
Computational Study on the Pd-Catalyzed Pathway for the Formation of (R)-Methyl-(2-Hydroxy-1-Phenylethyl)Carbamate

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Results
2.1. Identified Species Experimentally and Computationally
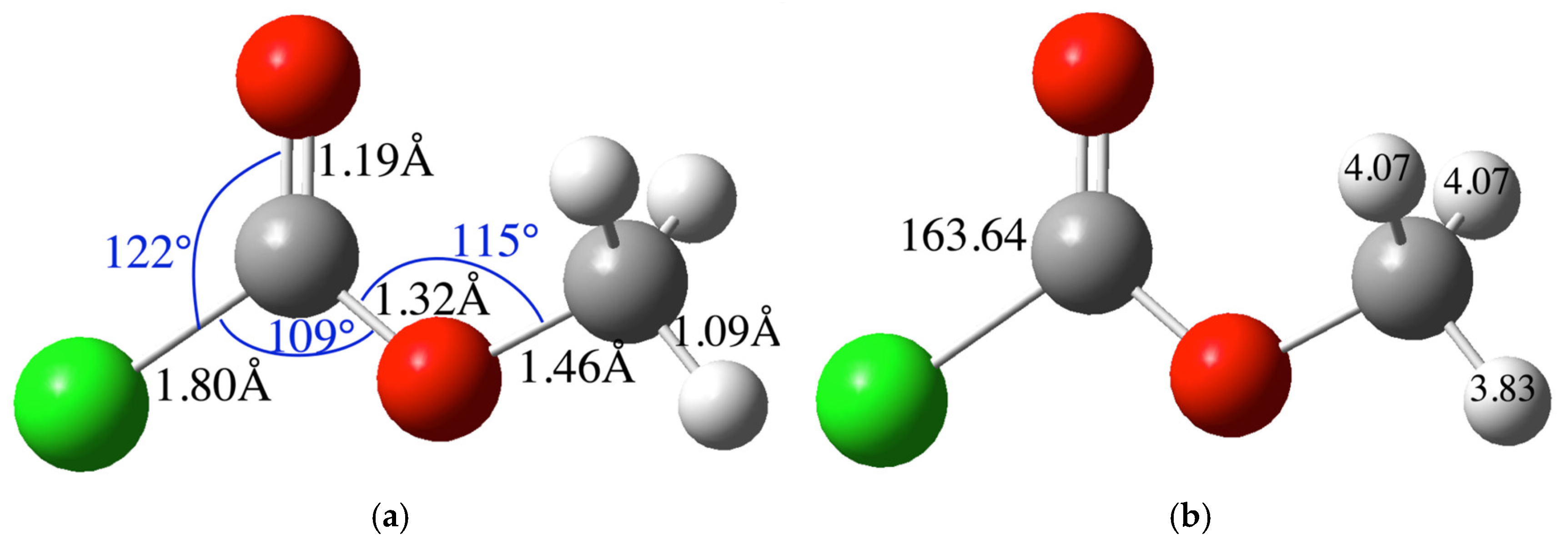
2.1.1. Reactant and Product Molecules
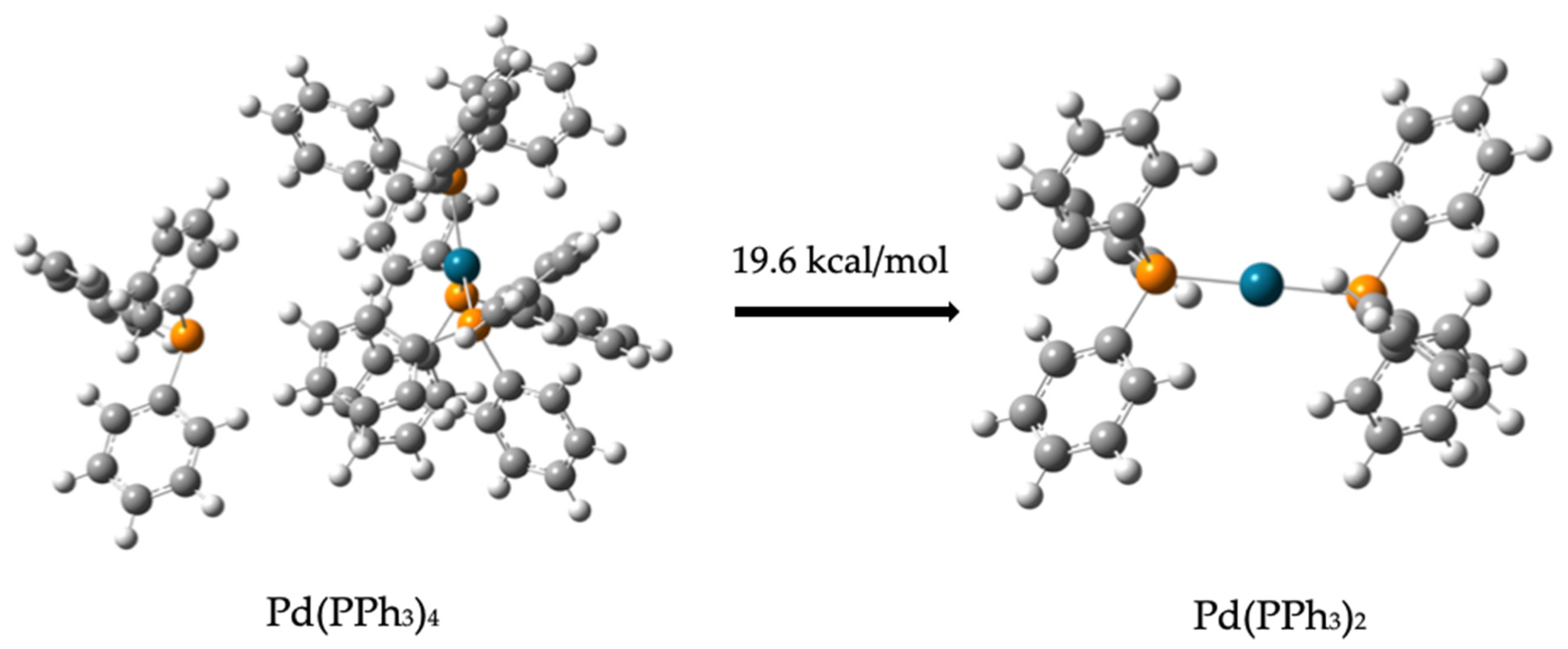
2.1.2. Tetratikis(trifenilfosfina)palladium Catalyst
2.1.3. Intermediates
2.2. Reaction Pathways
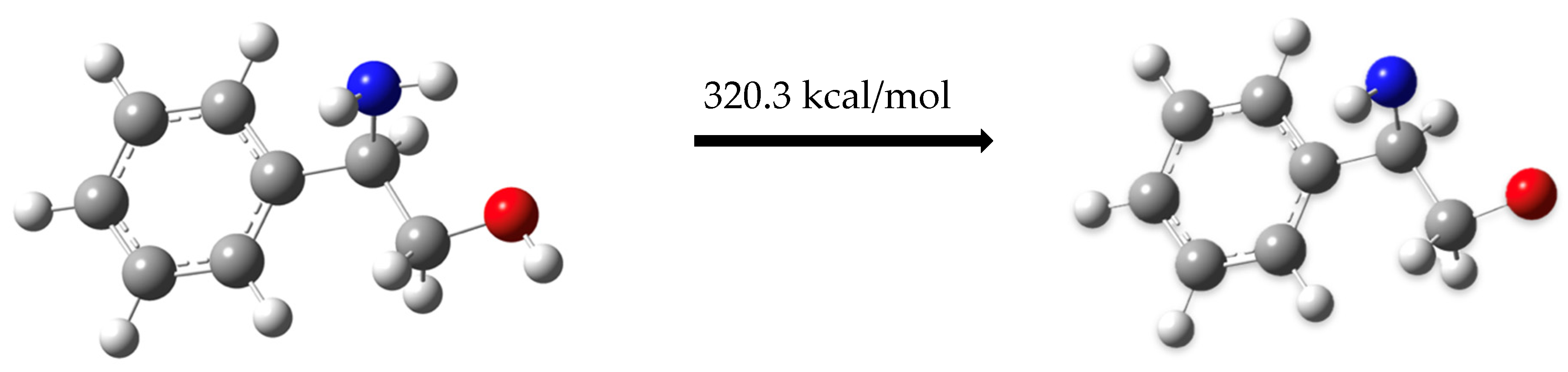
2.2.1. Pathway 1
Dehydrogenation of (R)-(-)-2-Phenylglycinol
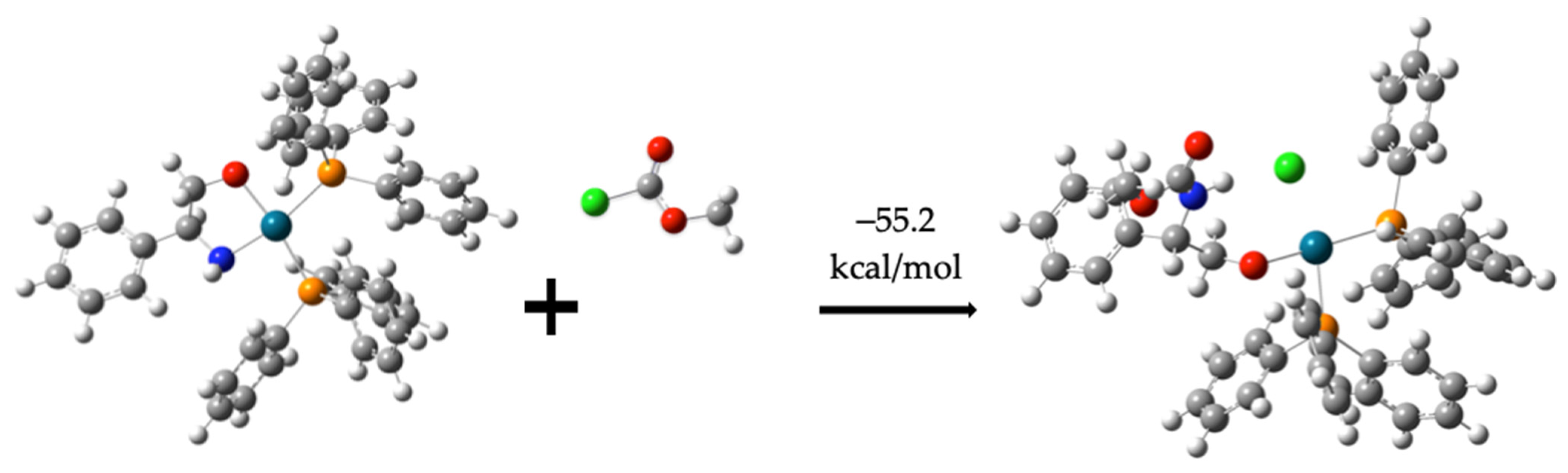
Formation of Compounds (7) and (8) as Intermediate Species
C6H5CH(NH)CH2O-Pd-(PPh3)2-C2H3ClO2 −55.2 kcal/mol
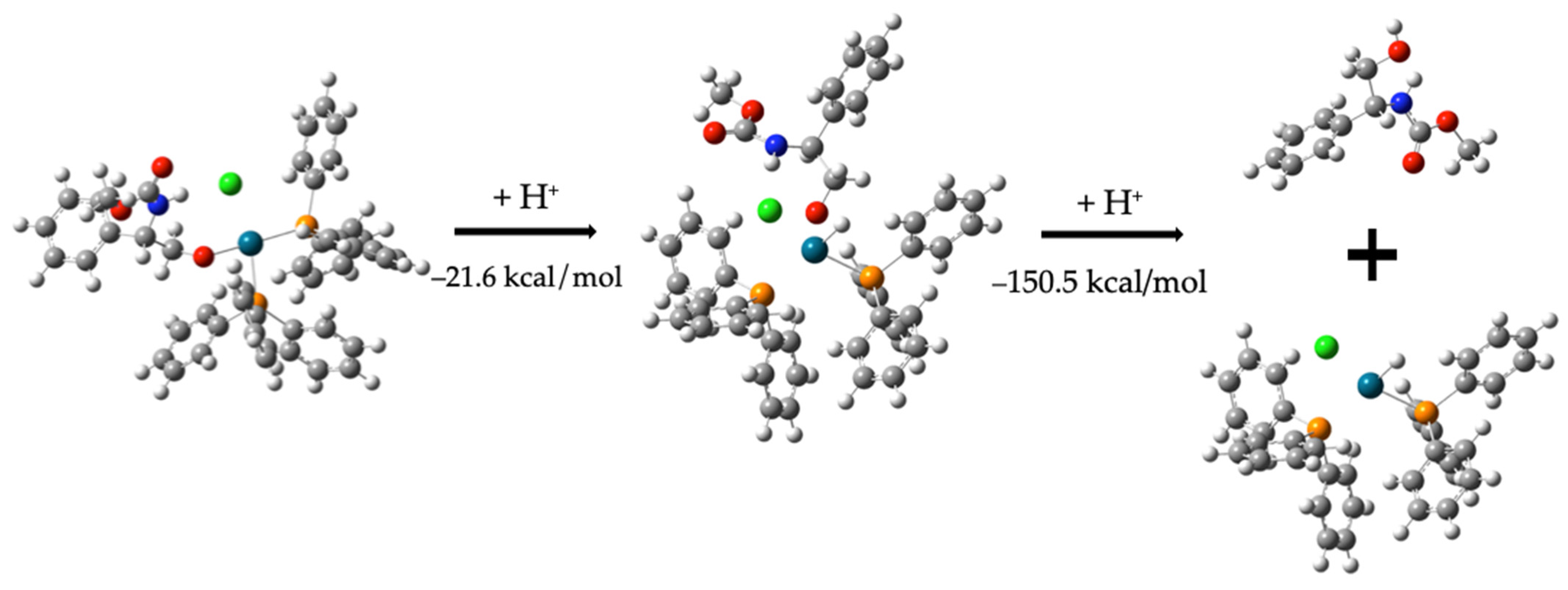
Formation of Compound (3)
H-Pd-(PPh3)2Cl + C6H5C2H3OH(NH)C2H3O2 −95.9 kcal/mol
Alternative Pathway: Formation of Compound (10)
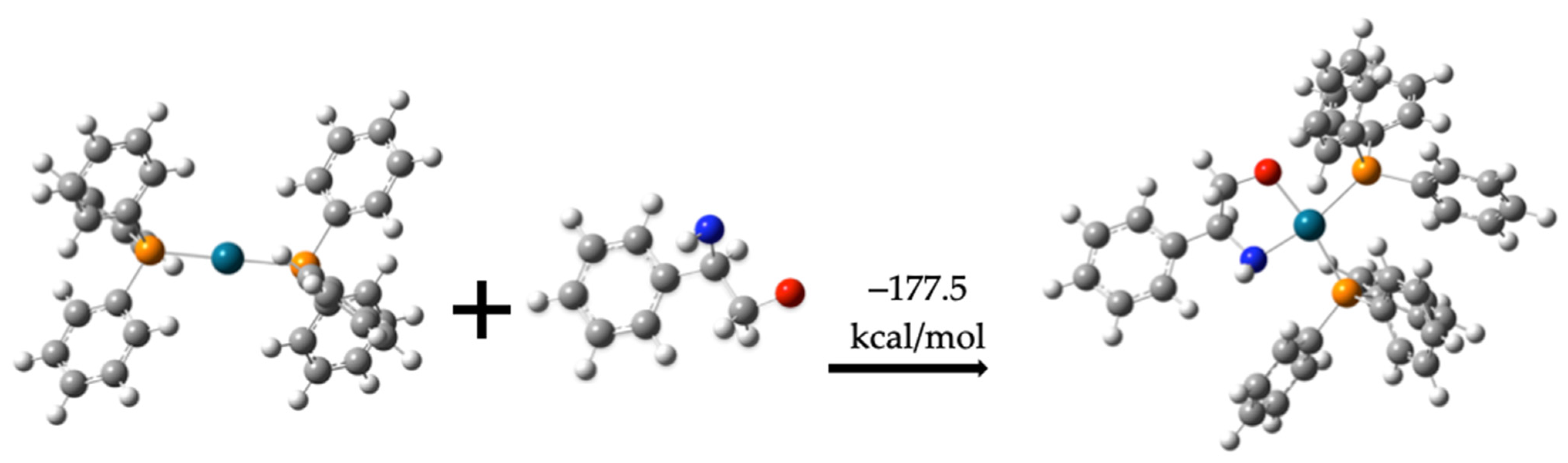
2.2.2. Pathway 2
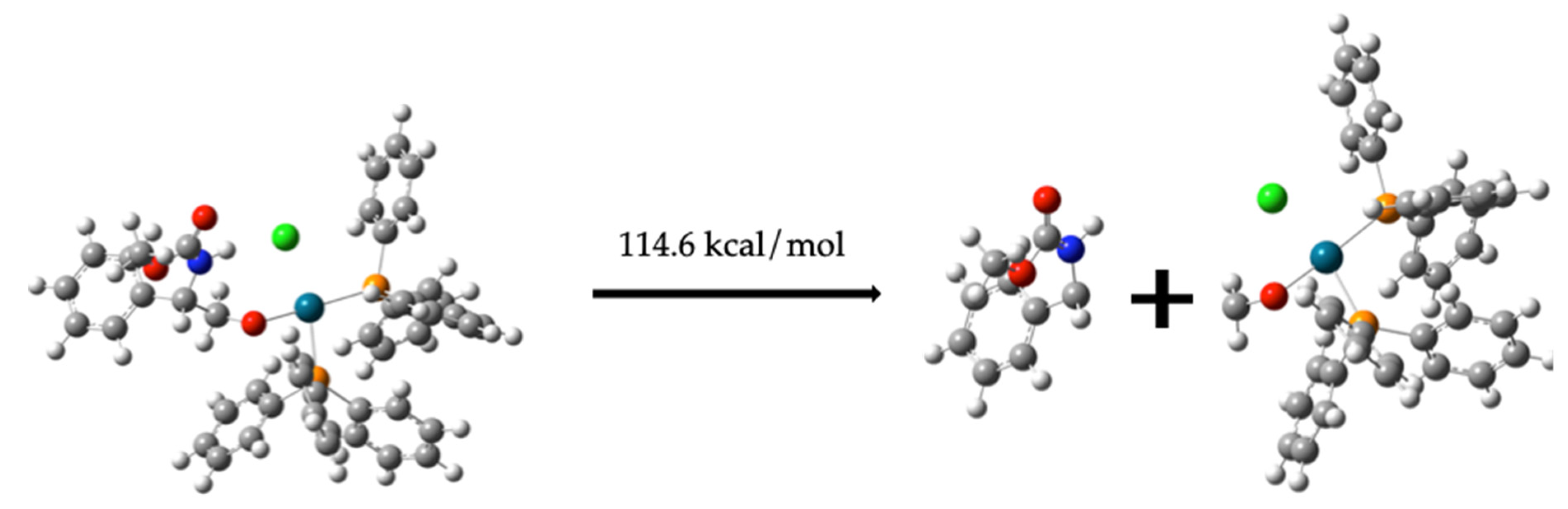
Formation of Compound (10) from Compound (8)
C6H5CH(NH)CH2OC = O + Pd-(PPh3)2-OCH2Cl 114.6 kcal/mol
Formation of Compound (11) from Compounds (7) and (10)
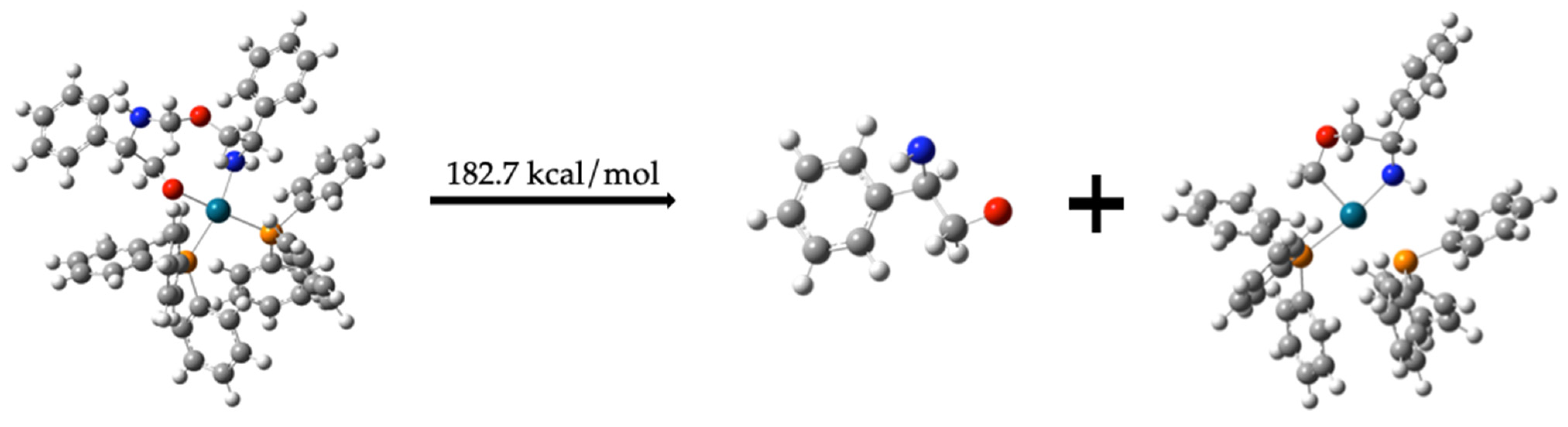
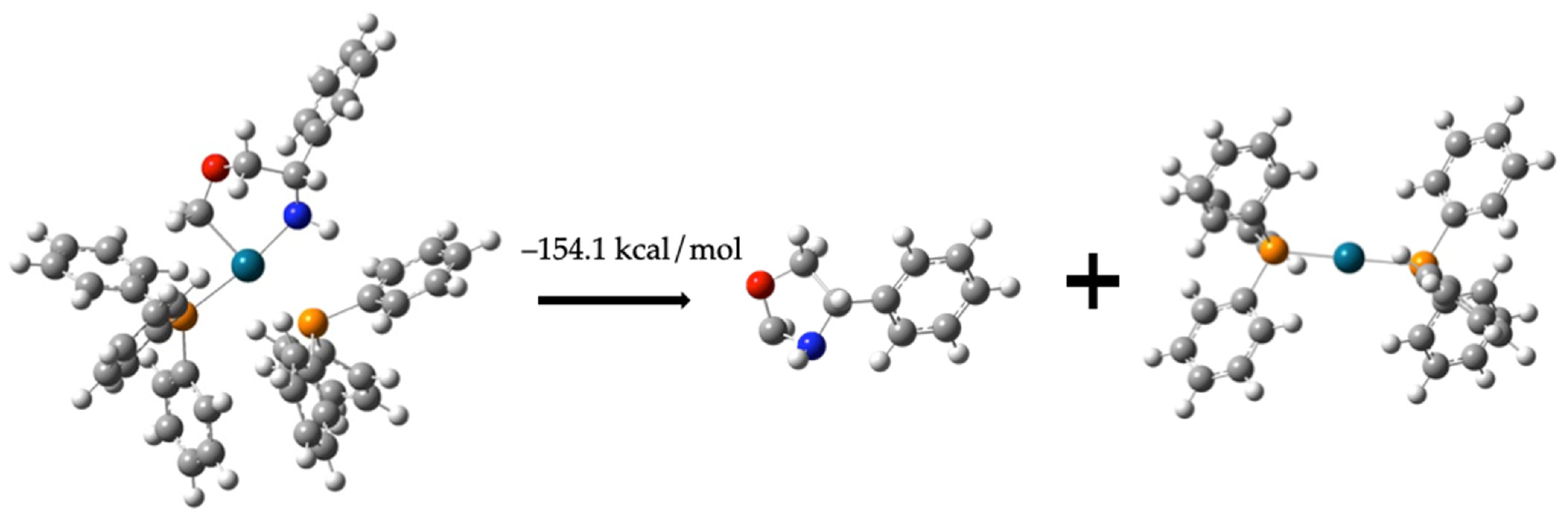
Regeneration of Dehydrogenated Compound (1) and Formation of Compound (6)
C6H5CH(NH)CH2OCH2 + Pd-(PPh3)2 −154.1 kcal/mol
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| PCM | Polarizable Continuum Model |
| SCF | Self-Consistent Field |
| TMS | Tetramethylsilane |
| THF | Tetrahydrofuran |
| NMR | Nuclear Magnetic Resonance |
| IR | Infrared |
References
- González, S.; Jaramillo-Fierro, X. Density Functional Theory Study of Methylene Blue Demethylation as a Key Step in Degradation Mediated by Reactive Oxygen Species. Int. J. Mol. Sci. 2025, 26, 1756. [Google Scholar] [CrossRef]
- King, K.; Hauser, A.T.; Melesina, J.; Sippl, W.; Jung, M. Carbamates as Potential Prodrugs and a New Warhead for HDAC Inhibition. Molecules 2018, 23, 321. [Google Scholar] [CrossRef]
- Cheshari, E.C.; Ren, X.; Li, X. Core–shell Ag-dual template molecularly imprinted composite for detection of carbamate pesticide residues. Chem. Pap. 2021, 75, 3679–3693. [Google Scholar] [CrossRef]
- Shenk, T.M.; Benjamin, K.M.; Winter, R.M. Impact of carbamate formation on the surface tension of epoxy-amine curing systems. Polym. Eng. Sci. 2023, 63, 1347–1358. [Google Scholar] [CrossRef]
- Chaudhary, J.; Jain, A.; Malik, G. Carbamate–drug conjugates in drug delivery: Structural and mechanistic considerations. In Polymer-Drug Conjugates; Academic Press: Cambridge, MA, USA, 2023; pp. 225–243. [Google Scholar] [CrossRef]
- Nasrabadi, M.; Nazarian, M.; Darroudi, M.; Marouzi, S.; Harifi-Mood, M.S.; Samarghandian, S.; Farkhondeh, T. Carbamate compounds induced toxic effects by affecting Nrf2 signaling pathways. Toxicol. Rep. 2024, 12, 148–157. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef]
- Singh, Y.P.; Kumar, N.; Chauhan, B.S.; Garg, P. Carbamate as a potential anti-Alzheimer’s pharmacophore: A review. Drug Dev. Res. 2023, 84, 1624–1651. [Google Scholar] [CrossRef]
- Nunes, D.; Loureiro, J.A.; Pereira, M.C. Drug Delivery Systems as a Strategy to Improve the Efficacy of FDA-Approved Alzheimer’s Drugs. Pharmaceutics 2022, 14, 2296. [Google Scholar] [CrossRef]
- Dhingra, H.; Choudhari, S.G. Alzheimer’s Disease: Understanding Its Novel Drug Delivery Systems and Treatments. Cureus 2022, 14, e31394. [Google Scholar] [CrossRef]
- Gupta, R.C.; Doss, R.B.; Yurdakok-Dikmen, B.; Malik, J.K.; Zaja-Milatovic, S.; Milatovic, D. Organophosphates and carbamates. In Reproductive and Developmental Toxicology; Academic Press: Cambridge, MA, USA, 2022; pp. 617–639. [Google Scholar] [CrossRef]
- Hanh, V.T.; Duong, N. Van Laboratory Validation of an LC-MS/MS Method for Simultaneous Determination of Carbamate Residues in Vegetables. Acta Sci. Pol. Technol. Aliment. 2023, 22, 385–394. [Google Scholar] [CrossRef]
- Flynn, S.; Polena, J.; Ngai, J.H.L.; Liu, H.T.; Li, X.; Wang, J.L.; Li, Y.N. Effects of replacing carbamate with alkyl side chains on the properties and temperature sensing performance of hemi-isoindigo-based polymers. Flex. Print. Electron 2022, 7, 044003. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y.; Fan, Y.; Rager, M.N.; Guérineau, V.; Bouteiller, L.; Li, M.H.; Thomas, C.M. Polymerization of Cyclic Carbamates: A Practical Route to Aliphatic Polyurethanes. Macromolecules 2019, 52, 2719–2724. [Google Scholar] [CrossRef]
- Perez Mellor, A.F.; Brazard, J.; Kozub, S.; Bürgi, T.; Szweda, R.; Adachi, T.B.M. Unveiling the Configurational Landscape of Carbamate: Paving the Way for Designing Functional Sequence-Defined Polymers. J. Phys. Chem. A 2023, 127, 7309–7322. [Google Scholar] [CrossRef]
- Chaturvedi, D. Perspectives on the synthesis of organic carbamates. Tetrahedron 2012, 68, 15–45. [Google Scholar] [CrossRef]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arh. Hig. Rada Toksikol. 2020, 71, 285–299. [Google Scholar] [CrossRef]
- Gong, Y.; Wang, Y.; Li, P.; Yuan, Y.; Kong, F. Chromatography-free synthesis of carbamates from CO2 catalyzed by a green and recyclable biobased ionic liquid/Cu2O system. J. CO2 Util. 2024, 79, 102656. [Google Scholar] [CrossRef]
- Mora Vargas, J.A.; Burtoloso, A.C.B. CO2-Based Carbamate Synthesis Utilizing Reusable Polymer-Supported DBU. ChemSusChem 2023, 16, e202300936. [Google Scholar] [CrossRef]
- Satheesh, A.; Usha, H.; Priya, D.S.; Hariharan, A.V.L.N.H.; Ramanjaneyulu, M.V.V. Greener Protocol for the Synthesis of Carbamates. J. Sci. Res. 2023, 15, 481–488. [Google Scholar] [CrossRef]
- Choudhary, S.K.; Trivedi, M.; Sogani, N. Synthesis and characterization of carbamates derivatives from 4-amino-1,2,4-triazole. Indian J. Chem. 2022, 61, 1113–1119. [Google Scholar] [CrossRef]
- Day, C.S.; Ton, S.J.; Kaussler, C.; Vrønning Hoffmann, D.; Skrydstrup, T. Low Pressure Carbonylation of Benzyl Carbonates and Carbamates for Applications in 13C Isotope Labeling and Catalytic CO2 Reduction. Angew. Chem. Int. Ed. 2023, 62, e202308238. [Google Scholar] [CrossRef]
- Herrera, M.d.C.M.; Cruz, M.S.; Díaz, L.M.P.; Márquez, J.A.R.; Flores, L.O.; Bernès, S. Methyl N-{(1R)-2-[(methoxycarbonyl)oxy]-1-phenylethyl}carbamate. IUCrData 2024, 9, x240222. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Y.; Achab, A.; Bharathan, I.; Hopkins, B.A.; Yu, W.; Zhang, H.; Sanyal, S.; Pu, Q.; Zhou, H.; et al. Carbamate and N-Pyrimidine Mitigate Amide Hydrolysis: Structure-Based Drug Design of Tetrahydroquinoline IDO1 Inhibitors. ACS Med. Chem. Lett. 2021, 12, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Gupta, G.; Ayyannan, S.R. Synthesis and evaluation of carbamate derivatives as fatty acid amide hydrolase and monoacylglycerol lipase inhibitors. Arch. Pharm. 2022, 355, 2200081. [Google Scholar] [CrossRef] [PubMed]
- Hellström, A.K.; Oskarsson, H.; Bordes, R. Formation, physicochemical and interfacial study of carbamate surfactants. J. Colloid Interface Sci. 2018, 511, 84–91. [Google Scholar] [CrossRef]
- Atmaca, U.; Aksoy, M.; Öztekin, A. A safe alternative synthesis of primary carbamates from alcohols; in vitro and in silico assessments as an alternative acetylcholinesterase inhibitors. J. Biomol. Struct. Dyn. 2023, 41, 8191–8200. [Google Scholar] [CrossRef]
- Ray, S.; Pathak, S.R.; Chaturvedi, D. Organic carbamates in drug development. Part II: Antimicrobial agents-Recent reports. Drugs Future 2005, 30, 161–180. [Google Scholar] [CrossRef]
- Prasher, P.; Mall, T.; Sharma, M. Cyclic carbamates in medicine: A clinical perspective. Drug Dev. Res. 2023, 84, 397–405. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016.
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 6.1.1; Semichem Inc.: Shawnee, KS, USA, 2019.
- Condon, J.B. Density functional theory (DFT). In Surface Area and Porosity Determinations by Physisorption; Elsevier Science: Amsterdam, The Netherlands, 2020; pp. 335–360. [Google Scholar] [CrossRef]
- Alhanzal, F.; Joudieh, N.; Hussein, K.; Chamoun, N. Comparison between PBE-D3, B3LYP, B3LYP-D3 and MP2 Methods for quantum mechanical calculations of polarizability and IR-NMR spectra in C24 isomers, including a novel isomer with D2d symmetry. Nano-Struct. Nano-Objects 2023, 36, 101036. [Google Scholar] [CrossRef]
- Lu, L.; Hu, H.; Hou, H.; Wang, B. An improved B3LYP method in the calculation of organic thermochemistry and reactivity. Comput. Theor. Chem. 2013, 1015, 64–71. [Google Scholar] [CrossRef]
- Singh, I.; El-Emam, A.A.; Pathak, S.K.; Srivastava, R.; Shukla, V.K.; Prasad, O.; Sinha, L. Experimental and theoretical DFT (B3LYP, X3LYP, CAM-B3LYP and M06-2X) study on electronic structure, spectral features, hydrogen bonding and solvent effects of 4-methylthiadiazole-5-carboxylic acid. Mol. Simul. 2019, 45, 1029–1043. [Google Scholar] [CrossRef]
- Benjamine, A.A.; Mawa, K.; Laye, A.L.; Lucie, B.A.; Soleymane, K.; Robert, N.B.; Sawaliho, B.E.H.; Thomas, N.Y. Density Functional Theory (B3LYP/6-311+G(d, p)) Study of Stability, Tautomerism and Acidity of 2-Thioxanthine in Gas and Aqueous Phases. Int. J. Comput. Theor. Chem. 2019, 7, 49–55. [Google Scholar] [CrossRef]
- Chelikowsky, J.R. Pseudopotential methods. In Encyclopedia of Condensed Matter Physics; Academic Press: Cambridge, MA, USA, 2024; pp. 833–848. [Google Scholar]
- Rinaldo, D.; Tian, L.; Harvey, J.N.; Friesner, R.A. Density functional localized orbital corrections for transition metals. J. Chem. Phys. 2008, 129, 164108. [Google Scholar] [CrossRef] [PubMed]
- Bimukhanov, A.; Aldongarov, A.A. A comparative study of DFT functionals and basis sets for describing the luminescent spectra of hexacoordinated silicon complexes. Вестник. Серия Физическая 2023, 84, 56–64. [Google Scholar] [CrossRef]
- Backes, A.C.; Schatz, J.; Siehl, H.-U. GIAO-DFT calculated and experimentally derived complexation-induced chemical shifts of calix[4]arene–solvent inclusion complexes. J. Chem. Soc. Perkin Trans. 2 2002, 3, 484–488. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Shift H (ppm) | Chemical Shift C (ppm) | Calculated IR Frequencies (cm−1) | ||
|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | |
| 2.61 | 2.4 | 67.84 | 71.56 | 3543–3465 (N-H) |
| 3.54 | 3.78 | 57.37 | 59.72 | 3802 (O-H) |
| 3.71 | 4.59 | 142.46 | 149.96 | 1537 (C=C) |
| 4.02 | 4.05 | 126.53–128.58 | 128.23–136.20 | 1070 (C-C) |
| 7.24–7.28 | 7.57 | 888 (C-N) | ||
| 7.29–7.35 | 7.24–8.19 | 1045 (C-O) | ||
| H Chemical Shift (ppm) | C Chemical Shift (ppm) | Calculated IR Frequencies (cm−1) | ||
|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | |
| 3.97 | 3.83–4.07 | 58.24 | 61.85 | 1152 (C-O) |
| 151.41 | 163.64 | 1844 (C=O) | ||
| 815 (O=C-O) | ||||
| 454 (C-Cl) | ||||
| H Chemical Shift (ppm) | C Chemical Shift (ppm) | IR Frequencies (cm−1) | |||
|---|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | Experimental | Calculated |
| 3.67 | 3.43, 3.62 | 52.35 | 53.64 | 3333 | 3602 (N-H) |
| 3.87 | 3.65, 3.90 | 66.52 | 71.06 | 1701 | 1772 (C=O) |
| 4.83 | 4.66 | 57.10 | 62.36 | 1541 | 1540 (-C=C-) |
| 0.88 | 1260 | 1264 (C-O) | |||
| 5.5 | 5.52 | 1028 | 1054 (C-O) | ||
| 7.29–7.38 | 7.61–7.79 | 128.5–128.8 | 130.57, 132.24 | 701 | 771 (=C-H) |
| 132.05 | 133.16, 133.25, 133.54 | ||||
| 132.15 | 148.39 | ||||
| 157.09 | 164.26 | ||||
| Bond | Experimental Bond Distance (Å) | Calculated Bond Distance (Å) | Angle | Experimental (°) | Calculated (°) |
|---|---|---|---|---|---|
| N-C | 1.430 | 1.461 | N-C-O | 109.7 | 110.1 |
| N-C | 1.336 | 1.363 | N-C-O | 126.1 | 125.8 |
| C=O | 1.185 | 1.215 | O-C-O | 124.2 | 124.1 |
| C-O | 1.353 | 1.358 | C-O-C | 116.1 | 115.5 |
| O-C | 1.414 | 1.436 | C-N-C | 122.5 | 120.7 |
| C-C | 1.521 | 1.531 | C-C-O | 111.7 | 108.0 |
| C-O(OH) | 1.441 | 1.426 |
| Chemical Shift H (ppm) | Chemical Shift C (ppm) | ||
|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated |
| 4.69 | 4.18,4.78 | 127.02 | 86.37 |
| 4.36 | 4.78 | 56.29 | 65.28 |
| 3.82 | 3.17,4.54 | 62.42 | 76.4 |
| 7.30–7.37 | 7.34–7.70 | 133.50 | 151.75 |
| 129.22–129.50 | 128.67–133.42 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, S.; Mendoza Herrera, C.; Pérez Díaz, L.M.; Orea Flores, L.; Rivera Márquez, J.A.; Jaramillo-Fierro, X. Computational Study on the Pd-Catalyzed Pathway for the Formation of (R)-Methyl-(2-Hydroxy-1-Phenylethyl)Carbamate. Molecules 2025, 30, 1781. https://doi.org/10.3390/molecules30081781
González S, Mendoza Herrera C, Pérez Díaz LM, Orea Flores L, Rivera Márquez JA, Jaramillo-Fierro X. Computational Study on the Pd-Catalyzed Pathway for the Formation of (R)-Methyl-(2-Hydroxy-1-Phenylethyl)Carbamate. Molecules. 2025; 30(8):1781. https://doi.org/10.3390/molecules30081781
Chicago/Turabian StyleGonzález, Silvia, Consuelo Mendoza Herrera, Lydia María Pérez Díaz, Laura Orea Flores, José Antonio Rivera Márquez, and Ximena Jaramillo-Fierro. 2025. "Computational Study on the Pd-Catalyzed Pathway for the Formation of (R)-Methyl-(2-Hydroxy-1-Phenylethyl)Carbamate" Molecules 30, no. 8: 1781. https://doi.org/10.3390/molecules30081781
APA StyleGonzález, S., Mendoza Herrera, C., Pérez Díaz, L. M., Orea Flores, L., Rivera Márquez, J. A., & Jaramillo-Fierro, X. (2025). Computational Study on the Pd-Catalyzed Pathway for the Formation of (R)-Methyl-(2-Hydroxy-1-Phenylethyl)Carbamate. Molecules, 30(8), 1781. https://doi.org/10.3390/molecules30081781