar] [CrossRef]
- Gong, G.; Ganesan, K.; Wang, Y.; Zhang, Z.; Liu, Y.; Wang, J.; Yang, F.; Zheng, Y. Ononin ameliorates depression-like behaviors by regulating BDNF-TrkB-CREB signaling in vitro and in vivo. J. Ethnopharmacol. 2024, 320, 117375. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, Z.; Chen, L.; Peng, X.; Luan, F.; Hu, J.; Xie, H.; Liu, R.; Zeng, N. Rosmarinic acid alleviate CORT-induced depressive-like behavior by promoting neurogenesis and regulating BDNF/TrkB/PI3K signaling axis. Biomed. Pharmacother. 2024, 170, 115994. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, D.; Kondo, S.; Ferdousi, F.; Mizuno, S.; Yada, A.; Tominaga, K.; Takahashi, S.; Isoda, H. A rare olive compound oleacein functions as a TrkB agonist and mitigates neuroinflammation both in vitro and in vivo. Cell Commun. Signal. 2024, 22, 309. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; Kubo, M.; Harada, K. The search for, and chemistry and mechanism of, neurotrophic natural products. J. Nat. Med. 2020, 74, 648–671. [Google Scholar] [CrossRef] [PubMed]
- Garibova, T.L.; Kraineva, V.A.; Kotel’nikova, S.O.; Povarnina, P.Y.; Gudasheva, T.A.; Seredenin, S.B. Behavioral Effects of Dimeric Dipeptide BDNF Mimetic GSB-106 in a Rat Model of Depressive-Like State. Bull. Exp. Biol. Med. 2020, 169, 286–289. [Google Scholar] [CrossRef]
- Gudasheva, T.A.; Tallerova, A.V.; Mezhlumyan, A.G.; Antipova, T.A.; Logvinov, I.O.; Firsova, Y.N.; Povarnina, P.Y.; Seredenin, S.B. Low-Molecular Weight BDNF Mimetic, Dimeric Dipeptide GSB-106, Reverses Depressive Symptoms in Mouse Chronic Social Defeat Stress. Biomolecules 2021, 11, 252. [Google Scholar] [CrossRef] [PubMed]
- Povarnina, P.Y.; Antipova, T.A.; Logvinov, I.O.; Gudasheva, T.A.; Seredenin, S.B. Chronically Administered BDNF Dipeptide Mimetic GSB-106 Prevents the Depressive-like Behavior and Memory Impairments after Transient Middle Cerebral Artery Occlusion in Rats. Curr. Pharm. Des. 2023, 29, 126–132. [Google Scholar] [CrossRef]
- Tiliwaerde, M.; Gao, N.; Yang, Y.; Jin, Z. A novel NMDA receptor modulator: The antidepressant effect and mechanism of GW043. CNS Neurosci. Ther. 2024, 30, e14598. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Xiao, L.; Liu, R.; Du, J.; Liu, N.; Yu, J.; Li, Y.; Lu, G. Antidepressant effect and mechanism of TMP269 on stress-induced depressive-like behavior in mice. Biochem. Pharmacol. 2024, 225, 116320. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Y.; Zhang, X.; Yu, Z.Z.; Wang, X.Y.; Zeng, Z.H.; Wei, M.X.; Qiu, M.T.; Wang, J.; Cheng, J.; Yi, L.T. Polygonatum sibiricum Polysaccharides Alleviate Depressive-like Symptoms in Chronic Restraint Stress-Induced Mice via Microglial Regulation in Prefrontal Cortex. Polymers 2024, 16, 2358. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Torazza, C.; Ciarla, C.; Bronzuoli, M.R.; Balbi, M.; Bonanno, G.; Popoli, M.; Steardo, L.; Milanese, M.; et al. Molecular signatures of astrocytes and microglia maladaptive responses to acute stress are rescued by a single administration of ketamine in a rodent model of PTSD. Transl. Psychiatry 2024, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, K.; Xu, S.X.; Xie, X.H.; Tang, Y.; Zhang, L.; Liu, Z. Investigating Neuroplasticity Changes Reflected by BDNF Levels in Astrocyte-Derived Extracellular Vesicles in Patients with Depression. Int. J. Nanomed. 2024, 19, 8971–8985. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhang, Y.; Tu, M.; Ye, Y.; Li, M.; Ran, R.; Zou, Z. Brain-derived neurotrophic factor levels across psychiatric disorders: A systemic review and network meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 131, 110954. [Google Scholar] [CrossRef]
- Liberona, A.; Jones, N.; Zúñiga, K.; Garrido, V.; Zelada, M.I.; Silva, H.; Nieto, R.R. Brain-Derived Neurotrophic Factor (BDNF) as a Predictor of Treatment Response in Schizophrenia and Bipolar Disorder: A Systematic Review. Int. J. Mol. Sci. 2024, 25, 11204. [Google Scholar] [CrossRef] [PubMed]
- Shkundin, A.; Halaris, A. Associations of BDNF/BDNF-AS SNPs with Depression, Schizophrenia, and Bipolar Disorder. J. Pers. Med. 2023, 13, 1395. [Google Scholar] [CrossRef] [PubMed]
- Gredicak, M.; Nikolac Perkovic, M.; Nedic Erjavec, G.; Uzun, S.; Kozumplik, O.; Svob Strac, D.; Pivac, N. Association between reduced plasma BDNF concentration and MMSE scores in both chronic schizophrenia and mild cognitive impairment. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 134, 111086. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, G. 7,8-Dihydroxyflavone and Neuropsychiatric Disorders: A Translational Perspective from the Mechanism to Drug Development. Curr. Neuropharmacol. 2022, 20, 1479–1497. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zhang, J.C.; Yao, W.; Yang, C.; Ishima, T.; Ren, Q.; Ma, M.; Dong, C.; Huang, X.F.; Hashimoto, K. Intake of 7,8-Dihydroxyflavone During Juvenile and Adolescent Stages Prevents Onset of Psychosis in Adult Offspring After Maternal Immune Activation. Sci. Rep. 2016, 6, 36087. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, B.; Dunn, A.; Sundram, S.; Hill, R.A. Investigating 7,8-Dihydroxyflavone to combat maternal immune activation effects on offspring gene expression and behaviour. Prog. Neuropsychopharmacol. Biol. Psychiatry 2024, 134, 111078. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, B.M.M.; Chaves Filho, A.J.M.; Costa, D.; de Menezes, A.T.; da Fonseca, A.C.C.; Gama, C.S.; Moura Neto, V.; de Lucena, D.F.; Vale, M.L.; Macêdo, D.S. N-3 polyunsaturated fatty acids and clozapine abrogates poly I: C-induced immune alterations in primary hippocampal neurons. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 90, 186–196. [Google Scholar] [CrossRef]
- Yang, Y.J.; Li, Y.K.; Wang, W.; Wan, J.G.; Yu, B.; Wang, M.Z.; Hu, B. Small-molecule TrkB agonist 7,8-dihydroxyflavone reverses cognitive and synaptic plasticity deficits in a rat model of schizophrenia. Pharmacol. Biochem. Behav. 2014, 122, 30–36. [Google Scholar] [CrossRef]
- Ben-Azu, B.; Fokoua, A.R.; Annafi, O.S.; Adebayo, O.G.; Del Re, E.C.; Okuchukwu, N.; Aregbesola, G.J.; Ejenavi, A.C.; Isiwele, D.M.; Efezino, A.J.; et al. Effective action of silymarin against ketamine-induced schizophrenia in male mice: Insight into the biochemical and molecular mechanisms of action. J. Psychiatr. Res. 2024, 179, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Khalid, I.; Saleem, U.; Ahmad, B.; Hawwal, M.F.; Mothana, R.A. NMDA receptor modulation by Esculetin: Investigating behavioral, biochemical and neurochemical effects in schizophrenic mice model. Saudi Pharm. J. 2024, 32, 101994. [Google Scholar] [CrossRef] [PubMed]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Shirani, A.; Okuda, D.T.; Stüve, O. Therapeutic Advances and Future Prospects in Progressive Forms of Multiple Sclerosis. Neurotherapeutics 2016, 13, 58–69. [Google Scholar] [CrossRef]
- Li, C.; Zhuo, C.; Ma, X.; Li, R.; Chen, X.; Li, Y.; Zhang, Q.; Yang, L.; Wang, L. Exploring the molecular targets of fingolimod and siponimod for treating the impaired cognition of schizophrenia using network pharmacology and molecular docking. Schizophrenia 2024, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Sykes, L.; Haddon, J.; Lancaster, T.M.; Sykes, A.; Azzouni, K.; Ihssen, N.; Moon, A.L.; Lin, T.E.; Linden, D.E.; Owen, M.J.; et al. Genetic Variation in the Psychiatric Risk Gene CACNA1C Modulates Reversal Learning Across Species. Schizophr. Bull. 2019, 45, 1024–1032. [Google Scholar] [CrossRef]
- Qi, X.; Yu, X.; Wei, L.; Jiang, H.; Dong, J.; Li, H.; Wei, Y.; Zhao, L.; Deng, W.; Guo, W.; et al. Novel α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR) potentiator LT-102: A promising therapeutic agent for treating cognitive impairment associated with schizophrenia. CNS Neurosci. Ther. 2024, 30, e14713. [Google Scholar] [CrossRef] [PubMed]
- Amidfar, M.; de Oliveira, J.; Kucharska, E.; Budni, J.; Kim, Y.K. The role of CREB and BDNF in neurobiology and treatment of Alzheimer’s disease. Life Sci. 2020, 257, 118020. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef]
- Wang, F.; Wei, X.X.; Chang, L.S.; Dong, L.; Wang, Y.L.; Li, N.N. Ultrasound Combined With Microbubbles Loading BDNF Retrovirus to Open BloodBrain Barrier for Treatment of Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 615104. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, A.H.; Mateling, M.; Kovacs, I.; Wang, L.; Eggert, S.; Rockenstein, E.; Koo, E.H.; Masliah, E.; Tuszynski, M.H. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci. 2013, 33, 15596–15602. [Google Scholar] [CrossRef] [PubMed]
- Braschi, C.; Capsoni, S.; Narducci, R.; Poli, A.; Sansevero, G.; Brandi, R.; Maffei, L.; Cattaneo, A.; Berardi, N. Intranasal delivery of BDNF rescues memory deficits in AD11 mice and reduces brain microgliosis. Aging Clin. Exp. Res. 2021, 33, 1223–1238. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.G.; Frey, W.H., 2nd. Delivery of neurotrophic factors to the central nervous system: Pharmacokinetic considerations. Clin. Pharmacokinet. 2001, 40, 907–946. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Belichenko, N.P.; Yang, T.; Condon, C.; Monbureau, M.; Shamloo, M.; Jing, D.; Massa, S.M.; Longo, F.M. A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J. Neurosci. 2013, 33, 18712–18727. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.T. Can Brain-derived Neurotrophic Factor (BDNF) Mimetics be a Way Out for Neurodegenerative Diseases? Curr. Pharm. Des. 2023, 29, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Liu, X.; Wang, Z.H.; Luo, S.; Liao, J.; Ye, K. Optimized TrkB Agonist Ameliorates Alzheimer’s Disease Pathologies and Improves Cognitive Functions via Inhibiting Delta-Secretase. ACS Chem. Neurosci. 2021, 12, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Gascon, S.; Jann, J.; Langlois-Blais, C.; Plourde, M.; Lavoie, C.; Faucheux, N. Peptides Derived from Growth Factors to Treat Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 6071. [Google Scholar] [CrossRef] [PubMed]
- Povarnina, P.Y.; Volkova, A.A.; Vorontsova, O.N.; Kamensky, A.A.; Gudasheva, T.A.; Seredenin, S.B. A Low-Molecular-Weight BDNF Mimetic, Dipeptide GSB-214, Prevents Memory Impairment in Rat Models of Alzheimer’s Disease. Acta Naturae 2022, 14, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ahn, E.H.; Kang, S.S.; Liu, X.; Alam, A.; Ye, K. Gut dysbiosis contributes to amyloid pathology, associated with C/EBPβ/AEP signaling activation in Alzheimer’s disease mouse model. Sci. Adv. 2020, 6, eaba0466. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chen, C.; Ahn, E.H.; Liu, X.; Li, H.; Edgington-Mitchell, L.E.; Lu, Z.; Ming, S.; Ye, K. Targeting both BDNF/TrkB pathway and delta-secretase for treating Alzheimer’s disease. Neuropharmacology 2021, 197, 108737. [Google Scholar] [CrossRef]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef]
- Nam, J.H.; Leem, E.; Jeon, M.T.; Jeong, K.H.; Park, J.W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) transduction of SNpc neurons: Neuroprotective mechanisms of hRheb(S16H) in a model of Parkinson’s disease. Mol. Neurobiol. 2015, 51, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Guillin, O.; Diaz, J.; Carroll, P.; Griffon, N.; Schwartz, J.C.; Sokoloff, P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 2001, 411, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, I.; Marusiak, J.; Toczyłowska, B.; Stępień, A.; Brodacki, B.; Langfort, J.; Chalimoniuk, M. Association between the Val66Met (rs6265) polymorphism of the brain-derived neurotrophic factor (BDNF) gene, BDNF protein level in the blood and the risk of developing early-onset Parkinson’s disease. Acta Neurobiol. Exp. 2024, 84, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Wang, Y.; Sun, Z. The Relationships Between Stress, Mental Disorders, and Epigenetic Regulation of BDNF. Int. J. Mol. Sci. 2020, 21, 1375. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef]
- Zuo, L.; Dai, C.; Yi, L.; Dong, Z. 7,8-dihydroxyflavone ameliorates motor deficits via regulating autophagy in MPTP-induced mouse model of Parkinson’s disease. Cell Death Discov. 2021, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Massaquoi, M.S.; Liguore, W.A.; Churchill, M.J.; Moore, C.; Melrose, H.L.; Meshul, C.K. Gait Deficits and Loss of Striatal Tyrosine Hydroxlase/Trk-B are Restored Following 7,8-Dihydroxyflavone Treatment in a Progressive MPTP Mouse Model of Parkinson’s Disease. Neuroscience 2020, 433, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, T.; Chandramohan, V.; Lalithamba, H.S.; Jayaraj, R.L.; Kumaradhas, P.; Sivanandam, M.; Hunday, G.; Vijayakumar, R.; Balakrishnan, R.; Manimaran, D.; et al. Design and Molecular dynamic Investigations of 7,8-Dihydroxyflavone Derivatives as Potential Neuroprotective Agents Against Alpha-synuclein. Sci. Rep. 2020, 10, 599. [Google Scholar] [CrossRef]
- Li, X.H.; Dai, C.F.; Chen, L.; Zhou, W.T.; Han, H.L.; Dong, Z.F. 7,8-dihydroxyflavone Ameliorates Motor Deficits Via Suppressing α-synuclein Expression and Oxidative Stress in the MPTP-induced Mouse Model of Parkinson’s Disease. CNS Neurosci. Ther. 2016, 22, 617–624. [Google Scholar] [CrossRef]
- Park, H.Y.; Park, C.; Hwang, H.J.; Kim, B.W.; Kim, G.Y.; Kim, C.M.; Kim, N.D.; Choi, Y.H. 7,8-Dihydroxyflavone attenuates the release of pro-inflammatory mediators and cytokines in lipopolysaccharide-stimulated BV2 microglial cells through the suppression of the NF-κB and MAPK signaling pathways. Int. J. Mol. Med. 2014, 33, 1027–1034. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alexiou, A.; Papadakis, M.; AlAseeri, A.A.; Alruwaili, M.; Saad, H.M.; Batiha, G.E. BDNF/TrkB activators in Parkinson’s disease: A new therapeutic strategy. J. Cell. Mol. Med. 2024, 28, e18368. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Wu, Z.; Liu, X.; Edgington-Mitchell, L.; Ye, K. Treating Parkinson’s Disease via Activation of BDNF/TrkB Signaling Pathways and Inhibition of Delta-Secretase. Neurotherapeutics 2022, 19, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Firouzan, B.; Iravanpour, F.; Abbaszadeh, F.; Akparov, V.; Zaringhalam, J.; Ghasemi, R.; Maghsoudi, N. Dipeptide mimetic of BDNF ameliorates motor dysfunction and striatal apoptosis in 6-OHDA-induced Parkinson’s rat model: Considering Akt and MAPKs signaling. Behav. Brain Res. 2023, 452, 114585. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Cannella, M.; Calza, S.; Poletti, B.; Frati, L.; Squitieri, F.; Silani, V. Low brain-derived neurotrophic factor (BDNF) levels in serum of Huntington’s disease patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 574–577. [Google Scholar] [CrossRef] [PubMed]
- Krzysztoń-Russjan, J.; Zielonka, D.; Jackiewicz, J.; Kuśmirek, S.; Bubko, I.; Klimberg, A.; Marcinkowski, J.T.; Anuszewska, E.L. A study of molecular changes relating to energy metabolism and cellular stress in people with Huntington’s disease: Looking for biomarkers. J. Bioenerg. Biomembr. 2013, 45, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Bithell, A.; Johnson, R.; Buckley, N.J. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington’s disease. Biochem. Soc. Trans. 2009, 37, 1270–1275. [Google Scholar] [CrossRef]
- Kumar, A.; Vaish, M.; Ratan, R.R. Transcriptional dysregulation in Huntington’s disease: A failure of adaptive transcriptional homeostasis. Drug Discov. Today 2014, 19, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Ginés, S.; Bosch, M.; Marco, S.; Gavaldà, N.; Díaz-Hernández, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur. J. Neurosci. 2006, 23, 649–658. [Google Scholar] [CrossRef]
- Cho, S.R.; Benraiss, A.; Chmielnicki, E.; Samdani, A.; Economides, A.; Goldman, S.A. Induction of neostriatal neurogenesis slows disease progression in a transgenic murine model of Huntington disease. J. Clin. Investig. 2007, 117, 2889–2902. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, S.; Lahaye, R.A.; Vitet, H.; Scaramuzzino, C.; Virlogeux, A.; Capellano, L.; Genoux, A.; Gershoni-Emek, N.; Geva, M.; Hayden, M.R.; et al. Pridopidine rescues BDNF/TrkB trafficking dynamics and synapse homeostasis in a Huntington disease brain-on-a-chip model. Neurobiol. Dis. 2022, 173, 105857. [Google Scholar] [CrossRef] [PubMed]
- Speidell, A.; Bin Abid, N.; Yano, H. Brain-Derived Neurotrophic Factor Dysregulation as an Essential Pathological Feature in Huntington’s Disease: Mechanisms and Potential Therapeutics. Biomedicines 2023, 11, 2275. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis 2021, 26, 52–70. [Google Scholar] [CrossRef]
- García-Díaz Barriga, G.; Giralt, A.; Anglada-Huguet, M.; Gaja-Capdevila, N.; Orlandi, J.G.; Soriano, J.; Canals, J.M.; Alberch, J. 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCγ1 pathway. Hum. Mol. Genet. 2017, 26, 3144–3160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Peng, Q.; Liu, X.; Jin, J.; Hou, Z.; Zhang, J.; Mori, S.; Ross, C.A.; Ye, K.; Duan, W. Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A. Modulating Neurotrophin Receptor Signaling as a Therapeutic Strategy for Huntington’s Disease. J. Huntingt. Dis. 2017, 6, 303–325. [Google Scholar] [CrossRef]
- Sada, N.; Fujita, Y.; Mizuta, N.; Ueno, M.; Furukawa, T.; Yamashita, T. Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death Dis. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Sartor, G.C.; Malvezzi, A.M.; Kumar, A.; Andrade, N.S.; Wiedner, H.J.; Vilca, S.J.; Janczura, K.J.; Bagheri, A.; Al-Ali, H.; Powell, S.K.; et al. Enhancement of BDNF Expression and Memory by HDAC Inhibition Requires BET Bromodomain Reader Proteins. J. Neurosci. 2019, 39, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Benn, C.L.; Franklin, S.A.; Smith, D.L.; Woodman, B.; Marks, P.A.; Bates, G.P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2011, 6, e27746. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Choi, J.; Sim, H.R.; Kim, J.; Jun, J.H.; Kyung, J.; Ha, N.; Kim, S.; Ryu, K.H.; Chung, S.S.; et al. A novel HDAC6 inhibitor, CKD-504, is effective in treating preclinical models of huntington’s disease. BMB Rep. 2023, 56, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Elifani, F.; Amico, E.; Pepe, G.; Capocci, L.; Castaldo, S.; Rosa, P.; Montano, E.; Pollice, A.; Madonna, M.; Filosa, S.; et al. Curcumin dietary supplementation ameliorates disease phenotype in an animal model of Huntington’s disease. Hum. Mol. Genet. 2019, 28, 4012–4021. [Google Scholar] [CrossRef]
- Kaur, K.; Al-Khazaleh, A.K.; Bhuyan, D.J.; Li, F.; Li, C.G. A Review of Recent Curcumin Analogues and Their Antioxidant, Anti-Inflammatory, and Anticancer Activities. Antioxidants 2024, 13, 1092. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Delivery of Biologics Across the Blood-Brain Barrier with Molecular Trojan Horse Technology. BioDrugs 2017, 31, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Kopec, B.M.; Zhao, L.; Rosa-Molinar, E.; Siahaan, T.J. Non-invasive Brain Delivery and Efficacy of BDNF in APP/PS1 Transgenic Mice as a Model of Alzheimer’s Disease. Med. Res. Arch. 2020, 8, 2043. [Google Scholar] [CrossRef] [PubMed]
- Pilakka-Kanthikeel, S.; Atluri, V.S.; Sagar, V.; Saxena, S.K.; Nair, M. Targeted brain derived neurotropic factors (BDNF) delivery across the blood-brain barrier for neuro-protection using magnetic nano carriers: An in-vitro study. PLoS ONE 2013, 8, e62241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pardridge, W.M. Blood-brain barrier targeting of BDNF improves motor function in rats with middle cerebral artery occlusion. Brain Res. 2006, 1111, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Boltaev, U.; Meyer, Y.; Tolibzoda, F.; Jacques, T.; Gassaway, M.; Xu, Q.; Wagner, F.; Zhang, Y.L.; Palmer, M.; Holson, E.; et al. Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Sci. Signal. 2017, 10, eaal1670. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numakawa, T.; Kajihara, R. The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders. Molecules 2025, 30, 848. https://doi.org/10.3390/molecules30040848
Numakawa T, Kajihara R. The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders. Molecules. 2025; 30(4):848. https://doi.org/10.3390/molecules30040848
Chicago/Turabian StyleNumakawa, Tadahiro, and Ryutaro Kajihara. 2025. "The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders" Molecules 30, no. 4: 848. https://doi.org/10.3390/molecules30040848
APA StyleNumakawa, T., & Kajihara, R. (2025). The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders. Molecules, 30(4), 848. https://doi.org/10.3390/molecules30040848