
PLM-ATG: Identification of Autophagy Proteins by Integrating Protein Language Model Embeddings with PSSM-Based Features


Abstract
1. Introduction
2. Results and Discussion
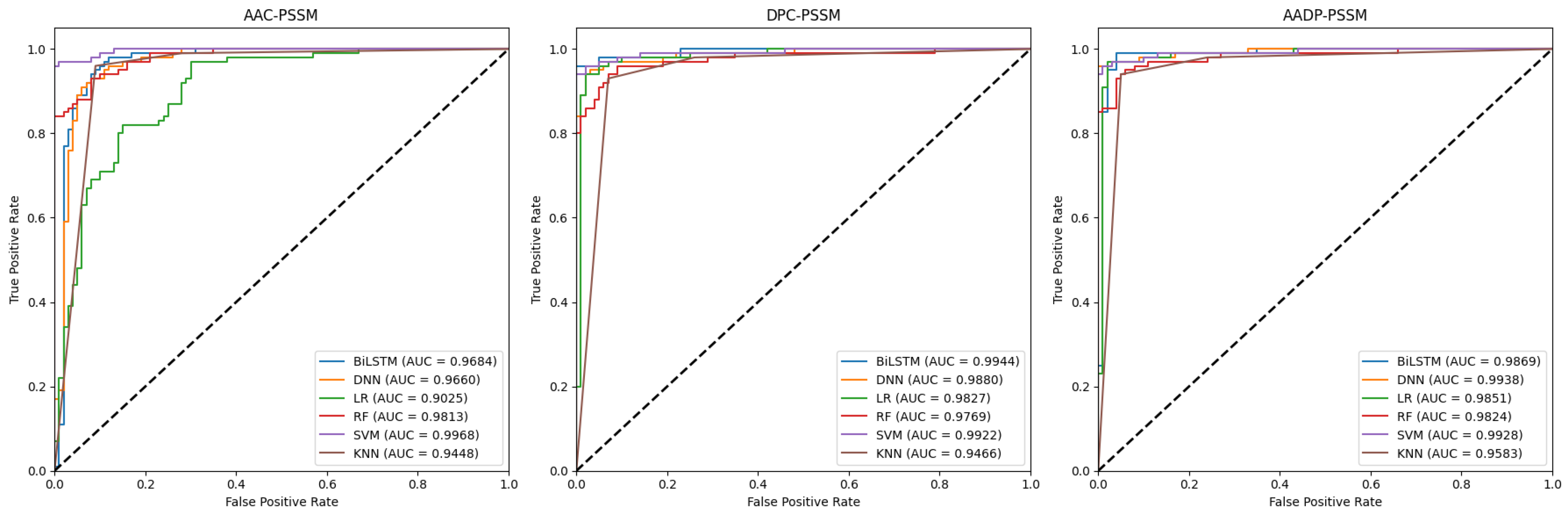
2.1. Performance Analysis of Baseline Models
2.2. Performance Comparison of Three PLM Embeddings
2.3. Performance Analysis of Feature Selection
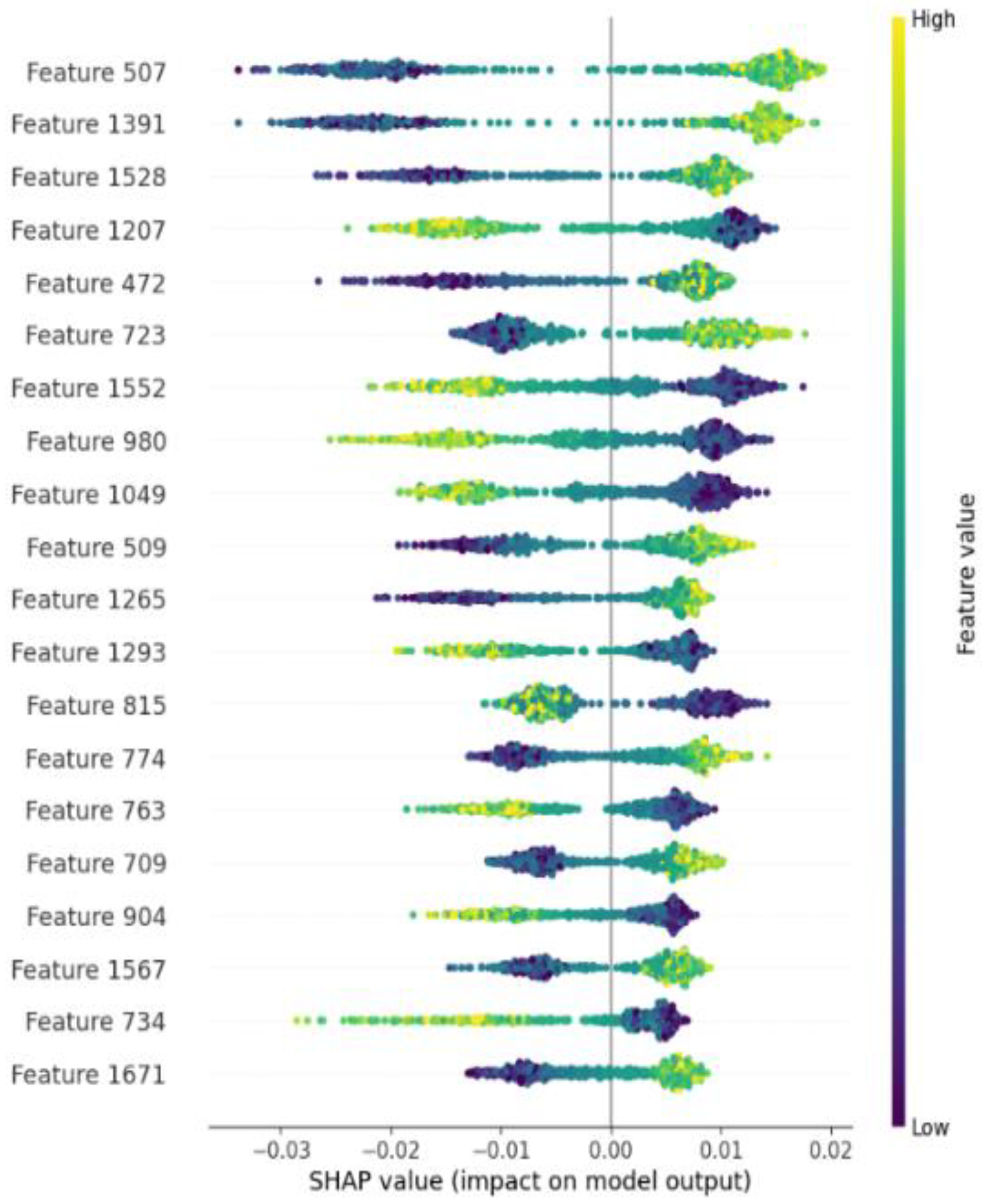
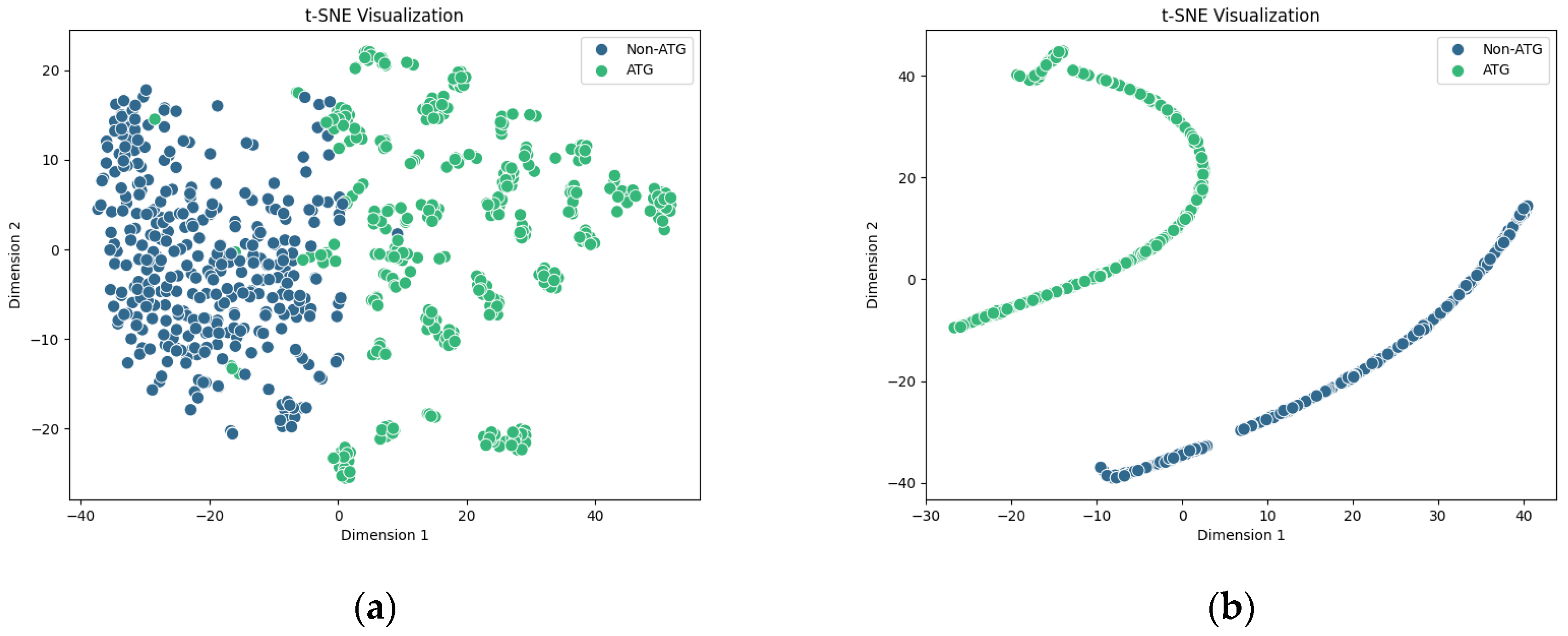
2.4. Interpretability of the PLM-ATG Model
2.5. Performance Comparison with Existing Models
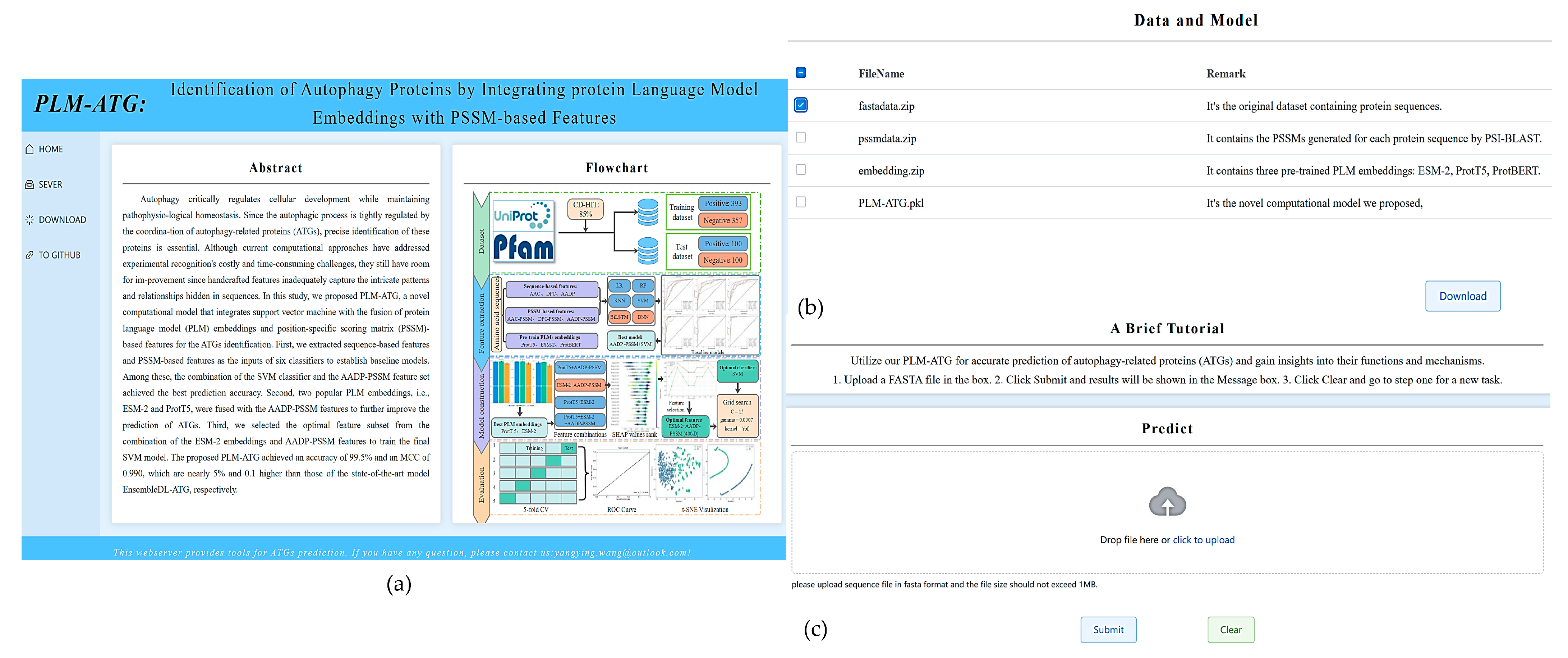
2.6. Web Server Implementation
3. Materials and Methods
3.1. Datasets
3.2. Feature Representation
3.2.1. PLM Embedding
3.2.2. Sequence-Based Features
3.2.3. PSSM-Based Features
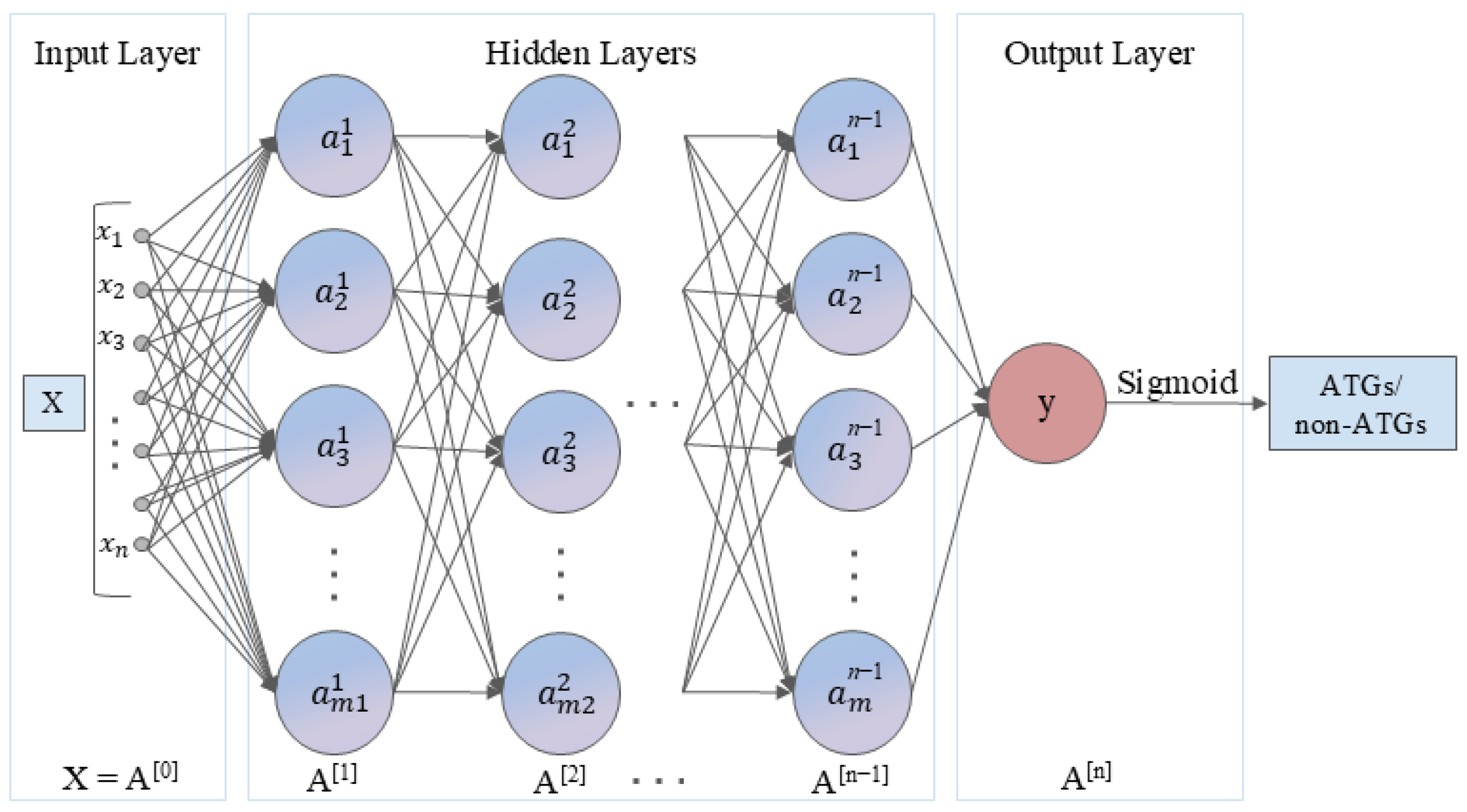
3.3. Model Architecture
3.4. Performance Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.-S. Autophagy-a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef]
- Rockel, J.S.; Kapoor, M. Autophagy: Controlling cell fate in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 517–531. [Google Scholar] [CrossRef]
- Nakahira, K.; Porras, M.A.P.; Choi, A.M.K. Autophagy in Pulmonary Diseases. Am. J. Respir. Crit. Care Med. 2016, 194, 1196–1207. [Google Scholar] [CrossRef]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Demaria, S.; Formenti, S.C.; Kroemer, G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat. Rev. Clin. Oncol. 2017, 14, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3-and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef]
- Martinet, W.; Timmermans, J.-P.; De Meyer, G.R. Methods to assess autophagy in situ—Transmission electron microscopy versus immunohistochemistry. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 543, pp. 89–114. [Google Scholar]
- Cheng, L.; Zeng, Y.; Hu, S.; Zhang, N.; Cheung, K.C.P.; Li, B.; Leung, K.-S.; Jiang, L. Systematic prediction of autophagy-related proteins using Arabidopsis thaliana interactome data. Plant J. 2021, 105, 708–720. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, Z.; Zhang, L.; Zhou, X.; Shi, L. ATGPred-FL: Sequence-based prediction of autophagy proteins with feature representation learning. Amino Acids 2022, 54, 799–809. [Google Scholar] [CrossRef]
- Ben-Hur, A.; Ong, C.S.; Sonnenburg, S.; Schoelkopf, B.; Raetsch, G. Support Vector Machines and Kernels for Computational Biology. PLoS Comput. Biol. 2008, 4, e1000173. [Google Scholar] [CrossRef]
- Yu, L.; Zhang, Y.; Xue, L.; Liu, F.; Jing, R.; Luo, J. EnsembleDL-ATG: Identifying autophagy proteins by integrating their sequence and evolutionary information using an ensemble deep learning framework. Comput. Struct. Biotechnol. J. 2023, 21, 4836–4848. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, H.; Wu, Y.; Hong, Z. Survey of Natural Language Processing Techniques in Bioinformatics. Comput. Math. Methods Med. 2015, 2015, 674296. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef]
- Asgari, E.; Mofrad, M.R.K. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef]
- Du, Z.; Ding, X.; Hsu, W.; Munir, A.; Xu, Y.; Li, Y. pLM4ACE: A protein language model based predictor for antihypertensive peptide screening. Food Chem. 2024, 431, 137162. [Google Scholar] [CrossRef]
- Han, J.; Kong, T.; Liu, J. PepNet: An interpretable neural network for anti-inflammatory and antimicrobial peptides prediction using a pre-trained protein language model. Commun. Biol. 2024, 7, 1198. [Google Scholar] [CrossRef]
- Thumuluri, V.; Armenteros, J.J.A.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef]
- Villegas-Morcillo, A.; Gomez, A.M.; Sanchez, V. An analysis of protein language model embeddings for fold prediction. Brief. Bioinform. 2022, 23, bbac142. [Google Scholar] [CrossRef]
- Qi, D.; Song, C.; Liu, T. PreDBP-PLMs: Prediction of DNA-binding proteins based on pre-trained protein language models and convolutional neural networks. Anal. Biochem. 2024, 694, 115603. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, T. PDNAPred: Interpretable prediction of protein-DNA binding sites based on pre-trained protein language models. Int. J. Biol. Macromol. 2024, 281, 136147. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jin, J.; Long, W.; Wei, L. PLPMpro: Enhancing promoter sequence prediction with prompt-learning based pre-trained language model. Comput. Biol. Med. 2023, 164, 107260. [Google Scholar] [CrossRef] [PubMed]
- Medina-Ortiz, D.; Contreras, S.; Fernandez, D.; Soto-Garcia, N.; Moya, I.; Cabas-Mora, G.; Olivera-Nappa, A. Protein Language Models and Machine Learning Facilitate the Identification of Antimicrobial Peptides. Int. J. Mol. Sci. 2024, 25, 8851. [Google Scholar] [CrossRef]
- Liu, T.; Zheng, X.; Wang, J. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie 2010, 92, 1330–1334. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; Candido, S. Language models of protein sequences at the scale of evolution enable accurate structure prediction. BioRxiv 2022, 2022, 500902. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Boateng, E.Y.; Abaye, D.A. A review of the logistic regression model with emphasis on medical research. J. Data Anal. Inf. Process. 2019, 7, 190. [Google Scholar] [CrossRef]
- Cutler, D.R.; Edwards, T.C., Jr.; Beard, K.H.; Cutler, A.; Hess, K.T.; Gibson, J.; Lawler, J.J. Random forests for classification in ecology. Ecology 2007, 88, 2783–2792. [Google Scholar] [CrossRef]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Zhou, P.; Shi, W.; Tian, J.; Qi, Z.; Li, B.; Hao, H.; Xu, B. Attention-based bidirectional long short-term memory networks for relation classification. In Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics, (Volume 2: Short Papers), Taiwan, China, 7–12 August 2016; Association for Computational Linguistics: Stroudsburg, PA, USA; pp. 207–212. [Google Scholar]
- Dhanuka, R.; Singh, J.P.; Tripathi, A. A Comprehensive Survey of Deep Learning Techniques in Protein Function Prediction. IEEE-Acm Trans. Comput. Biol. Bioinform. 2023, 20, 2291–2301. [Google Scholar] [CrossRef]
- Lv, H.; Dao, F.-Y.; Zulfiqar, H.; Su, W.; Ding, H.; Liu, L.; Lin, H. A sequence-based deep learning approach to predict CTCF-mediated chromatin loop. Brief. Bioinform. 2021, 22, bbab031. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, G.; Jin, S.; Li, Y.; Wang, Y. Predicting human microRNA-disease associations based on support vector machine. Int. J. Data Min. Bioinform. 2013, 8, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, D.; Wang, Y.; Zhang, X.; Su, M.; Wang, C.; Sun, Z.; Jiang, Q.; Sun, B.; Zhang, Y. Prediction of transcription factors binding events based on epigenetic modifications in different human cells. Epigenomics 2020, 12, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32 (Suppl. S1), D138–D141. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; UniProt, C. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef]
- Collobert, R.; Weston, J.; Bottou, L.; Karlen, M.; Kavukcuoglu, K.; Kuksa, P. Natural Language Processing (Almost) from Scratch. J. Mach. Learn. Res. 2011, 12, 2493–2537. [Google Scholar]
- Tran, C.; Khadkikar, S.; Porollo, A. Survey of Protein Sequence Embedding Models. Int. J. Mol. Sci. 2023, 24, 3775. [Google Scholar] [CrossRef]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language models enable zero-shot prediction of the effects of mutations on protein function. In Proceedings of the 35th Annual Conference on Neural Information Processing Systems (NeurIPS), Viture, 6–14 December 2021. [Google Scholar]
- Raffel, C.; Shazeer, N.; Roberts, A.; Lee, K.; Narang, S.; Matena, M.; Zhou, Y.; Li, W.; Liu, P.J. Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer. J. Mach. Learn. Res. 2020, 21, 1–67. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K.; Assoc Computat, L. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. In Proceedings of the Conference of the North-American-Chapter of the Association-for-Computational-Linguistics—Human Language Technologies (NAACL-HLT), Minneapolis, MN, USA, 2–7 February 2019; pp. 4171–4186. [Google Scholar]
- Rao, R.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.F.; Abbeel, P.; Sercu, T.; Rives, A. MSA Transformer. In Proceedings of the International Conference on Machine Learning (ICML), Viture, 18–24 July 2021. [Google Scholar]
- Rao, R.; Meier, J.; Sercu, T.; Ovchinnikov, S.; Rives, A. Transformer protein language models are unsupervised structure learners. Biorxiv 2020, 2020, 422761. [Google Scholar]
- Zhang, Y.; Yu, S.; Xie, R.; Li, J.; Leier, A.; Marquez-Lago, T.T.; Akutsu, T.; Smith, A.I.; Ge, Z.; Wang, J.; et al. PeNGaRoo, a combined gradient boosting and ensemble learning framework for predicting non-classical secreted proteins. Bioinformatics 2020, 36, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, Z.-C.; Su, W.; Yang, Y.-H.; Lv, H.; Yang, H.; Lin, H. iCarPS: A computational tool for identifying protein carbonylation sites by novel encoded features. Bioinformatics 2021, 37, 171–177. [Google Scholar] [CrossRef]
- Liu, J.; Su, R.; Zhang, J.; Wei, L. Classification and gene selection of triple-negative breast cancer subtype embedding gene connectivity matrix in deep neural network. Brief. Bioinform. 2021, 22, bbaa395. [Google Scholar] [CrossRef]
- Luo, J.; Yu, L.; Guo, Y.; Li, M. Functional classification of secreted proteins by position specific scoring matrix and auto covariance. Chemom. Intell. Lab. Syst. 2012, 110, 163–167. [Google Scholar] [CrossRef]
- Yu, L.; Liu, F.; Li, Y.; Luo, J.; Jing, R. DeepT3_4: A Hybrid Deep Neural Network Model for the Distinction Between Bacterial Type III and IV Secreted Effectors. Front. Microbiol. 2021, 12, 605782. [Google Scholar] [CrossRef]
- Yu, L.; Xue, L.; Liu, F.; Li, Y.; Jing, R.; Luo, J. The applications of deep learning algorithms on in silico druggable proteins identification. J. Adv. Res. 2022, 41, 219–231. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Liu, X. Deep Recurrent Neural Network for Protein Function Prediction from Sequence. arXiv 2017, arXiv:1701.08318. [Google Scholar]
- Yu, Y.; Si, X.; Hu, C.; Zhang, J. A review of recurrent neural networks: LSTM cells and network architectures. Neural Comput. 2019, 31, 1235–1270. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Classifier | Acc | Pre | Sen | Spe | F1-Score | MCC |
|---|---|---|---|---|---|---|---|
| AAC | LR | 0.7150 | 0.6937 | 0.7700 | 0.6600 | 0.7299 | 0.4326 |
| RF | 0.7950 | 0.7706 | 0.8400 | 0.7500 | 0.8038 | 0.5924 | |
| SVM | 0.8450 | 0.8224 | 0.8800 | 0.8100 | 0.8502 | 0.6917 | |
| KNN | 0.8100 | 0.8523 | 0.7500 | 0.8700 | 0.7979 | 0.6245 | |
| BiLSTM | 0.7150 | 0.6720 | 0.8400 | 0.5900 | 0.7467 | 0.4441 | |
| DNN | 0.7250 | 0.6772 | 0.8600 | 0.5900 | 0.7577 | 0.4674 | |
| DPC | LR | 0.8150 | 0.8247 | 0.8000 | 0.8300 | 0.8122 | 0.6303 |
| RF | 0.8200 | 0.7807 | 0.8900 | 0.7500 | 0.8318 | 0.6464 | |
| SVM | 0.8450 | 0.8224 | 0.8800 | 0.8100 | 0.8502 | 0.6917 | |
| KNN | 0.7200 | 0.8667 | 0.5200 | 0.9200 | 0.6500 | 0.4801 | |
| BiLSTM | 0.7800 | 0.8111 | 0.7300 | 0.8300 | 0.7684 | 0.5628 | |
| DNN | 0.7900 | 0.8295 | 0.7300 | 0.8500 | 0.7766 | 0.5842 | |
| AADP | LR | 0.8150 | 0.8247 | 0.8000 | 0.8300 | 0.8122 | 0.6303 |
| RF | 0.8400 | 0.8036 | 0.9000 | 0.7800 | 0.8491 | 0.6849 | |
| SVM | 0.8450 | 0.8224 | 0.8800 | 0.8100 | 0.8502 | 0.6917 | |
| KNN | 0.6900 | 0.8276 | 0.4800 | 0.9000 | 0.6076 | 0.4187 | |
| BiLSTM | 0.7600 | 0.8250 | 0.6600 | 0.8600 | 0.7333 | 0.5307 | |
| DNN | 0.8000 | 0.8659 | 0.7100 | 0.8900 | 0.7802 | 0.6100 |
| Feature | Classifier | Acc | Pre | Sen | Spe | F1-Score | MCC |
|---|---|---|---|---|---|---|---|
| AAC-PSSM | LR | 0.8250 | 0.7826 | 0.9000 | 0.7500 | 0.8372 | 0.6574 |
| RF | 0.9200 | 0.9200 | 0.9200 | 0.9200 | 0.9200 | 0.8400 | |
| SVM | 0.9700 | 0.9700 | 0.9700 | 0.9700 | 0.9700 | 0.9400 | |
| KNN | 0.9350 | 0.9143 | 0.9600 | 0.9100 | 0.9366 | 0.8711 | |
| BiLSTM | 0.9300 | 0.9057 | 0.9600 | 0.9000 | 0.9320 | 0.8616 | |
| DNN | 0.9200 | 0.8889 | 0.9600 | 0.8800 | 0.9231 | 0.8427 | |
| DPC-PSSM | LR | 0.9350 | 0.9065 | 0.9700 | 0.9000 | 0.9372 | 0.8721 |
| RF | 0.9350 | 0.9307 | 0.9400 | 0.9300 | 0.9353 | 0.8700 | |
| SVM | 0.9700 | 0.9796 | 0.9600 | 0.9800 | 0.9697 | 0.9402 | |
| KNN | 0.9300 | 0.9300 | 0.9300 | 0.9300 | 0.9300 | 0.8600 | |
| BiLSTM | 0.9650 | 0.9515 | 0.9800 | 0.9500 | 0.9500 | 0.9304 | |
| DNN | 0.9400 | 0.9783 | 0.9000 | 0.9800 | 0.9375 | 0.8828 | |
| AADP-PSSM | LR | 0.9500 | 0.9327 | 0.9700 | 0.9300 | 0.9510 | 0.9007 |
| RF | 0.9400 | 0.9314 | 0.9500 | 0.9300 | 0.9406 | 0.8802 | |
| SVM | 0.9750 | 0.9798 | 0.9700 | 0.9800 | 0.9749 | 0.9500 | |
| KNN | 0.9450 | 0.9495 | 0.9400 | 0.9500 | 0.9447 | 0.8900 | |
| BiLSTM | 0.9750 | 0.9612 | 0.9900 | 0.9600 | 0.9754 | 0.9504 | |
| DNN | 0.9450 | 0.9238 | 0.9700 | 0.9200 | 0.9463 | 0.8911 |
| Feature | Dimension | Acc | Pre | Sen | Spe | F1-Score | MCC |
|---|---|---|---|---|---|---|---|
| ProtT5 | 1024 | 0.9800 | 0.9706 | 0.9900 | 0.9700 | 0.9802 | 0.9602 |
| ESM-2 | 1280 | 0.9850 | 0.9900 | 0.9700 | 0.9900 | 0.9848 | 0.9704 |
| ProtT5 + AADP-PSSM | 1444 | 0.9800 | 0.9898 | 0.9700 | 0.9900 | 0.9798 | 0.9602 |
| ESM-2 + AADP-PSSM | 1700 | 0.9900 | 0.9900 | 0.9900 | 0.9900 | 0.9900 | 0.9800 |
| ProtT5 + ESM-2 | 2304 | 0.9800 | 0.9706 | 0.9900 | 0.9700 | 0.9802 | 0.9602 |
| ProtT5 + ESM-2 + AADP-PSSM | 2724 | 0.9800 | 0.9706 | 0.9900 | 0.9700 | 0.9802 | 0.9602 |
| Dataset Type | Positive (ATGs) | Negative (Non-ATGs) |
|---|---|---|
| Training | 393 | 357 |
| Independent test | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, C. PLM-ATG: Identification of Autophagy Proteins by Integrating Protein Language Model Embeddings with PSSM-Based Features. Molecules 2025, 30, 1704. https://doi.org/10.3390/molecules30081704
Wang Y, Wang C. PLM-ATG: Identification of Autophagy Proteins by Integrating Protein Language Model Embeddings with PSSM-Based Features. Molecules. 2025; 30(8):1704. https://doi.org/10.3390/molecules30081704
Chicago/Turabian StyleWang, Yangying, and Chunhua Wang. 2025. "PLM-ATG: Identification of Autophagy Proteins by Integrating Protein Language Model Embeddings with PSSM-Based Features" Molecules 30, no. 8: 1704. https://doi.org/10.3390/molecules30081704
APA StyleWang, Y., & Wang, C. (2025). PLM-ATG: Identification of Autophagy Proteins by Integrating Protein Language Model Embeddings with PSSM-Based Features. Molecules, 30(8), 1704. https://doi.org/10.3390/molecules30081704