
Electrocatalytic Oxidation of HMF to FDCA over Multivalent Ruthenium in Neutral Electrolyte

 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
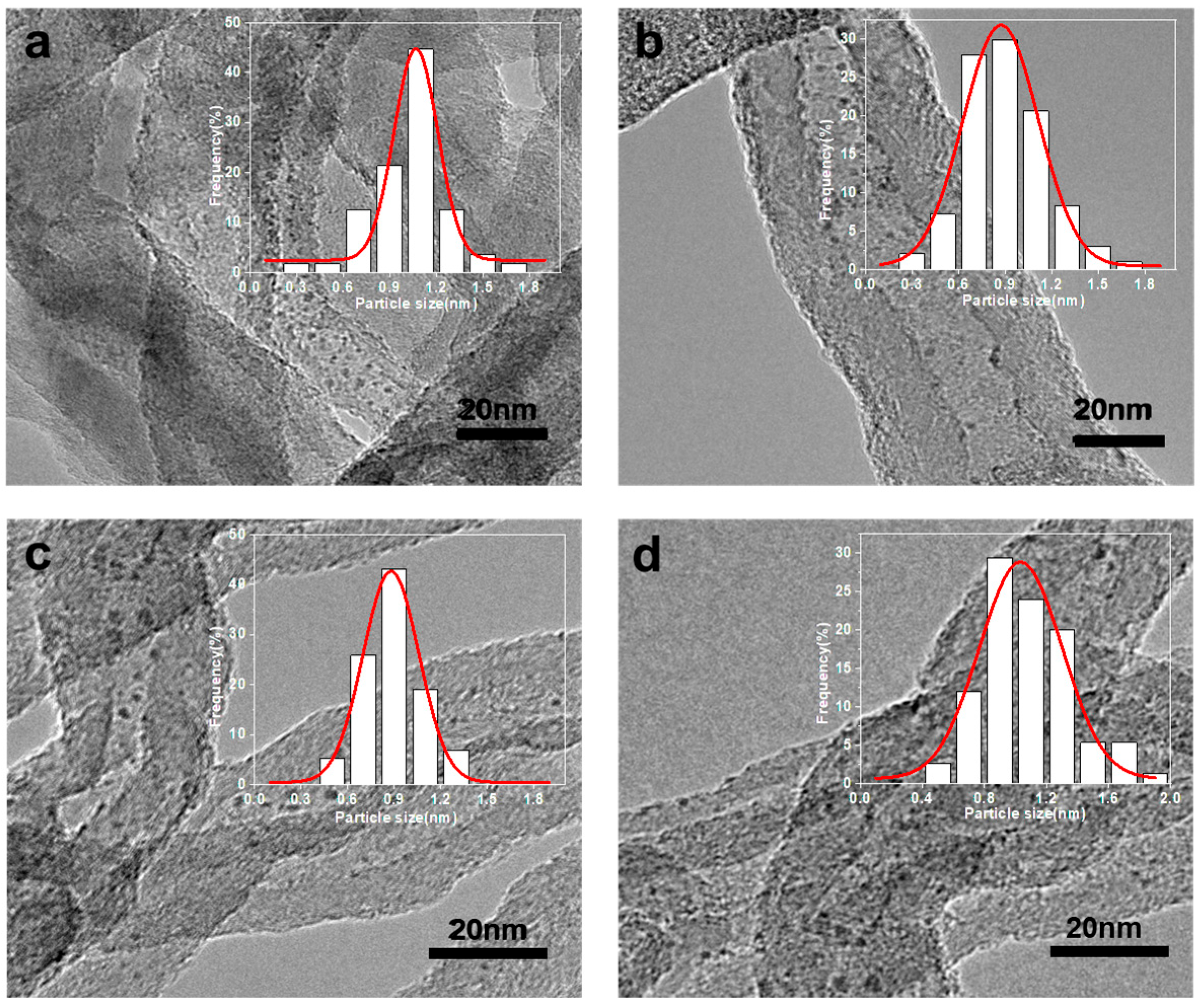
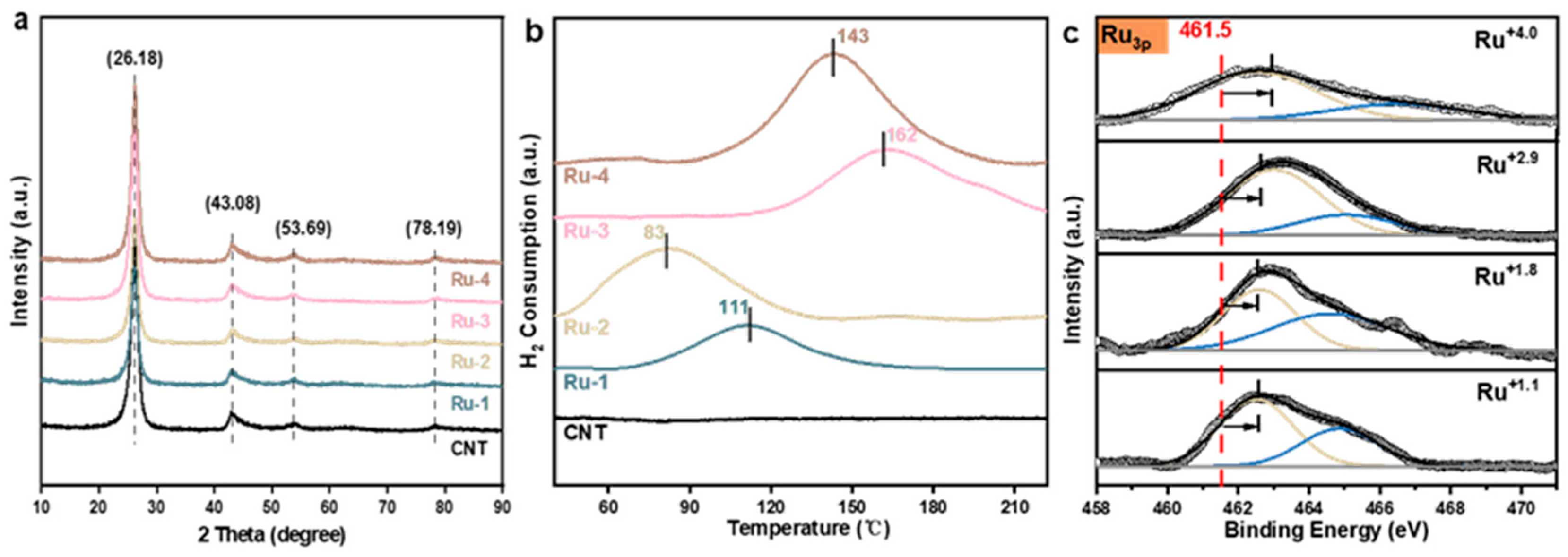
2.1. Fabrication and Characterization of the Ru Catalysts
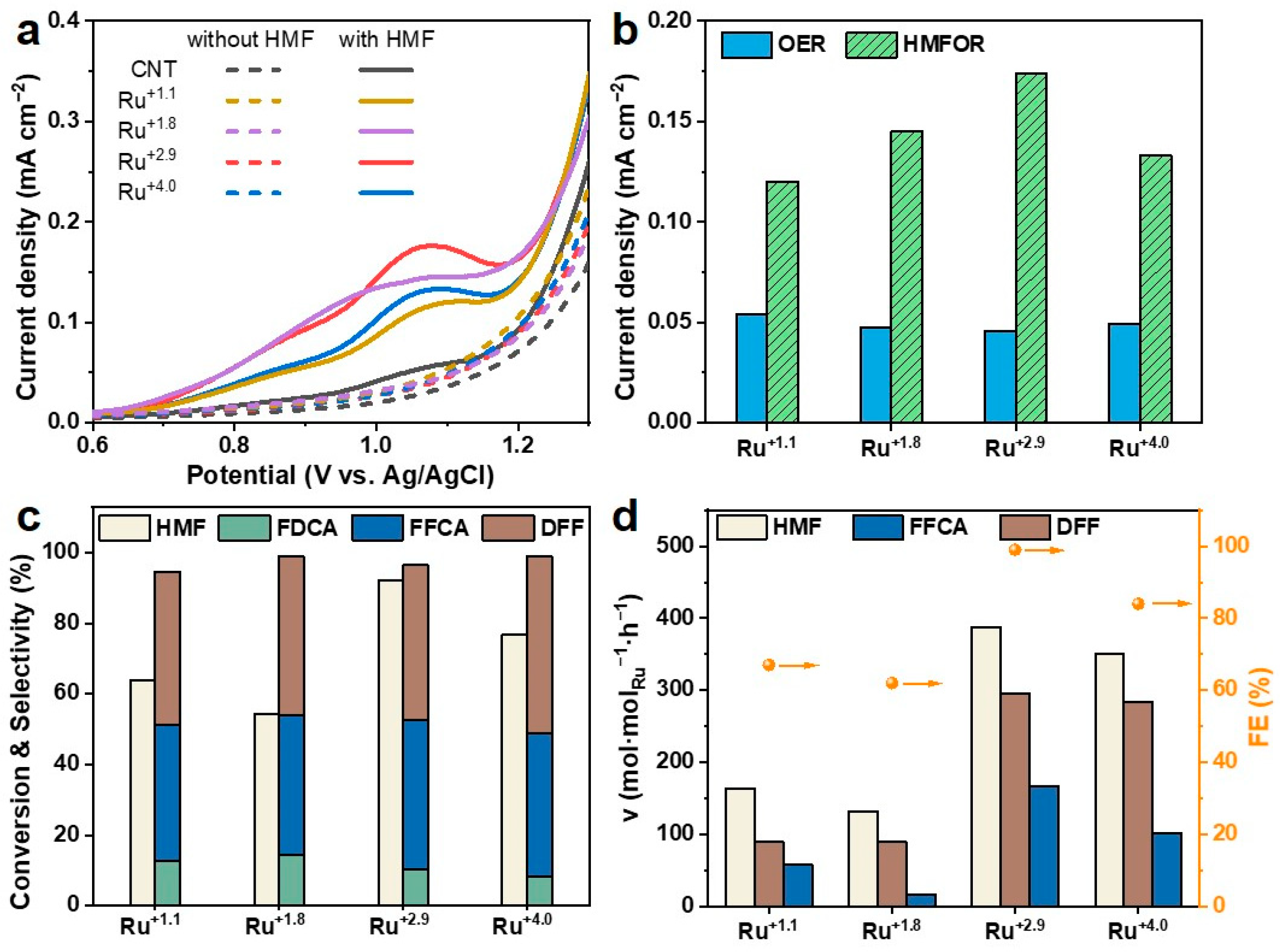
2.2. Electrooxidation of HMF over Ru Catalysts
2.3. Optimization of Ru+2.9 Catalyst Reaction Parameters
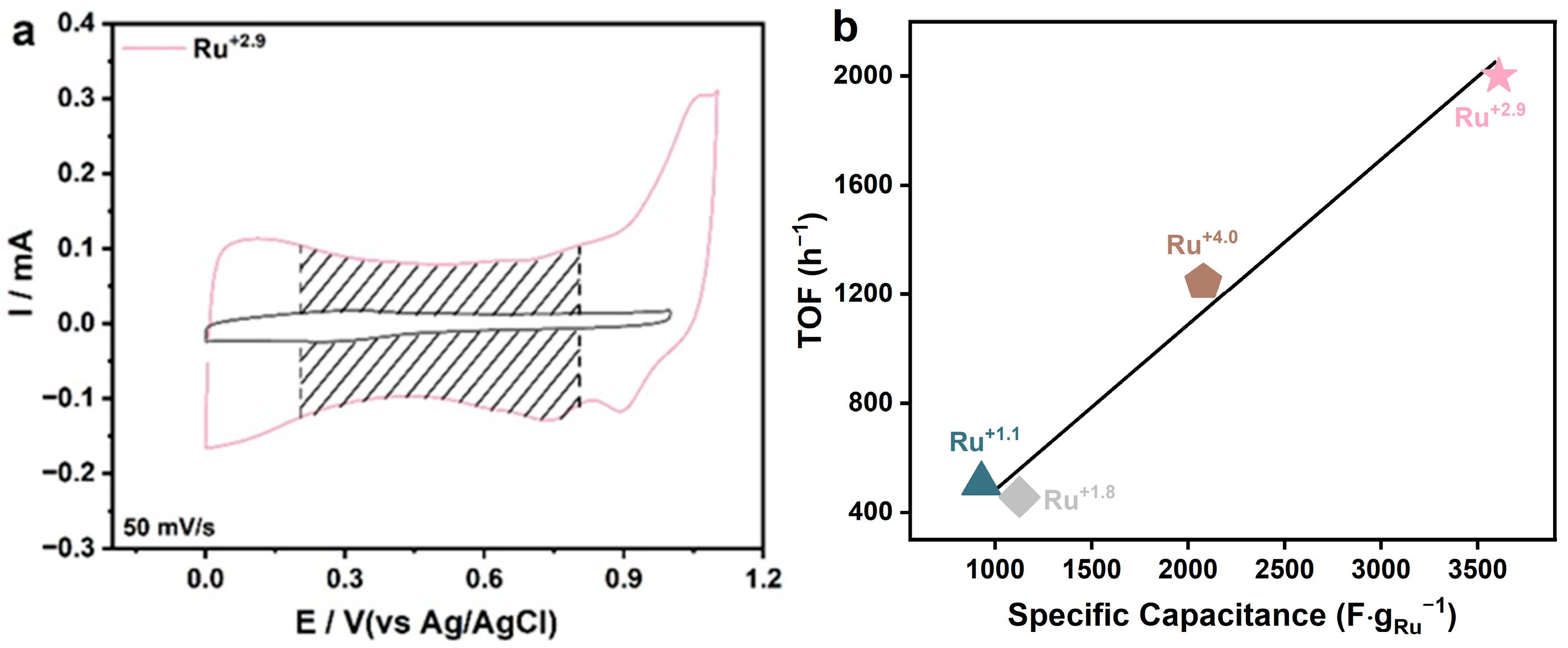
2.4. The Catalytic Ability of Ru+2.9 Catalyst
2.5. Reaction Mechanism of HMF Electrooxidation
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.2.1. CNT Purification [38]
3.2.2. RuO2/CNT Catalyst Preparation
- Ru-CNT-H2 catalyst preparation [39]
- RuCl3-CNT catalyst preparation [42]
- RuO2-CNT catalyst preparation
3.2.3. Catalyst Characterization
3.2.4. Electrochemical Catalysis Oxidation Reaction
3.2.5. Analyses of Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wan, Y.; Lee, J.-M. Toward Value-Added Dicarboxylic Acids from Biomass Derivatives via Thermocatalytic Conversion. ACS Catal. 2021, 11, 2524–2560. [Google Scholar] [CrossRef]
- Popp, J.; Kovács, S.; Oláh, J.; Divéki, Z.; Balázs, E. Bioeconomy: Biomass and biomass-based energy supply and demand. New Biotechnol. 2021, 60, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Pan, Y.; Zhou, H.; Tang, Y.; Ren, G.; Yu, Z.; Li, J.; Zhang, R.; Li, X.; Qiao, Y.; et al. Catalytic Systems for 5-Hydroxymethylfurfural Preparation from Different Biomass Feedstocks: A Review. Catalysts 2023, 14, 30. [Google Scholar] [CrossRef]
- Chacón-Huete, F.; Messina, C.; Cigana, B.; Forgione, P. Diverse Applications of Biomass-Derived 5-Hydroxymethylfurfural and Derivatives as Renewable Starting Materials. ChemSusChem 2022, 15, e202200328. [Google Scholar] [CrossRef]
- Zhou, C.; Shi, W.; Wan, X.; Meng, Y.; Yao, Y.; Guo, Z.; Dai, Y.; Wang, C.; Yang, Y. Oxidation of 5-hydroxymethylfurfural over a magnetic iron oxide decorated rGO supporting Pt nanocatalyst. Catal. Today 2019, 330, 92–100. [Google Scholar] [CrossRef]
- Kong, X.; Zhu, Y.; Fang, Z.; Kozinski, J.A.; Butler, I.S.; Xu, L.; Song, H.; Wei, X. Catalytic conversion of 5-hydroxymethylfurfural to some value-added derivatives. Green Chem. 2018, 20, 3657–3682. [Google Scholar] [CrossRef]
- Partenheimer, W.; Grushin, V.V. Synthesis of 2,5-diformylfuran and furan-2,5-dicarboxylic acid by catalytic air-oxidation of 5-hydroxymethylfurfural. Unexpectedly selective aerobic oxidation of benzyl alcohol to benzaldehyde with metal/bromide catalysts. Adv. Synth. Catal. 2001, 343, 102–111. [Google Scholar] [CrossRef]
- Saha, B.; Dutta, S.; Abu-Omar, M.M. Aerobic oxidation of 5-hydroxylmethylfurfural with homogeneous and nanoparticulate catalysts. Catal. Sci. Technol. 2012, 2, 79–81. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, B.; Bian, L.; Tang, Q.; Cao, Q.; Fang, W. Influence of NiO-calcination on Pt-supported catalyst for selective oxidation of 5-hydroxymethylfurfural. Catal. Commun. 2023, 184, 106791. [Google Scholar] [CrossRef]
- Siyo, B.; Schneider, M.; Radnik, J.; Pohl, M.-M.; Langer, P.; Steinfeldt, N. Influence of support on the aerobic oxidation of HMF into FDCA over preformed Pd nanoparticle based materials. Appl. Catal. A Gen. 2014, 478, 107–116. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, Y.; Chen, Y.; Wang, F.; Cao, Y.; Guan, W.; Li, X. Crystal Faces-Tailored Oxygen Vacancy in Au/CeO2 Catalysts for Efficient Oxidation of HMF to FDCA. ChemSusChem 2021, 15, e202101983. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Kaur, M.; Singh, V. Mild and selective catalytic oxidation of 5-HMF to 2,5-FDCA over metal loaded biomass derived graphene oxide using hydrogen peroxide in aqueous media. Mol. Catal. 2023, 546, 113223. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, L.; Li, B.; Bian, S.; Wang, J.; Zhang, H.; Zhao, C. Base metal catalyzed oxidation of 5-hydroxy-methyl-furfural to 2,5-furan-dicarboxylic acid: A review. Catal. Today 2023, 408, 64–72. [Google Scholar] [CrossRef]
- Le, T.K.T.; Kongparakul, S.; Zhang, H.; Zhao, J.; Guan, G.; Chanlek, N.; Tran, T.T.V.; Samart, C. Highly efficient liquid-phase oxidation of 5-hydroxymethylfurfural over Co-Cu/activated carbon catalysts. Mol. Catal. 2023, 539, 113017. [Google Scholar] [CrossRef]
- Lu, H.-z.; Bai, J.-f.; Yan, F.; Zhang, X.-y.; Jin, Y.; Wang, J.-y.; Chen, P.; Zhou, M.-d. Oxidation of 5-hydroxylmethylfurfural to 2,5-furandicarboxylic acid catalyzed by magnetic MnO2-Fe3O4 composite oxides. J. Fuel Chem. Technol. 2021, 49, 312–321. [Google Scholar] [CrossRef]
- Li, H.; Huang, X.; Lv, Y.; Zhang, J.; Li, W. Highly efficient electrooxidation of 5-hydroxymethylfurfural (HMF) by Cu regulated Co carbonate hydroxides boosting hydrogen evolution reaction. Int. J. Hydrogen Energy 2023, 48, 38279–38295. [Google Scholar] [CrossRef]
- Lei, J.; Zhang, H.J.; Yang, J.; Ran, J.; Ning, J.Q.; Wang, H.Y.; Hu, Y. Structural designs and mechanism insights into electrocatalytic oxidation of 5-hydroxymethylfurfural. J. Energy Chem. 2025, 100, 792–814. [Google Scholar] [CrossRef]
- Chen, C.L.; Lv, M.X.; Hu, H.L.; Huai, L.Y.; Zhu, B.; Fan, S.L.; Wang, Q.G.; Zhang, J. 5-Hydroxymethylfurfural and its downstream chemicals: A review of catalytic routes. Adv. Mater. 2024, 36, 2311464. [Google Scholar] [CrossRef]
- Duan, Y.F.; Lu, X.B.; Fan, O.Y.; Xu, H.C.; Zhang, Z.X.; Si, C.L.; Xu, T.; Du, H.S.; Li, X.Y. Non-noble metal catalysts for electrooxidation of 5-hydroxymethylfurfural. ChemSusChem 2025, 18, e202401487. [Google Scholar] [CrossRef]
- Zhu, B.; Li, Y.; Huai, L.; Cui, J.; Chen, G.; Huang, Q.; Zhang, J.; Chen, C. Enhanced Single-atom Cobalt Layer in MAX Phase for Biomass Electrooxidation Integrated with Hydrogen Evolution. Chem. Eng. J. 2024, 499, 155891. [Google Scholar] [CrossRef]
- Grabowski, G.; Lewkowski, J.; Skowroński, R. The electrochemical oxidation of 5-hydroxymethylfurfural with the nickel oxide/hydroxide electrode. Electrochim. Acta 1991, 36, 1995. [Google Scholar] [CrossRef]
- Chadderdon, D.J.; Xin, L.; Qi, J.; Qiu, Y.; Krishna, P.; More, K.L.; Li, W. Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles. Green Chem. 2014, 16, 3778–3786. [Google Scholar] [CrossRef]
- You, B.; Jiang, N.; Liu, X.; Sun, Y. Simultaneous H2 Generation and Biomass Upgrading in Water by an Efficient Noble-Metal-Free Bifunctional Electrocatalyst. Angew. Chem. Int. Ed. Engl. 2016, 55, 9913–9917. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; You, B.; Boonstra, R.; Terrero Rodriguez, I.M.; Sun, Y. Integrating Electrocatalytic 5-Hydroxymethylfurfural Oxidation and Hydrogen Production via Co–P-Derived Electrocatalysts. ACS Energy Lett. 2016, 1, 386–390. [Google Scholar] [CrossRef]
- Barwe, S.; Weidner, J.; Cychy, S.; Morales, D.M.; Dieckhöfer, S.; Hiltrop, D.; Masa, J.; Muhler, M.; Schuhmann, W. Electrocatalytic Oxidation of 5-(Hydroxymethyl)furfural Using High-Surface-Area Nickel Boride. Angew. Chem. Int. Ed. Engl. 2018, 57, 11460–11464. [Google Scholar] [CrossRef]
- Weidner, J.; Barwe, S.; Sliozberg, K.; Piontek, S.; Masa, J.; Apfel, U.-P.; Schuhmann, W. Cobalt–metalloid alloys for electrochemical oxidation of 5-hydroxymethylfurfural as an alternative anode reaction in lieu of oxygen evolution during water splitting. Beilstein J. Org. Chem. 2018, 14, 1436–1445. [Google Scholar] [CrossRef]
- Wang, T.; Song, Y.; Zhao, W.; Zhou, C.; Jin, Y.; Wan, X.; Dai, Y.; Yang, Y. Electro-catalytic oxidation of HMF to FDCA over RuO2/MnO2/CNT catalysts in base-free solution. New J. Chem. 2021, 45, 21285–21292. [Google Scholar] [CrossRef]
- Wang, Q.; De Brito Mendes, C.M.; Safonova, O.V.; Baaziz, W.; Urbina-Blanco, C.A.; Wu, D.; Marinova, M.; Ersen, O.; Capron, M.; Khodakov, A.Y.; et al. Tunable catalysis by reversible switching between Ru(III) single sites and Ru0 clusters in solid micelles. J. Catal. 2023, 426, 336–344. [Google Scholar] [CrossRef]
- Xi, W.; Jin, L.; Mahmood, A.; Zhang, W.; Li, Y.; Li, H.; An, P.; Zhang, J.; Ma, T.; Liu, S.; et al. Accelerating Ru0/Ru4+ Adjacent Dual Sites Construction by Copper Switch for Efficient Alkaline Hydrogen Evolution. Adv. Energy Mater. 2023, 13, 2302668. [Google Scholar] [CrossRef]
- Song, W.; Chen, Z.; Lai, W.; Rodríguez-Ramos, I.; Yi, X.; Weng, W.; Fang, W. Effect of lanthanum promoter on the catalytic performance of levulinic acid hydrogenation over Ru/carbon fiber catalyst. Appl. Catal. A Gen. 2017, 540, 21–30. [Google Scholar] [CrossRef]
- Yu, H.; Fu, X.; Zhou, C.; Peng, F.; Wang, H.; Yang, J. Capacitance dependent catalytic activity of RuO2·xH2O/CNT nanocatalysts for aerobic oxidation of benzyl alcohol. Chem. Commun. 2009, 17, 2408. [Google Scholar] [CrossRef] [PubMed]
- Al Ghatta, A.; Zhou, X.Y.; Casarano, G.; Wilton-Ely, J.D.E.T.; Hallett, J.P. Characterization and Valorization of Humins Produced by HMF Degradation in Ionic Liquids: A Valuable Carbonaceous Material for Antimony Removal. ACS Sustain. Chem. Eng. 2021, 9, 2212–2223. [Google Scholar] [CrossRef]
- Zhu, B.; Wang, Q.; Wang, J.; Yu, X.; Zhang, J.; Chen, C. Co@NC chainmail nanowires for thermo- and electrocatalytic oxidation of 2,5-bis(hydroxymethyl)furan to 2,5-furandicarboxylic acid. ChemSusChem 2025, 18, e202401422. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Kang, X.; Wang, F.; Zhang, J.; Zhang, T.; Ran, F. Capacitive contribution matters in facilitating high power battery materials toward fast-charging alkali metal ion batteries. Mater. Sci. Eng. R Rep. 2023, 154, 100737. [Google Scholar] [CrossRef]
- Buonaiuto, M.; De Crisci, A.G.; Jaramillo, T.F.; Waymouth, R.M. Electrooxidation of Alcohols with Electrode-Supported Transfer Hydrogenation Catalysts. ACS Catal. 2015, 5, 7343–7349. [Google Scholar] [CrossRef]
- Nikaidou, F.; Ushiyama, H.; Yamaguchi, K.; Yamashita, K.; Mizuno, N. Theoretical and Experimental Studies on Reaction Mechanism for Aerobic Alcohol Oxidation by Supported Ruthenium Hydroxide Catalysts. J. Phys. Chem. C 2010, 114, 10873–10880. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mizuno, N. Supported Ruthenium Catalyst for the Heterogeneous Oxidation of Alcohols with Molecular Oxygen. Angew. Chem. Int. Ed. Engl. 2002, 41, 4538–4542. [Google Scholar] [CrossRef]
- Zhou, C.; Peng, F.; Wang, H.; Yu, H.; Peng, C.; Yang, J. Development of stable PtRu catalyst coated with manganese dioxide for electrocatalytic oxidation of methanol. Electrochem. Commun. 2010, 12, 1210–1213. [Google Scholar] [CrossRef]
- Xie, J.; Nie, J.; Liu, H. Aqueous-phase selective aerobic oxidation of 5-hydroxymethylfurfural on Ru/C in the presence of base. Chin. J. Catal. 2014, 35, 937–944. [Google Scholar] [CrossRef]
- Yu, W.; Liu, M.; Liu, H.; Ma, X.; Liu, Z. Preparation, Characterization, and Catalytic Properties of Polymer-Stabilized Ruthenium Colloids. J. Colloid Interf. Sci. 1998, 208, 439–444. [Google Scholar] [CrossRef]
- Gao, T.; Yin, Y.; Fang, W.; Cao, Q. Highly dispersed ruthenium nanoparticles on hydroxyapatite as selective and reusable catalyst for aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid under base-free conditions. Mol. Catal. 2018, 450, 55–64. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, H.; Yan, Y.; Jia, X.; Liu, C.-j.; Yang, Y. Argon plasma reduced Pt nanocatalysts supported on carbon nanotube for aqueous phase benzyl alcohol oxidation. Catal. Today 2013, 211, 104–108. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte | Initial Reaction Rate (mol∙molRu−1∙h−1) | |||
|---|---|---|---|---|
| HMF | DFF | HMFCA | FFCA | |
| K2SO4 | 388 | 296 | / | 167 |
| H2SO4 | 161 | 52 | / | 40 |
| KOH | 156 | 558 | 117 | 49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Jin, X.; Zhu, B.; Yang, D.; Wan, X.; Dai, Y.; Zhou, C.; Jin, Y.; Yang, Y. Electrocatalytic Oxidation of HMF to FDCA over Multivalent Ruthenium in Neutral Electrolyte. Molecules 2025, 30, 1780. https://doi.org/10.3390/molecules30081780
Yang S, Jin X, Zhu B, Yang D, Wan X, Dai Y, Zhou C, Jin Y, Yang Y. Electrocatalytic Oxidation of HMF to FDCA over Multivalent Ruthenium in Neutral Electrolyte. Molecules. 2025; 30(8):1780. https://doi.org/10.3390/molecules30081780
Chicago/Turabian StyleYang, Shiying, Xin Jin, Bin Zhu, Dan Yang, Xiaoyue Wan, Yihu Dai, Chunmei Zhou, Yuguang Jin, and Yanhui Yang. 2025. "Electrocatalytic Oxidation of HMF to FDCA over Multivalent Ruthenium in Neutral Electrolyte" Molecules 30, no. 8: 1780. https://doi.org/10.3390/molecules30081780
APA StyleYang, S., Jin, X., Zhu, B., Yang, D., Wan, X., Dai, Y., Zhou, C., Jin, Y., & Yang, Y. (2025). Electrocatalytic Oxidation of HMF to FDCA over Multivalent Ruthenium in Neutral Electrolyte. Molecules, 30(8), 1780. https://doi.org/10.3390/molecules30081780