

A Comprehensive Guide to Enzyme Immobilization: All You Need to Know



Abstract
:1. Introduction
2. Classical Non-Specific Immobilization
2.1. Classical Non-Covalent Immobilization
2.1.1. Entrapment
2.1.2. Encapsulation
2.1.3. Adsorption
2.1.4. Ionic Binding
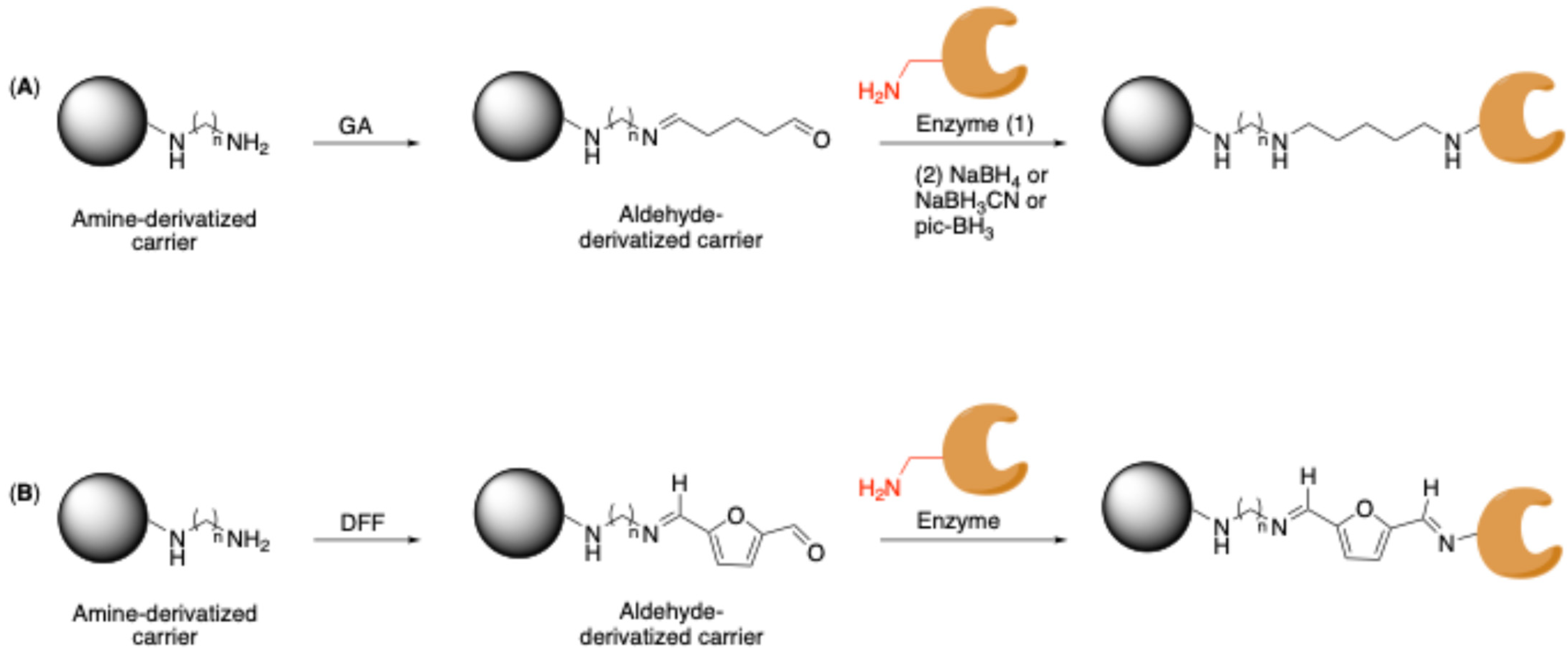
2.2. Classical Covalent Immobilization
Carrier-Bound
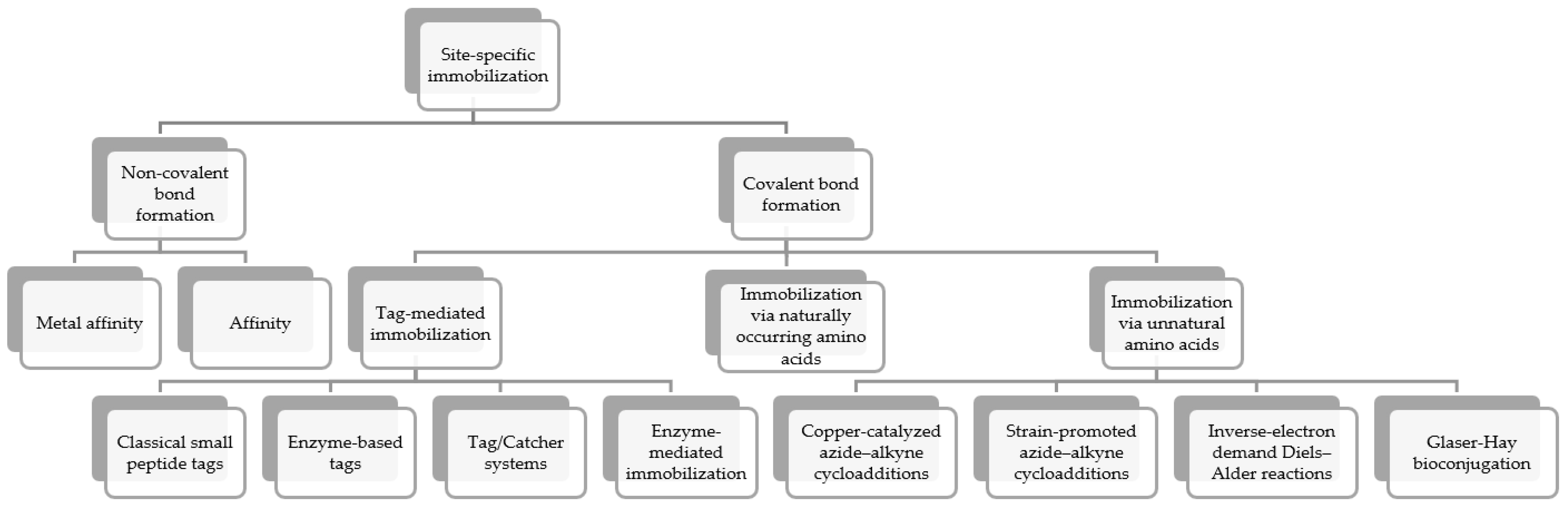
3. Site-Specific Immobilization
3.1. Site-Specific Non-Covalent Immobilization
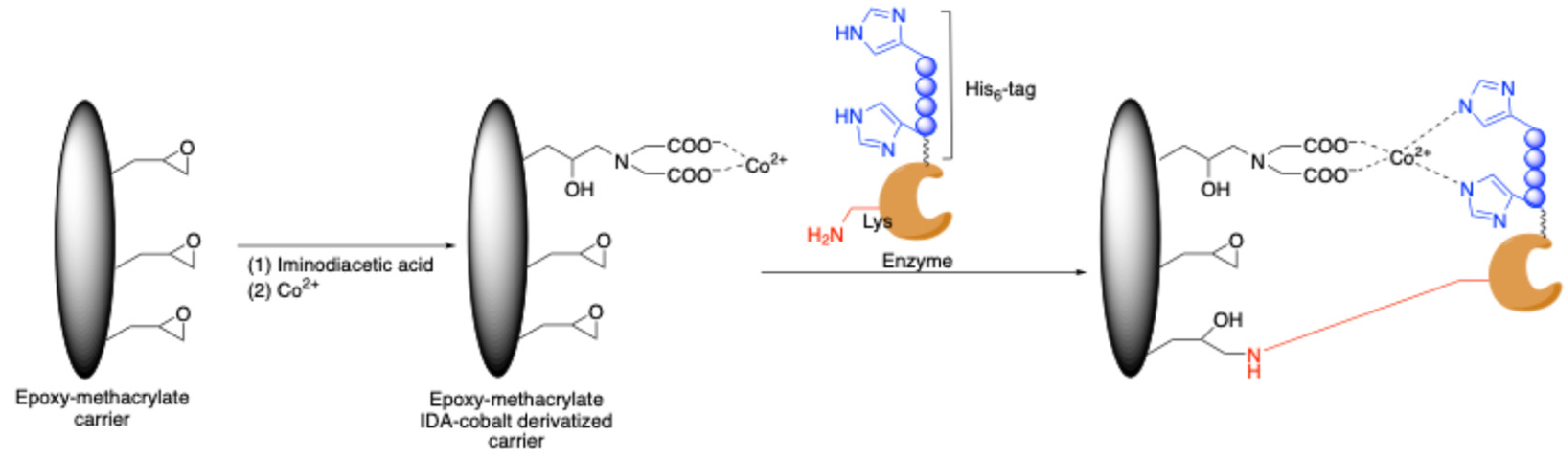
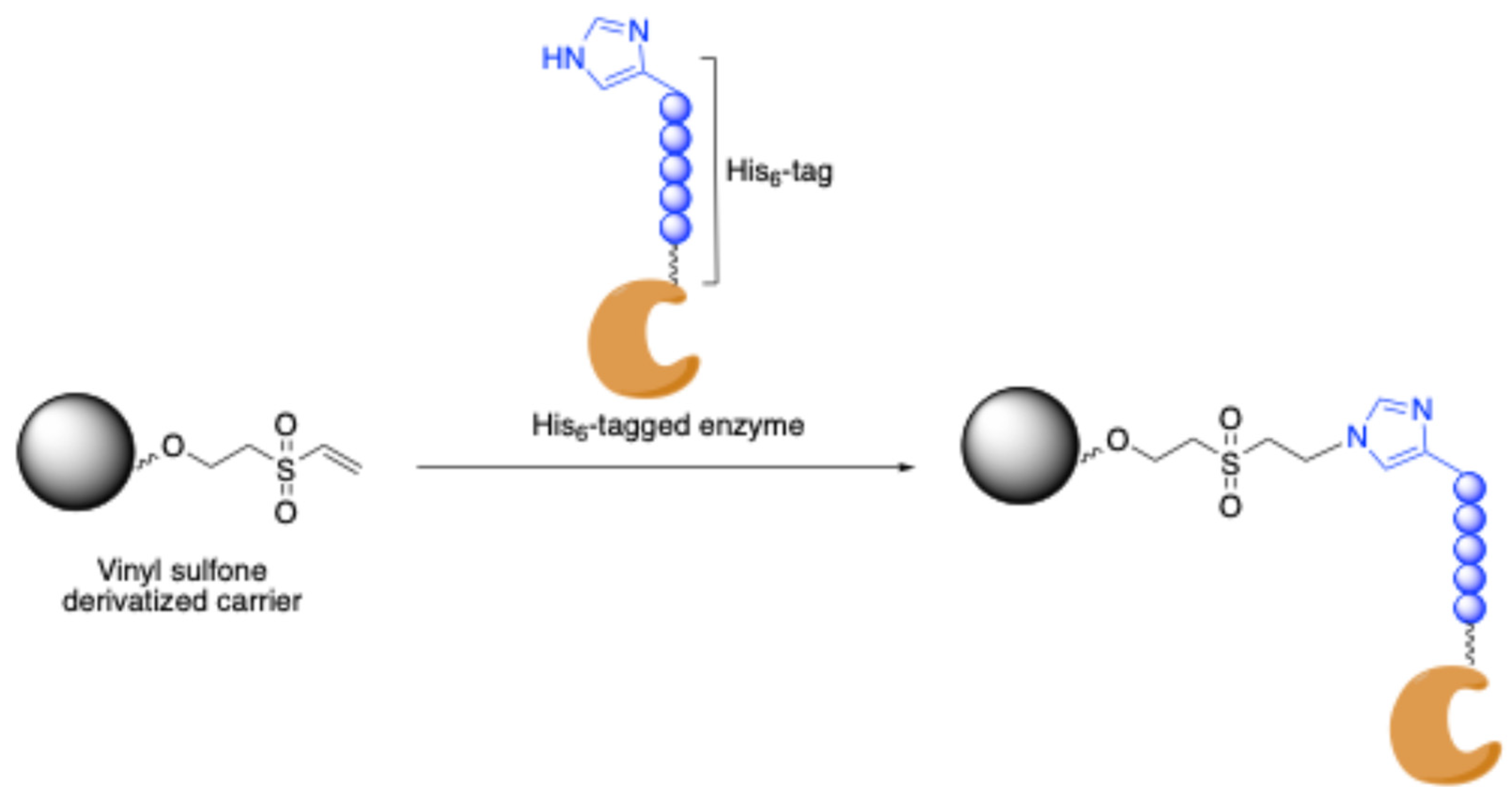
3.1.1. Site-Specific Non-Covalent Immobilization via Metal Affinity
3.1.2. Site-Specific Non-Covalent Immobilization via Biotin-(Strept)Avidin Interaction
3.2. Site-Specific Covalent Immobilization
3.2.1. Tag-Mediated Bio-Orthogonal Covalent Immobilization
Classical Small Peptide Tags
Self-Labeling Protein Tags
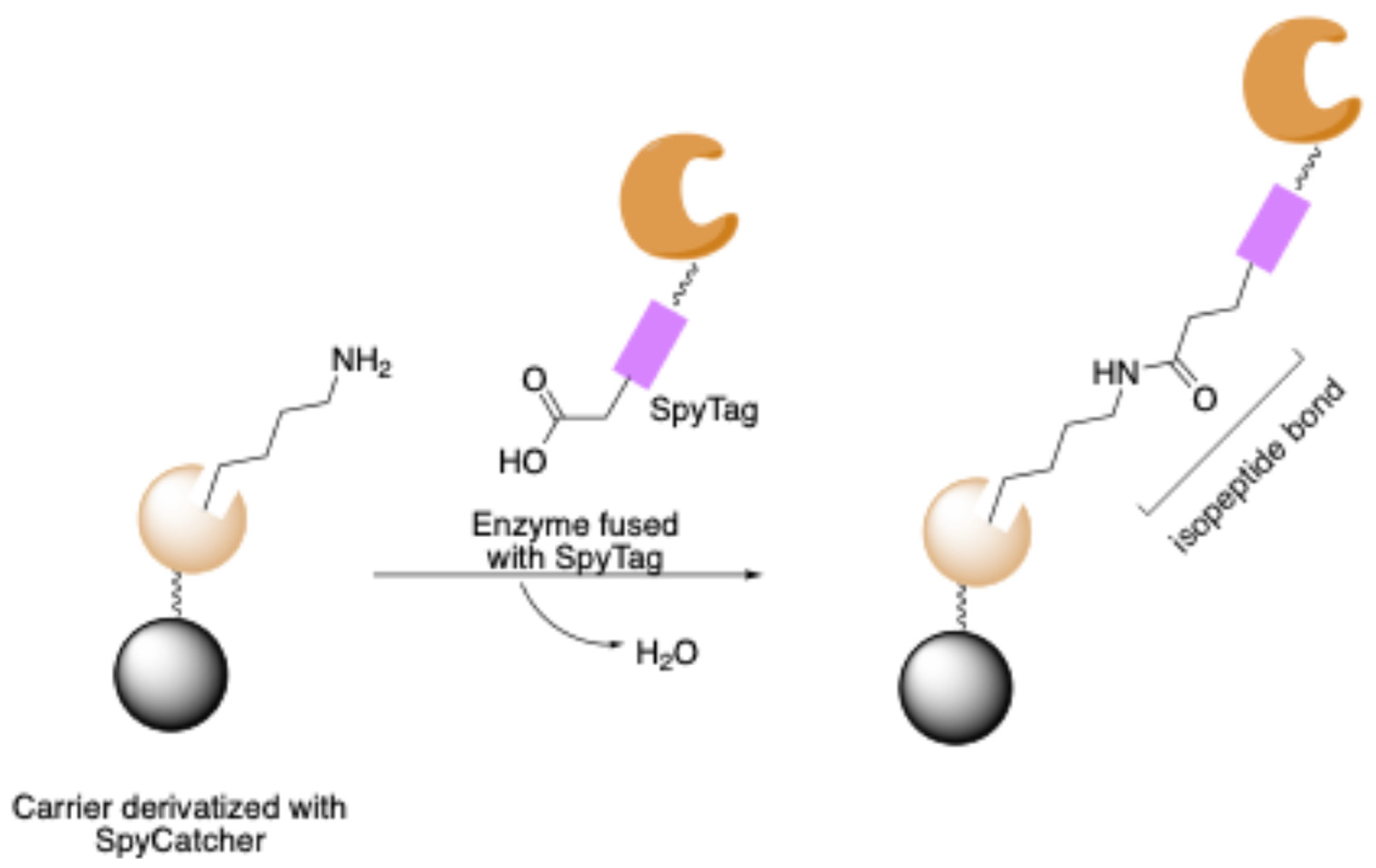
Catcher/Tag Systems
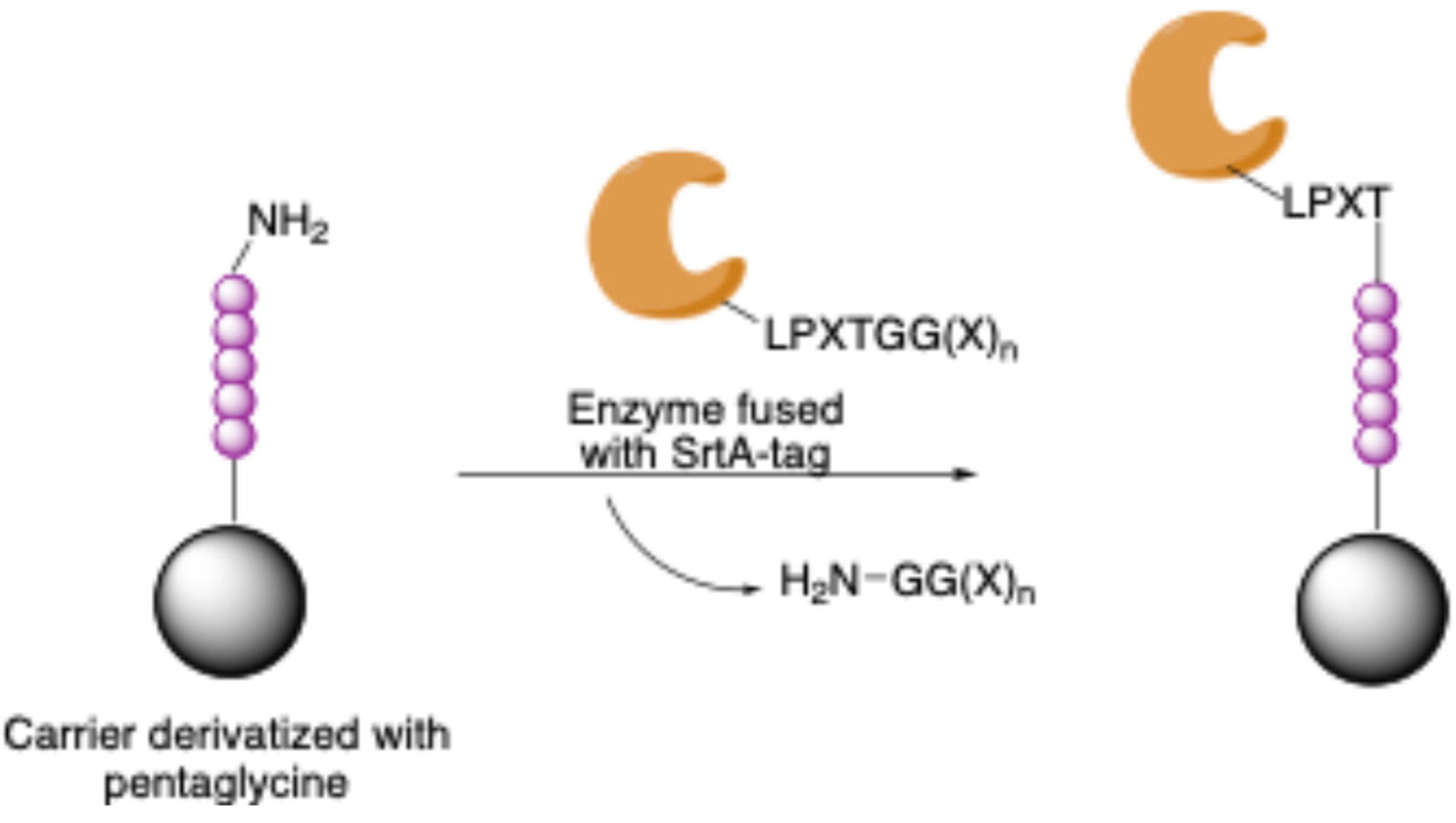
Enzyme-Mediated Immobilization Driven by Tag Sequences
3.2.2. Sequence Engineering-Mediated Bio-Orthogonal Covalent Immobilization
Incorporation of Naturally Reactive Occurring Amino Acids by Site-Specific Mutagenesis
Incorporation of Unnatural Amino Acids
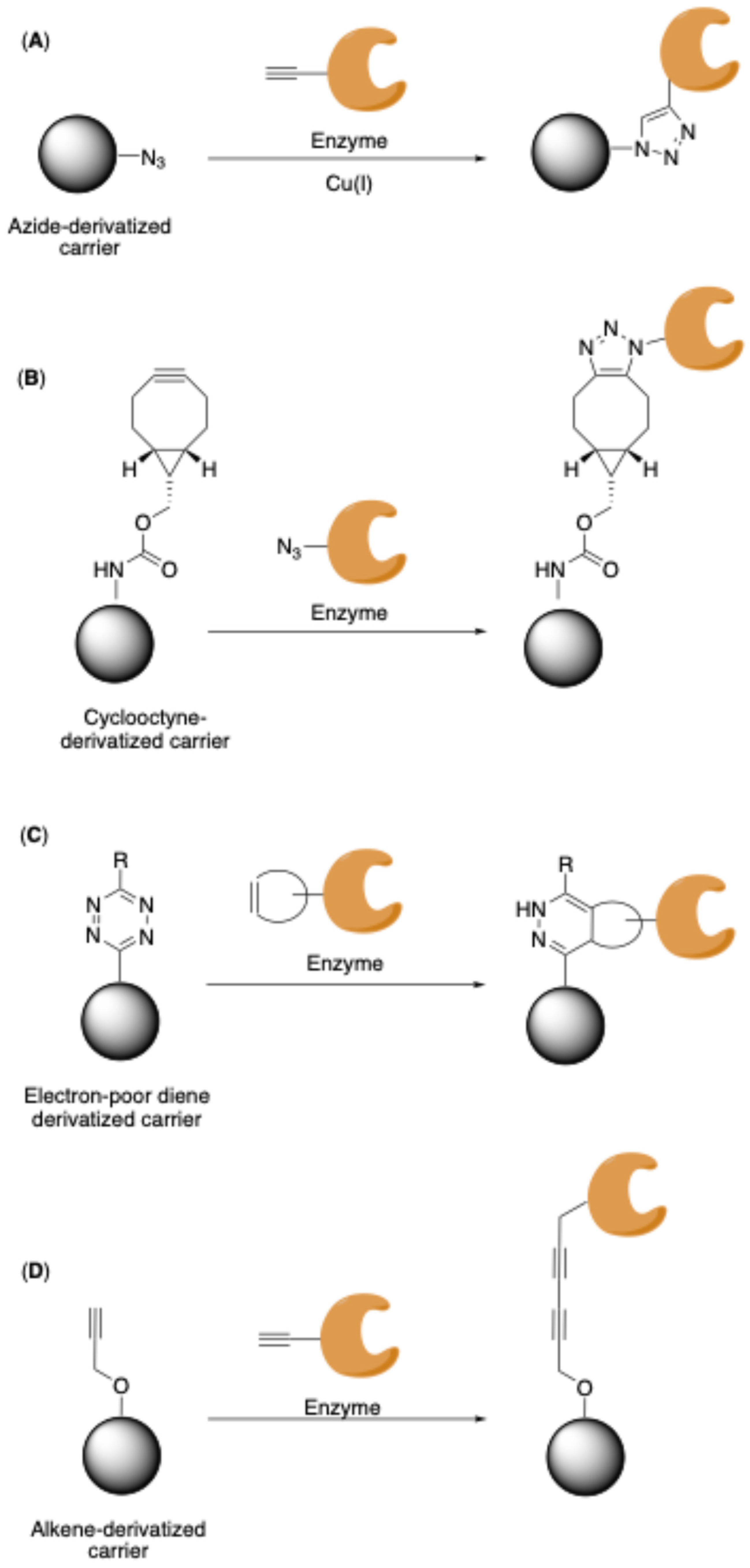
Copper-Catalyzed Azide–Alkyne Cycloadditions (CuAAC)
Strain-Promoted Azide-Alkyne Cycloadditions (SPAAC)
Inverse-Electron-Demand Diels-Alder Reactions (IEDDA)
Glaser-Hay
4. Evaluation of Immobilization Outcome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boudrant, J.; Woodley, J.M.; Fernandez-Lafuente, R. Parameters Necessary to Define an Immobilized Enzyme Preparation. Process Biochem. 2020, 90, 66–80. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D. Broadening the Scope of Biocatalysis in Sustainable Organic Synthesis. ChemSusChem 2019, 12, 2859–2881. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, A.; Barry, A.; Burke, A.J.; Hutton, A.E.; Bell, E.L.; Green, A.P.; O’Reilly, E. Biocatalysis: Landmark Discoveries and Applications in Chemical Synthesis. Chem. Soc. Rev. 2024, 53, 2828. [Google Scholar] [CrossRef]
- Rossino, G.; Robescu, M.S.; Licastro, E.; Tedesco, C.; Martello, I.; Maffei, L.; Vincenti, G.; Bavaro, T.; Collina, S. Biocatalysis: A Smart and Green Tool for the Preparation of Chiral Drugs. Chirality 2022, 34, 1403–1418. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Yi, D.; Bayer, T.; Badenhorst, C.P.S.; Wu, S.; Doerr, M.; Hohne, M.; Bornscheuer, U.T. Recent Trends in Biocatalysis. Chem. Soc. Rev. 2021, 50, 8003. [Google Scholar] [CrossRef] [PubMed]
- Bié, J.; Sepodes, B.; Fernandes, P.C.B.; Ribeiro, M.H.L. Enzyme Immobilization and Co-Immobilization: Main Framework, Advances and Some Applications. Processes 2022, 10, 494. [Google Scholar] [CrossRef]
- Basso, A.; Serban, S. Industrial Applications of Immobilized Enzymes—A Review. Mol. Catal. 2019, 479, 110607. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef] [PubMed]
- Romero-Fernandez, M.; Paradisi, F. General Overview on Immobilization Techniques of Enzymes for Biocatalysis. In Catalysts Immobilization; John Wiley & Sons, Ltd.: New York, NY, USA, 2019; pp. 409–435. [Google Scholar]
- Bilal, M.; Iqbal, H.M.N. Tailoring Multipurpose Biocatalysts via Protein Engineering Approaches: A Review. Catal. Lett. 2019, 149, 2204–2217. [Google Scholar] [CrossRef]
- Bernal, C.; Rodriguez, K.; Martinez, R. Integrating Enzyme Immobilization and Protein Engineering: An Alternative Path for the Development of Novel and Improved Industrial Biocatalysts. Biotechnol. Adv. 2018, 36, 1470–1480. [Google Scholar] [CrossRef]
- Redeker, E.S.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein Engineering for Directed Immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Guisan, J.M. Immobilization of Enzymes and Cells: Methods and Protocols, Methods in Molecular Biology; Springer Science Business Media, LLC., part of Springer Nature: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Bolivar, J.M.; Woodley, J.M.; Fernandez-Lafuente, R. Is Enzyme Immobilization a Mature Discipline? Some Critical Considerations to Capitalize on the Benefits of Immobilization. Chem. Soc. Rev. 2022, 51, 6251–6290. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Basso, A.; Brady, D. New Frontiers in Enzyme Immobilisation: Robust Biocatalysts for a Circular Bio-Based Economy. Chem. Soc. Rev. 2021, 50, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient Immobilisation of Industrial Biocatalysts: Criteria and Constraints for the Selection of Organic Polymeric Carriers and Immobilisation Methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme Immobilisation in Biocatalysis: Why, What and How. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and Its Derivatives as Supports for Enzyme Immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Maghraby, Y.R.; El-Shabasy, R.M.; Ibrahim, A.H.; Azzazy, H.M.E.-S. Enzyme Immobilization Technologies and Industrial Applications. ACS Omega 2023, 8, 5184–5196. [Google Scholar] [CrossRef]
- Kumari, A.; Kaur, B.; Srivastava, R.; Sangwan, R.S. Isolation and Immobilization of Alkaline Protease on Mesoporous Silica and Mesoporous ZSM-5 Zeolite Materials for Improved Catalytic Properties. Biochem. Biophys. Rep. 2015, 2, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.; Kaur, R.; Kaur, S.; Kaur, P.S. Immobilization of Laccase-ABTS System for the Development of a Continuous Flow Packed Bed Bioreactor for Decolorization of Textile Effluent. Int. J. Biol. Macromol. 2018, 117, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Kim, M. An Overview of Techniques in Enzyme Immobilization. Appl. Sci. Converg. Technol. 2017, 26, 157–163. [Google Scholar] [CrossRef]
- Luo, J.; Wan, Y.; Song, S.; Zhang, H.; Zhang, H.; Zhang, J.; Wan, Y. Biocatalytic Membrane: Go Far beyond Enzyme Immobilization. Eng. Life Sci. 2020, 20, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.-K.; Liu, Y.-X.; Xue, Y.-P.; Zheng, Y.-G. Immobilization of Enzymes in/on Membranes and Their Applications. Adv. Synth. Catal. 2019, 361, 5500–5515. [Google Scholar] [CrossRef]
- Brena, B.; Gonzalez-Pombo, P.; Batista-Viera, F. Immobilization of Enzymes: A Literature Survey. In Immobilization of Enzymes and Cells; Methods in Molecular Biology; Guisan, J., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 1051. [Google Scholar]
- Pantić, N.; Prodanović, R.; Ðurđić, K.I.; Polović, N.; Spasojević, M.; Prodanović, O. Optimization of Phenol Removal with Horseradish Peroxidase Encapsulated within Tyramine-Alginate Micro-Beads. Environ. Technol. Innov. 2021, 21, 101211. [Google Scholar] [CrossRef]
- Guan, T.; Liu, B.; Wang, R.; Huang, Y.; Luo, J.; Li, Y. The Enhanced Fatty Acids Flavor Release for Low-Fat Cheeses by Carrier Immobilized Lipases on O/W Pickering Emulsions. Food Hydrocoll. 2021, 116, 106651. [Google Scholar] [CrossRef]
- Rother, C.; Nidetzky, B. Enzyme Immobilization by Microencapsulation: Methods, Materials, and Technological Applications. Encycl. Ind. Biotechnol. 2014, 7, 1–21. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “Perfect” Lipase Immobilized Biocatalyst? Catal. Sci. Technol. 2019, 9, 2380. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Virgen-Ortiz, J.J.; dos Santos, J.C.S.; Berenguer-Murcia, Á.; Alcantara, A.R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R. Immobilization of Lipases on Hydrophobic Supports: Immobilization Mechanism, Advantages, Problems, and Solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef]
- Semproli, R.; Robescu, M.S.; Sangiorgio, S.; Pargoletti, E.; Bavaro, T.; Rabuffetti, M.; Cappelletti, G.; Speranza, G.; Ubiali, D. From Lactose to Alkyl Galactoside Fatty Acid Esters as Non-ionic Biosurfactants: A Two-step Enzymatic Approach to Cheese Whey Valorization. ChemPlusChem 2023, 88, e202200331. [Google Scholar] [CrossRef]
- Kumar, R.; Maikhuri, V.K.; Mathur, D.; Kumar, M.; Singh, N.; Prasad, A.K. Novozyme-435: Perfect Catalyst for Chemo- and Regio-Selective Synthesis of Modified Carbohydrates—A Review. Biocatal. Biotransform. 2023, 42, 19–33. [Google Scholar] [CrossRef]
- Hoyos, P.; Perona, A.; Bavaro, T.; Berini, F.; Marinelli, F.; Terreni, M.; Hernáiz, M.J. Biocatalyzed Synthesis of Glycostructures with Anti-Infective Activity. Acc. Chem. Res. 2022, 55, 2409–2424. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, L.; Robescu, M.S.; Marzatico, S.; Recca, T.; Zhang, Y.; Terreni, M.; Bavaro, T. Developing a Library of Mannose-Based Mono- and Disaccharides: A General Chemoenzymatic Approach to Monohydroxylated Building Blocks. Molecules 2020, 25, 5764. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bavaro, T.; Tengattini, S.; Bernardini, R.; Mattei, M.; Annunziata, F.; Cole, R.B.; Zheng, C.; Sollogoub, M.; Tamborini, L.; et al. Chemoenzymatic Synthesis of Arabinomannan (AM) Glycoconjugates as Potential Vaccines for Tuberculosis. Eur. J. Med. Chem. 2020, 204, 112578–112589. [Google Scholar] [CrossRef] [PubMed]
- Robescu, M.S.; Ubiali, D.; Semproli, R.; Bavaro, T. Stepping into the Biocatalysis Lab for Undergraduate Students: From Enzyme Immobilization to Product Isolation. J. Chem. Educ. 2024, 101, 2790–2795. [Google Scholar] [CrossRef]
- Truppo, M.D.; Strotman, H.; Hughes, G. Development of an Immobilized Transaminase Capable of Operating in Organic Solvent. ChemCatChem 2012, 4, 1071–1074. [Google Scholar] [CrossRef]
- Antecka, K.; Zdarta, J.; Siwińska-Stefańska, K.; Sztuk, G.; Jankowska, E.; Oleskowicz-Popiel, P.; Jesionowski, T. Synergistic Degradation of Dye Wastewaters Using Binary or Ternary Oxide Systems with Immobilized Laccase. Catalysts 2018, 8, 402. [Google Scholar] [CrossRef]
- Bruni, M.; Robescu, M.S.; Ubiali, D.; Marrubini, G.; Vanna, R.; Morasso, C.; Benucci, I.; Speranza, G.; Bavaro, T. Immobilization of g-glutamyl Transpeptidase from Equine Kidney for the Synthesis of Kokumi Compounds. ChemCatChem 2020, 12, 210–218. [Google Scholar] [CrossRef]
- Liese, A.; Seelbach, K.; Wandrey, C. (Eds.) Industrial Biotransformations; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Rocchietti, S.; Ubiali, D.; Terreni, M.; Albertini, A.M.; Fernández-Lafuente, R.; Guisán, J.M.; Pregnolato, M. Immobilization and Stabilization of Recombinant Multimeric Uridine and Purine Nucleoside Phosphorylases from Bacillus subtilis. Biomacromolecules 2004, 5, 2195–2200. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Characteristic Features and Biotechnological Applications of Cross-Linked Enzyme Aggregates (CLEAs). Appl. Microbiol. Biotechnol. 2011, 92, 467–477. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding Enzyme Immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in Bio-Catalysts Design: A Useful Crosslinker and a Versatile Tool in Enzyme Immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Danielli, C.; van Langen, L.; Boes, D.; Asaro, F.; Anselmi, S.; Provenza, F.; Renzi, M.; Gardossi, L. 2,5-Furandicarboxaldehyde as a Bio-Based Crosslinking Agent Replacing Glutaraldehyde for Covalent Enzyme Immobilization. RSC Adv. 2022, 12, 35676. [Google Scholar] [CrossRef] [PubMed]
- Orrego, A.H.; Romero-Fern, M.; Millan-Linares, M.d.C.; del Mar Yust, M.; Guisan, J.M.; Rocha-Martin, J. Stabilization of Enzymes by Multipoint Covalent Attachment on Aldehyde-Supports: 2-Picoline Borane as an Alternative Reducing Agent. Catalysts 2018, 8, 333. [Google Scholar] [CrossRef]
- Guisan, J.M. Aldehyde-Agarose Gels as Activated Supports for Immobilization-Stabilization of Enzymes. Enzyme Microb. Technol. 1988, 10, 375–382. [Google Scholar] [CrossRef]
- Ho, J.; Al-Deen, F.M.N.; Al-Abboodi, A.; Selomulya, C.; Xiang, S.D.; Plebanski, M.; Forde, G.M. N,N′-Carbonyldiimidazole-Mediated Functionalization of Superparamagnetic Nanoparticles as Vaccine Carrier. Colloids Surf. B Biointerfaces 2011, 83, 83–90. [Google Scholar] [CrossRef]
- Mateo, C.; Palomo, J.M.; Fuentes, M.; Betancor, L.; Grazu, V.; López-Gallego, F.; Pessela, B.C.C.; Hidalgo, A.; Fernández-Lorente, G.; Fernández-Lafuente, R.; et al. Glyoxyl Agarose: A Fully Inert and Hydrophilic Support for Immobilization and High Stabilization of Proteins. Enzyme Microb. Technol. 2006, 39, 274–280. [Google Scholar] [CrossRef]
- Rueda, N.; dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved Performance of Lipases Immobilized on Heterofunctional Octyl-Glyoxyl Agarose Beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Palomo, J.M.; Filice, M.; Fernandez-Lafuente, R.; Terreni, M.; Guisan, J.M. Regioselective Hydrolysis of Different Peracetylated Β-monosaccharides by Immobilized Lipases from Different Sources. Key Role of the Immobilization. Adv. Synth. Catal. 2007, 349, 1969–1976. [Google Scholar] [CrossRef]
- Babaki, M.; Yousefi, M.; Habibi, Z.; Brask, J.; Mohammadi, M. Preparation of Highly Reusable Biocatalysts by Immobilization of Lipases on Epoxy-Functionalized Silica for Production of Biodiesel from Canola Oil. Biochem. Eng. J. 2015, 101, 23–31. [Google Scholar] [CrossRef]
- Semproli, R.; Vaccaro, G.; Ferrandi, E.E.; Vanoni, M.; Bavaro, T.; Marrubini, G.; Annunziata, F.; Conti, P.; Speranza, G.; Monti, D.; et al. Use of Immobilized Amine Transaminase from Vibrio fluvialis under Flow Conditions for the Synthesis of (S)-1-(5-fluoropyrimidin-2-yl)-ethanamine. ChemCatChem 2020, 12, 1359–1367. [Google Scholar] [CrossRef]
- Mallin, H.; Muschiol, J.; Bystrom, E.; Bornscheuer, U.T. Efficient Biocatalysis with Immobilized Enzymes or Encapsulated Whole Cell Microorganism by Using the SpinChem Reactor System. ChemCatChem 2013, 5, 3529–3532. [Google Scholar] [CrossRef]
- Crotti, M.; Robescu, M.S.; Bolivar, J.M.; Ubiali, D.; Wilson, L.; Contente, M.L. What’s New in Flow Biocatalysis? A Snapshot of 2020–2022. Front. Catal. 2023, 3, 1154452. [Google Scholar] [CrossRef]
- Robescu, M.S.; Annunziata, F.; Somma, V.; Calvio, C.; Morelli, C.F.; Speranza, G.; Tamborini, L.; Ubiali, D.; Pinto, A.; Bavaro, T. From Batch to Continuous Flow Bioprocessing: Use of an Immobilized γ-Glutamyl Transferase from B. subtilis for the Synthesis of Biologically Active Peptide Derivatives. J. Agric. Food Chem. 2022, 70, 13692–13699. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, J.C.S.; Rueda, N.; Barbosa, O.; Fernandez-Sanchez, J.F.; Medina-Castillo, A.L.; Ramon-Marquez, T.; Arias-Martos, M.C.; Millan-Linares, C.; Pedroche, J.; del Mar Yust, M.; et al. Characterization of Supports Activated with Divinyl Sulfone as a Tool to Immobilize and Stabilize Enzymes via Multipoint Covalent Attachment. Application to Chymotrypsin. RSC Adv. 2015, 5, 20639. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Sebastian, E.S.; Ríos-Lombardía, N.; Morís, F.; Gonzµlez-Sabín, J.; López-Gallego, F. Asymmetric Reduction of Prochiral Ketones by Using Self-sufficient Heterogeneous Biocatalysts Based on NADPH-dependent Ketoreductases. Chem. Eur. J. 2017, 23, 16843–16852. [Google Scholar] [CrossRef]
- Stalder, K.; Benítez-Mateos, A.I.; Paradisi, F. Biocatalytic Synthesis of L-pipecolic Acid by a Lysine Cyclodeaminase: Batch and Flow Reactors. ChemCatChem 2024, 16, e202301671. [Google Scholar] [CrossRef]
- Kohn, J.; Wilchek, M. A New Approach (Cyano-Transfer) for Cyanogen Bromide Activation of Sepharose at Neutral pH, Which Yields Activated Resins Free of Interfering Nitrogen Derivatives. Biochem. Biophys. Res. Commun. 1982, 107, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Bavaro, T.; Tengattini, S.; Rezwan, R.; Chiesa, E.; Temporini, C.; Dorati, R.; Massolini, G.; Conti, B.; Ubiali, D.; Terreni, M. Design of Epidermal Growth Factor Immobilization on 3D Biocompatible Scaffolds to Promote Tissue Repair and Regeneration. Sci. Rep. 2021, 11, 2629. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.J.; Medina, M.B. Covalent Immobilization of Enzymes Using Commercially Available CDI-Activated Agarose. In Immobilization of Enzymes and Cells; Methods in Biotechnology; Bickerstaff, G.F., Ed.; Humana Press: Totowa, NJ, USA, 1997; Volume 1. [Google Scholar]
- Hernandez, K.; Fernandez-Lafuente, R. Control of Protein Immobilization: Coupling Immobilization and Site-Directed Mutagenesis to Improve Biocatalyst or Biosensor Performance. Enzyme Microb. Technol. 2011, 48, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, N.; Liu, L. Boronic Acid-Based Approach for Separation and Immobilization of Glycoproteins and Its Application in Sensing. Int. J. Mol. Sci. 2013, 14, 20890–20912. [Google Scholar] [CrossRef] [PubMed]
- Abad, M.; Velez, M.; Santamaria, C.; Guisan, J.M.; Matheus, P.R.; Vazquez, L.; Gazaryan, I.; Gorton, L.; Gibson, T.; Fernandez, V.M. Immobilization of Peroxidase Glycoprotein on Gold Electrodes Modified with Mixed Epoxy-Boronic Acid Monolayers. J. Am. Chem. Soc. 2002, 124, 12845–12853. [Google Scholar] [CrossRef] [PubMed]
- Gutarra, M.L.E.; Mateo, C.; Freire, D.M.G.; Torres, F.A.G.; Castro, A.M.; Guisan, J.M.; Palomo, J.M. Oriented Irreversible Immobilization of a Glycosylated Candida antarctica B Lipase on Heterofunctional Organoborane-Aldehyde Support. Catal. Sci. Technol. 2011, 1, 260–266. [Google Scholar] [CrossRef]
- Nazemi, S.A.; Olesinska, M.; Pezzella, C.; Varriale, S.; Lin, C.-W.; Corvini, P.F.-X.; Shahgaldian, P. Immobilisation and Stabilisation of Glycosylated Enzymes on Boronic Acid-Functionalised Silica Nanoparticles. Chem. Commun. 2021, 57, 11960–11963. [Google Scholar] [CrossRef]
- Rao, S.V.; Anderson, K.W.; Bachas, L.G. Oriented Immobilization of Proteins. Mikrochim. Acta 1998, 128, 127–143. [Google Scholar] [CrossRef]
- Gupta, P.; Maqbool, T.; Saleemuddin, M. Oriented Immobilization of Stem Bromelain via the Lone Histidine on a Metal Affinity Support. J. Mol. Catal. B Enzym. 2007, 45, 78–83. [Google Scholar] [CrossRef]
- Freitas, A.I.; Domingues, L.; Aguiar, T.Q. Tag-Mediated Single-Step Purification and Immobilization of Recombinant Proteins toward Protein-Engineered Advanced Materials. J. Adv. Res. 2022, 36, 249–264. [Google Scholar] [CrossRef]
- Sunbul, M.; Yin, J. Site Specific Protein Labeling by Enzymatic Posttranslational Modification. Org. Biomol. Chem. 2009, 7, 3361–3371. [Google Scholar] [CrossRef]
- Young, T.S.; Schultz, P.C. Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef]
- Pellis, A.; Vastano, M.; Quartinello, F.; Acero, E.H.; Guebitz, G.M. His-tag Immobilization of Cutinase 1 from Thermobifida cellulosilytica for Solvent-free Synthesis of Polyesters. Biotechnol. J. 2017, 12, 1700322. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.P.; Derrington, S.R.; Heath, R.S.; Porter, J.L.; Mangas-Sanchez, J.; Devine, P.N.; Truppo, M.D.; Turner, N.J. A Generic Platform for the Immobilisation of Engineered Biocatalysts. Tetrahedron 2019, 75, 327–334. [Google Scholar] [CrossRef]
- Böhmer, W.; Knaus, T.; Volkov, A.; Slot, T.K.; Shiju, N.R.; Cassimjee, K.E.; Mutti, F.G. Highly Efficient Production of Chiral Amines in Batch and Continuous Flow by Immobilized ω-Transaminases on Controlled Porosity Glass Metal-Ion Affinity Carrier. J. Biotechnol. 2019, 291, 52–60. [Google Scholar] [CrossRef]
- Tentori, F.; Bavaro, T.; Brenna, E.; Colombo, D.; Monti, D.; Semproli, R.; Ubiali, D. Immobilization of Old Yellow Enzymes via Covalent or Coordination Bonds. Catalysts 2020, 10, 260. [Google Scholar] [CrossRef]
- de Andrade, B.C.; Gennari, A.; Renard, G.; Nervis, B.D.R.; Benvenutti, E.V.; Costa, T.M.H.; Nicolodi, S.; Pesce da Silveira, N.; Chies, J.M.; Volpato, G.; et al. Synthesis of Magnetic Nanoparticles Functionalized with Histidine and Nickel to Immobilize His-Tagged Enzymes Using β-Galactosidase as a Model. Int. J. Biol. Macromol. 2021, 184, 159–169. [Google Scholar] [CrossRef]
- Robescu, M.S.; Tengattini, S.; Rabuffetti, M.; Speranza, G.; Terreni, M.; Bavaro, T. β-Mannosidase from Cellulomonas fimi: Immobilization Study and Application in the β-Mannoside Synthesis. Catalysts 2023, 13, 1399. [Google Scholar] [CrossRef]
- Cassimjee, K.E.; Kadow, M.; Wikmark, Y.; Svedendahl Humble, M.; Rothstein, M.L.; Rothstein, D.M.; Backvall, J.-E. A General Protein Purification and Immobilization Method on Controlled Porosity Glass: Biocatalytic Applications. Chem. Commun. 2014, 50, 9134. [Google Scholar] [CrossRef] [PubMed]
- Zeballos, N.; Comino, N.; Andrés-Sanz, D.; Santiago-Arcos, J.; Azkargorta, M.; Elortza, F.; Diamanti, E.; López-Gallego, F. Region-Directed Enzyme Immobilization through Engineering Protein Surface with Histidine Clusters. ACS Appl. Mater. Interfaces 2024, 16, 833–846. [Google Scholar] [CrossRef]
- Mateo, C.; Fernandez-Lorente, G.; Cortes, E.; Garcia, J.L.; Fernandez-Lafuente, R.; Guisan, J.M. One-step Purification, Covalent Immobilization, and Additional Stabilization of poly-His-tagged Proteins Using Novel Heterofunctional Chelate-Epoxy Supports. Biotechnol. Bioeng. 2001, 76, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cheng, F.; Li, H.; Jin, W.; Chen, C.; He, W.; Cheng, G.; Wang, Q. Site-Specific and Covalent Immobilization of His-Tagged Proteins via Surface Vinyl Sulfone–Imidazole Coupling. Langmuir 2019, 35, 16466–16475. [Google Scholar] [CrossRef]
- Long, J.; Li, X.; Liu, X.; Jin, Z.; Xie, Z.; Xu, X.; Lu, C. Preparation of Streptavidin-Coated Magnetic Nanoparticles for Specific Immobilization of Enzymes with High Activity and Enhanced Stability. Ind. Eng. Chem. Res. 2021, 60, 1542–1552. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Xie, Z.; Zhou, X.; Chen, L.; Qui, C.; Lu, C.; Jin, Z.; Long, J. Comparative Study on Different Immobilization Sites of Immobilized β-Agarase Based on the Biotin/Streptavidin System. Int. J. Biol. Macromol. 2024, 261, 129807. [Google Scholar] [CrossRef] [PubMed]
- Cronan, J.E. Biotination of Proteins in vivo. A Post-Translational Modification to Label, Purify, and Study Proteins. J. Biol. Chem. 1990, 265, 10327–10333. [Google Scholar] [CrossRef] [PubMed]
- Cull, M.G.; Schatz, P.J. Biotinylation of Proteins in vivo and in vitro Using Small Peptide Tags. Methods Enzymol. 2000, 326, 430–440. [Google Scholar] [CrossRef]
- Vishwanath, S.; Bhattacharyya, D.; Huang, W.; Bachas, L.G. Site-Directed and Random Enzyme Immobilization on Functionalized Membranes: Kinetic Studies and Models. J. Membr. Sci. 1995, 108, 1–13. [Google Scholar] [CrossRef]
- Alftrén, J.; Ottow, K.E.; Hobley, T.J. In vivo Biotinylation of Recombinant Beta-Glucosidase Enables Simultaneous Purification and Immobilization on Streptavidin Coated Magnetic Particles. J. Mol. Catal. B Enzym. 2013, 94, 29–35. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Paradisi, F. Sustainable Flow-synthesis of (Bulky) Nucleoside Drugs by a Novel and Highly Stable Nucleoside Phosphorylase Immobilized on Reusable Supports. ChemSusChem 2022, 15, e202102030. [Google Scholar] [CrossRef] [PubMed]
- Hoelzel, C.A.; Zhang, X. Visualizing and Manipulating Biological Processes by Using HaloTag and SNAP-Tag Technologies. ChemBioChem 2020, 21, 1935–1946. [Google Scholar] [CrossRef]
- Döbber, J.; Pohl, M. HaloTagTM: Evaluation of a Covalent One-Step Immobilization for Biocatalysis. J. Biotechnol. 2017, 241, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Döbber, J.; Gerlach, T.; Offermann, H.; Rother, D.; Pohl, M. Closing the Gap for Efficient Immobilization of Biocatalysts in Continuous Processes: HaloTagTM Fusion Enzymes for a Continuous Enzymatic Cascade towards a Vicinal Chiral Diol. Green Chem. 2018, 20, 544. [Google Scholar] [CrossRef]
- Seide, S.; Arnold, L.; Wetzels, S.; Bregu, M.; Gätgens, J.; Pohl, M. From Enzyme to Preparative Cascade Reactions with Immobilized Enzymes: Tuning Fe(II)/α-Ketoglutarate-Dependent Lysine Hydroxylases for Application in Biotransformations. Catalysts 2022, 12, 354. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide Tag Forming a Rapid Covalent Bond to a Protein, through Engineering a Bacterial Adhesin. Proc. Natl. Acad. Sci. USA 2012, 24, E690–E697. [Google Scholar] [CrossRef] [PubMed]
- Keeble, A.H.; Banerjee, A.; Ferla, M.P.; Reddington, S.C.; Khairil Anuar, I.N.A.; Howarth, M. Evolving Accelerated Amidation by SpyTag/SpyCatcher to Analyze Membrane Dynamics. Angew. Chem. Int. Ed. 2017, 56, 16521–16525. [Google Scholar] [CrossRef]
- Veggiani, G.; Nakamura, T.; Brenner, M.D.; Gayet, R.V.; Yan, J.; Robinson, C.V.; Howarth, M. Programmable Polyproteams Built Using Twin Peptide Superglues. Proc. Natl. Acad. Sci. USA 2016, 113, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.L.; Hoon, S.S.; Wong, F.T. Kinetic Controlled Tag-Catcher Interactions for Directed Covalent Protein Assembly. PLoS ONE 2016, 11, e0165074. [Google Scholar] [CrossRef]
- Khairil Anuar, I.N.A.; Banerjee, A.; Keeble, A.H.; Carella, A.; Nikov, G.I.; Howarth, M. Spy&Go Purification of SpyTag-Proteins Using Pseudo-SpyCatcher to Access an Oligomerization Toolbox. Nat. Commun. 2019, 10, 1734. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, Q.; Li, J.; Pistolozzi, M.; Zhao, L.; Yang, X.; Ye, Y. Spy Chemistry-enabled Protein Directional Immobilization and Protein Purification. Biotechnol. Bioeng. 2020, 117, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wei, C.; Qi, H.; Li, C.; Lu, F.; Qin, H.-M. Directional Immobilization of D-Allulose 3-Epimerase Using SpyTag/SpyCatcher Strategy as a Robust Biocatalyst for Synthesizing D-Allulose. Food Chem. 2023, 402, 134199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Yang, Y.; Liu, X.; Zhu, W.; Pi, L.; Liu, X.; Wang, S. Spontaneous and Site-Specific Immobilization of PNGase F via Spy Chemistry. RSC Adv. 2023, 13, 28493. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, R.; Subramanian, S.; Boder, E.T. Sortase A as a Novel Molecular “Stapler” for Sequence-Specific Protein Conjugation. Bioconjug. Chem. 2007, 18, 469–476. [Google Scholar] [CrossRef]
- Hata, Y.; Matsumoto, T.; Tanaka, T.; Kondo, A. C-Terminal-oriented Immobilization of Enzymes Using Sortase A-mediated Technique. Macromol. Biosci. 2015, 15, 1375–1380. [Google Scholar] [CrossRef] [PubMed]
- Qafari, S.M.; Ahmadian, G.; Mohammadi, M. One-Step Purification and Oriented Attachment of Protein A on Silica and Graphene Oxide Nanoparticles Using Sortase-Mediated Immobilization. RSC Adv. 2017, 7, 56006. [Google Scholar] [CrossRef]
- Moosavi, F.; Ahrari, F.; Ahmadian, G.; Mohammadi, M. Sortase-Mediated Immobilization of Candida antarctica Lipase B (CalB) on Graphene Oxide; Comparison with Chemical Approach. Biotechnol. Rep. 2022, 34, e00733. [Google Scholar] [CrossRef]
- Ito, T.; Sadamoto, R.; Naruchi, K.; Togame, H.; Takemoto, H.; Kondo, H.; Nishimura, S.-I. Highly Oriented Recombinant Glycosyltransferases: Site-Specific Immobilization of Unstable Membrane Proteins by Using Staphylococcus aureus Sortase A. Biochemistry 2010, 49, 2604–2614. [Google Scholar] [CrossRef]
- Strop, P. Versatility of Microbial Transglutaminase. Bioconjug. Chem. 2014, 25, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Oteng-Pabi, S.K.; Clouthier, C.M.; Keillor, J.W. Design of a Glutamine Substrate Tag Enabling Protein Labelling Mediated by Bacillus subtilis Transglutaminase. PLoS ONE 2018, 13, e0197956. [Google Scholar] [CrossRef]
- Steffen, W.; Ko, F.C.; Patel, J.; Lyamichev, V.; Albert, T.J.; Benz, J.; Rudolph, M.G.; Bergmann, F.; Streidl, T.; Kratzsch, P.; et al. Discovery of a Microbial Transglutaminase Enabling Highly Site-Specific Labeling of Proteins. J. Biol. Chem. 2017, 292, 15622–15635. [Google Scholar] [CrossRef]
- Kamiya, N.; Doi, S.; Tanaka, Y.; Ichinose, H.; Goto, M. Functional Immobilization of Recombinant Alkaline Phosphatases Bearing a Glutamyl Donor Substrate Peptide of Microbial Transglutaminase. J. Biosci. Bioeng. 2007, 104, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Sung, K.; Goto, M.; Kamiya, N. Immobilization of Alkaline Phosphatase on Magnetic Particles by Site-Specific and Covalent Cross-Linking Catalyzed by Microbial Transglutaminase. J. Biosci. Bioeng. 2011, 111, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-M.; Zhou, H.; Zhang, W.-F.; Yang, H.-M.; Tang, J.-B. Site-Specific, Covalent Immobilization of BirA by Microbial Transglutaminase: A Reusable Biocatalyst for in vitro Biotinylation. Anal. Biochem. 2016, 511, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Tang, M.-Z.; Yu, X.-T.; Xu, C.-M.; Yang, H.-M.; Tang, J.-B. Site-Specific, Covalent Immobilization of an Engineered Enterokinase onto Magnetic Nanoparticles through Transglutaminase-Catalyzed Bioconjugation. Colloids Surf. B Biointerfaces 2019, 117, 506–511. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, W.; Yang, Y.; Zhu, W.; Li, P.; Wang, S.; Liu, X. Site-Specific, Covalent Immobilization of PNGase F on Magnetic Particles Mediated by Microbial Transglutaminase. Anal. Chim. Acta 2023, 1250, 340972. [Google Scholar] [CrossRef]
- Serra, I.; Ubiali, D.; Cecchini, D.A.; Calleri, E.; Albertini, A.M.; Terreni, M.; Temporini, C. Assessment of Immobilized PGA Orientation via the LC-MS Analysis of Tryptic Digests of the Wild Type and Its 3K-PGA Mutant Assists in the Rational Design of a High-Performance Biocatalyst. Anal. Bioanal. Chem. 2013, 405, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Pagolu, R.; Singh, R.; Shanmugam, R.; Kondaveeti, S.; Patel, S.K.S.; Kalia, V.C.; Lee, K.-K. Site-Directed Lysine Modification of Xylanase for Oriented Immobilization onto Silicon Dioxide Nanoparticles. Bioresour. Technol. 2021, 331, 125063. [Google Scholar] [CrossRef]
- Grazu, V.; Lopez-Gallego, F.; Montes, T.; Abian, O.; Gonzalez, R.; Hermoso, J.A.; Garcia, J.L.; Mateo, C.; Guisan, J.M. Promotion of Multipoint Covalent Immobilization through Different Regions of Genetically Modified Penicillin G Acylase from E. coli. Process Biochem. 2010, 45, 390–398. [Google Scholar] [CrossRef]
- Hong, V.; Presolski, S.I.; Ma, C.; Finn, M.G. Analysis and Optimization of Copper-Catalyzed Azide–Alkyne Cycloaddition for Bioconjugation. Angew. Chem. Int. Ed. 2009, 48, 9879–9883. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.Y.; Hutchings, C.H.; Lindsay, M.J.; Werner, C.J.; Bundy, B.C. Enhanced Enzyme Stability through Site-Directed Covalent Immobilization. J. Biotechnol. 2015, 193, 83–90. [Google Scholar] [CrossRef]
- Wang, A.; Du, F.; Pei, X.; Chen, C.; Wu, S.G.; Zheng, Y. Rational Immobilization of Lipase by Combining the Structure Analysis and Unnatural Amino Acid Insertion. J. Mol. Catal. B Enzym. 2016, 132, 54–60. [Google Scholar] [CrossRef]
- Guan, D.; Kurra, Y.; Liu, W.; Chen, Z. A Click Chemistry Approach to Site-Specific Immobilization of a Small Laccase Enables Efficient Direct Electron Transfer in a Biocathode. Chem. Commun. 2015, 51, 2522. [Google Scholar] [CrossRef]
- Bednar, R.M.; Golbek, T.W.; Kean, K.M.; Brown, W.J.; Jana, S.; Baio, J.E.; Karplus, P.A.; Mehl, R.A. Immobilization of Proteins with Controlled Load and Orientation. ACS Appl. Mater. Interfaces 2019, 11, 36391–36398. [Google Scholar] [CrossRef] [PubMed]
- Lampkowski, J.S.; Villa, J.K.; Young, T.S.; Young, D.D. Development and Optimization of Glaser–Hay Bioconjugations. Angew. Chem. Int. Ed. 2015, 54, 9343–9346. [Google Scholar] [CrossRef]
- Switzer, H.J.; Howard, C.A.; Halonski, J.F.; Peairs, E.M.; Smith, N.; Zamecnik, M.P.; Verma, S.; Young, D.D. Employing Non-Canonical Amino Acids towards the Immobilization of a Hyperthermophilic Enzyme to Increase Protein Stability. RSC Adv. 2023, 13, 8496. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactive Group | Structure | Amino Acid |
|---|---|---|
| Primary amine | 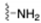 | N-terminus ε-amino groups of Lysine |
| Carboxylic acid | 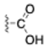 | C-terminus Glutamic acid Aspartic acid |
| Thiol | 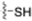 | Cysteine |
| Imidazole | 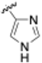 | Histidine |
| Functional Group | Structure | Binding | Reactive Group of the Enzyme |
|---|---|---|---|
| Epoxy |  | Nucleophilic attach and epoxy ring opening | Nucleophilic groups (-NH2 and -SH) |
| Amino |  | Activation with a dialdehyde; Schiff base formation | Primary amines (terminal-NH2 and Lys side chains) |
| Hydroxy | 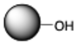 | Activation with a dialdehyde; Schiff base formation | Primary amines (terminal-NH2 and Lys side chains) |
| Activation with DVS; C-X bond formation | Imidazole, thiol and primary amines (depending on the pH) | ||
| Activation with CnBr; imidocarbonate bond formation | Primary amines (terminal-NH2 at mild pH) | ||
| Activation with CDI; carbamate bond formation | Primary amines (terminal-NH2 and Lys side chains) | ||
| Activation with epoxy groups (GLYMO); Nucleophilic attach and epoxy ring opening | Nucleophilic groups (-NH2 and -SH) | ||
| Activation with thiol groups; thiol exchange (S-S bond) | -SH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robescu, M.S.; Bavaro, T. A Comprehensive Guide to Enzyme Immobilization: All You Need to Know. Molecules 2025, 30, 939. https://doi.org/10.3390/molecules30040939
Robescu MS, Bavaro T. A Comprehensive Guide to Enzyme Immobilization: All You Need to Know. Molecules. 2025; 30(4):939. https://doi.org/10.3390/molecules30040939
Chicago/Turabian StyleRobescu, Marina Simona, and Teodora Bavaro. 2025. "A Comprehensive Guide to Enzyme Immobilization: All You Need to Know" Molecules 30, no. 4: 939. https://doi.org/10.3390/molecules30040939
APA StyleRobescu, M. S., & Bavaro, T. (2025). A Comprehensive Guide to Enzyme Immobilization: All You Need to Know. Molecules, 30(4), 939. https://doi.org/10.3390/molecules30040939