
Multifunctional 3D-Printable Photocurable Elastomer with Self-Healing Capability Derived from Waste Cooking Oil


Abstract

1. Introduction
2. Result and Discussion
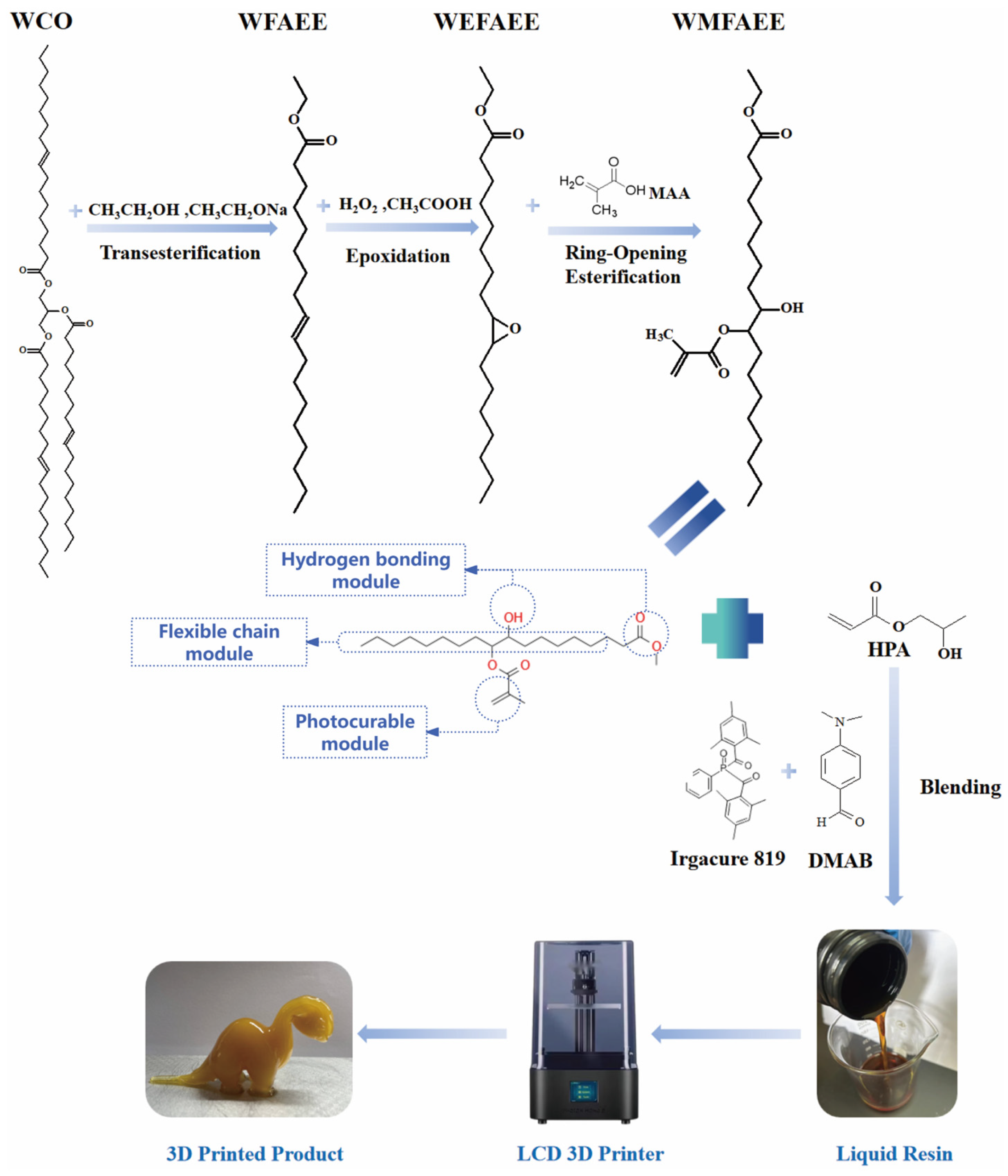
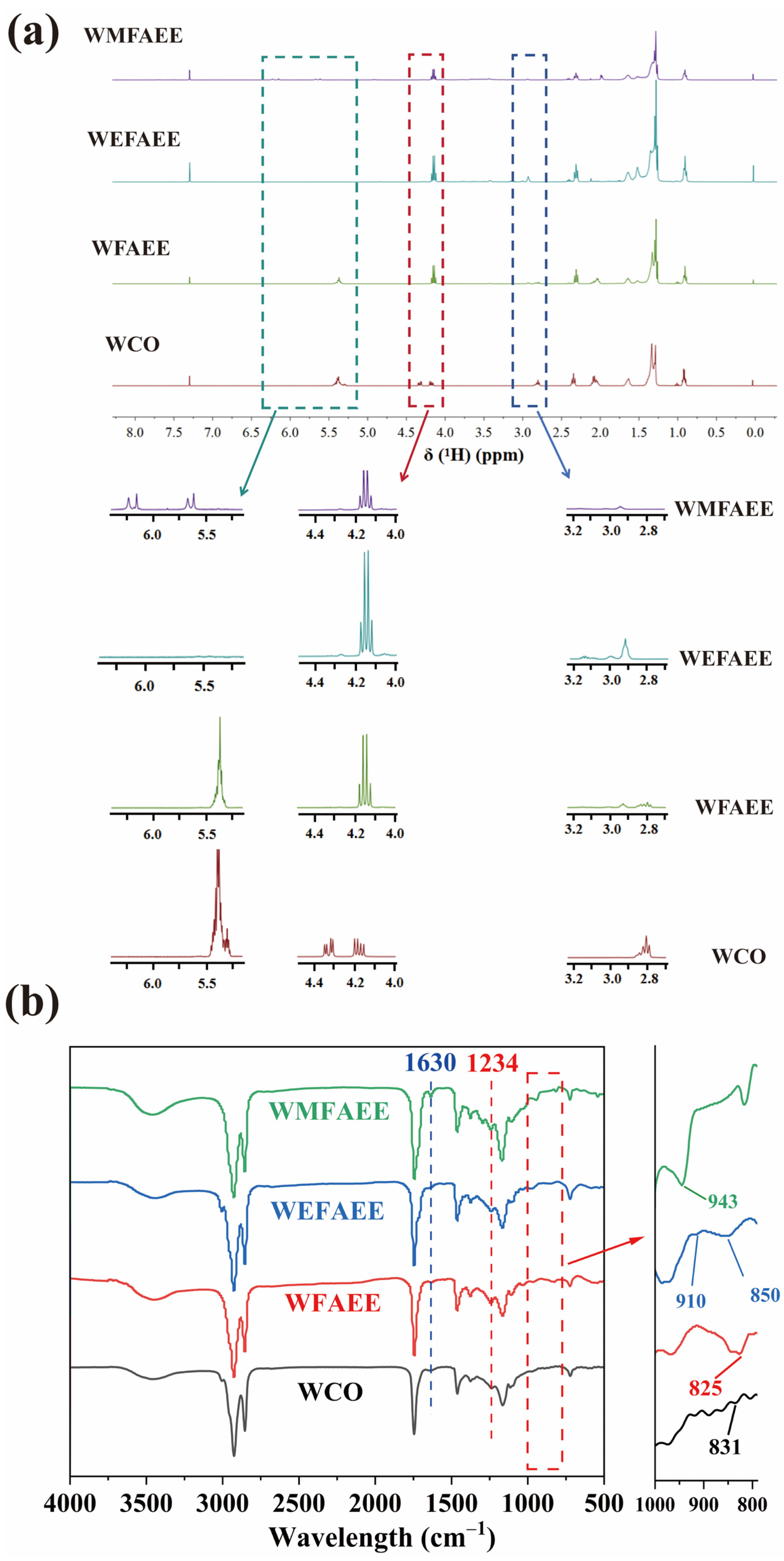
2.1. Spectroscopic Analysis of WMFAEE Monomer
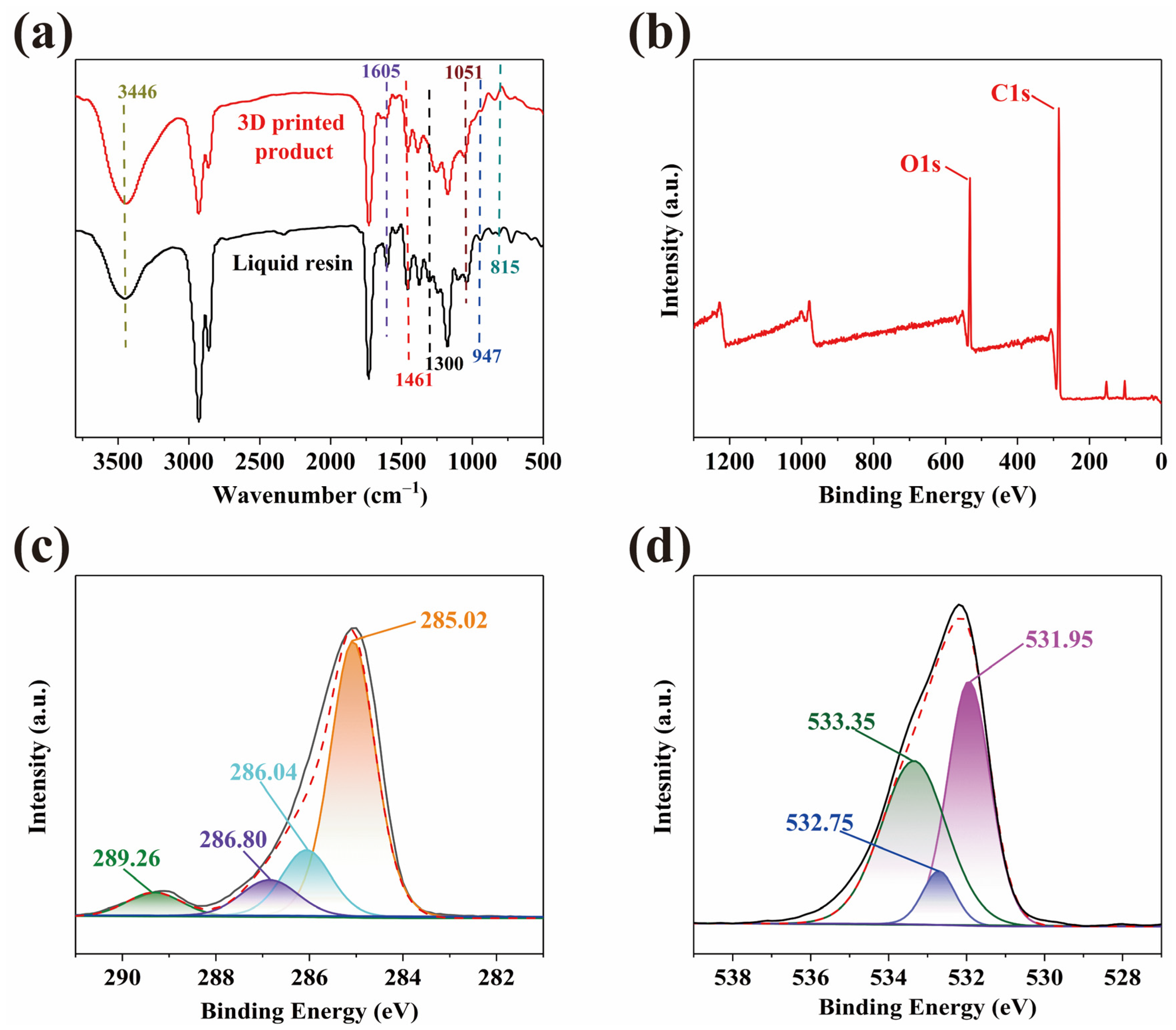
2.2. Spectroscopic Analysis of WCO-Based Photocurable Elastomer
2.3. Thermal Analysis
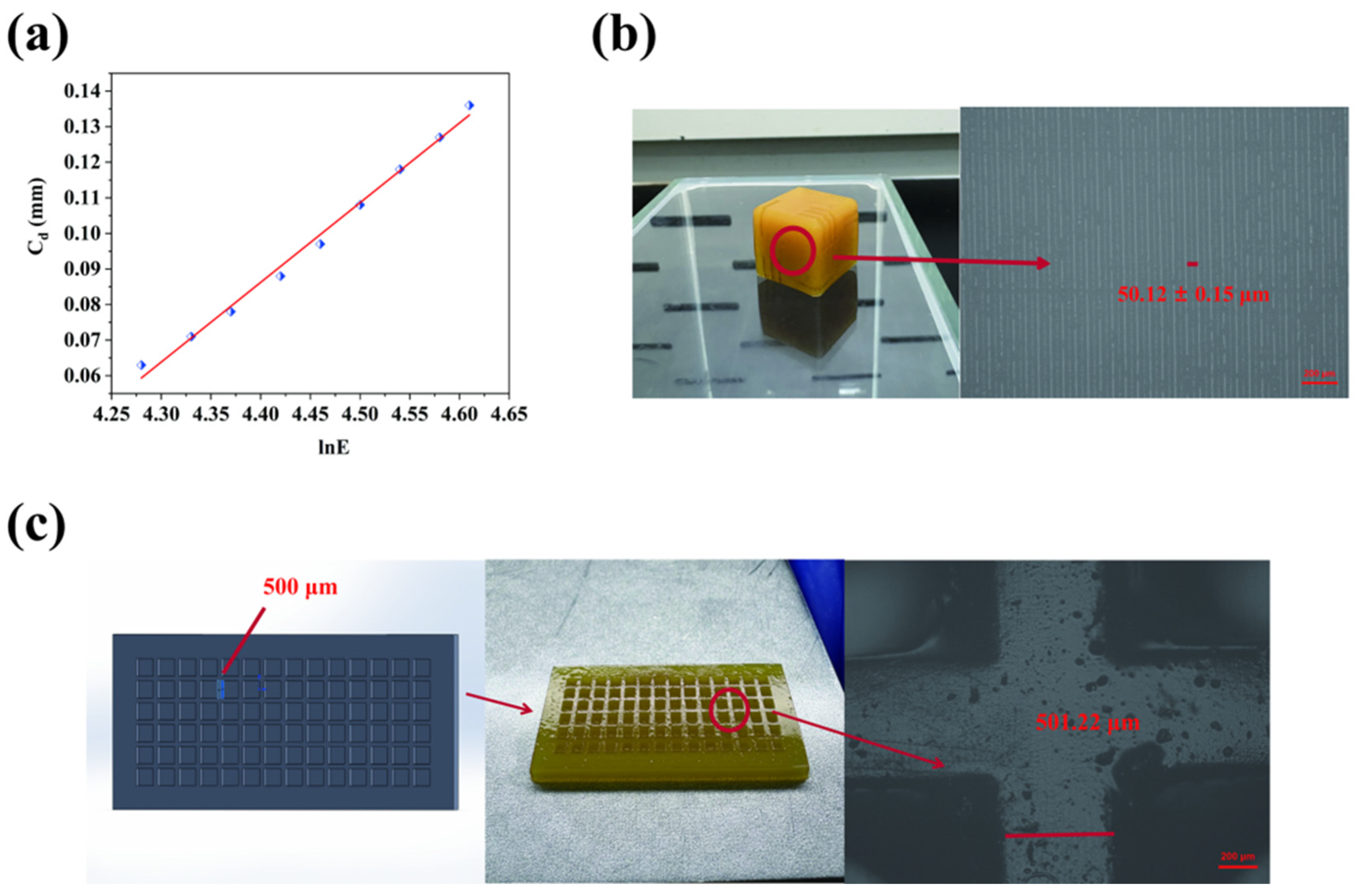
2.4. Three-Dimensional Printing Behavior
2.5. Mechanical Properties
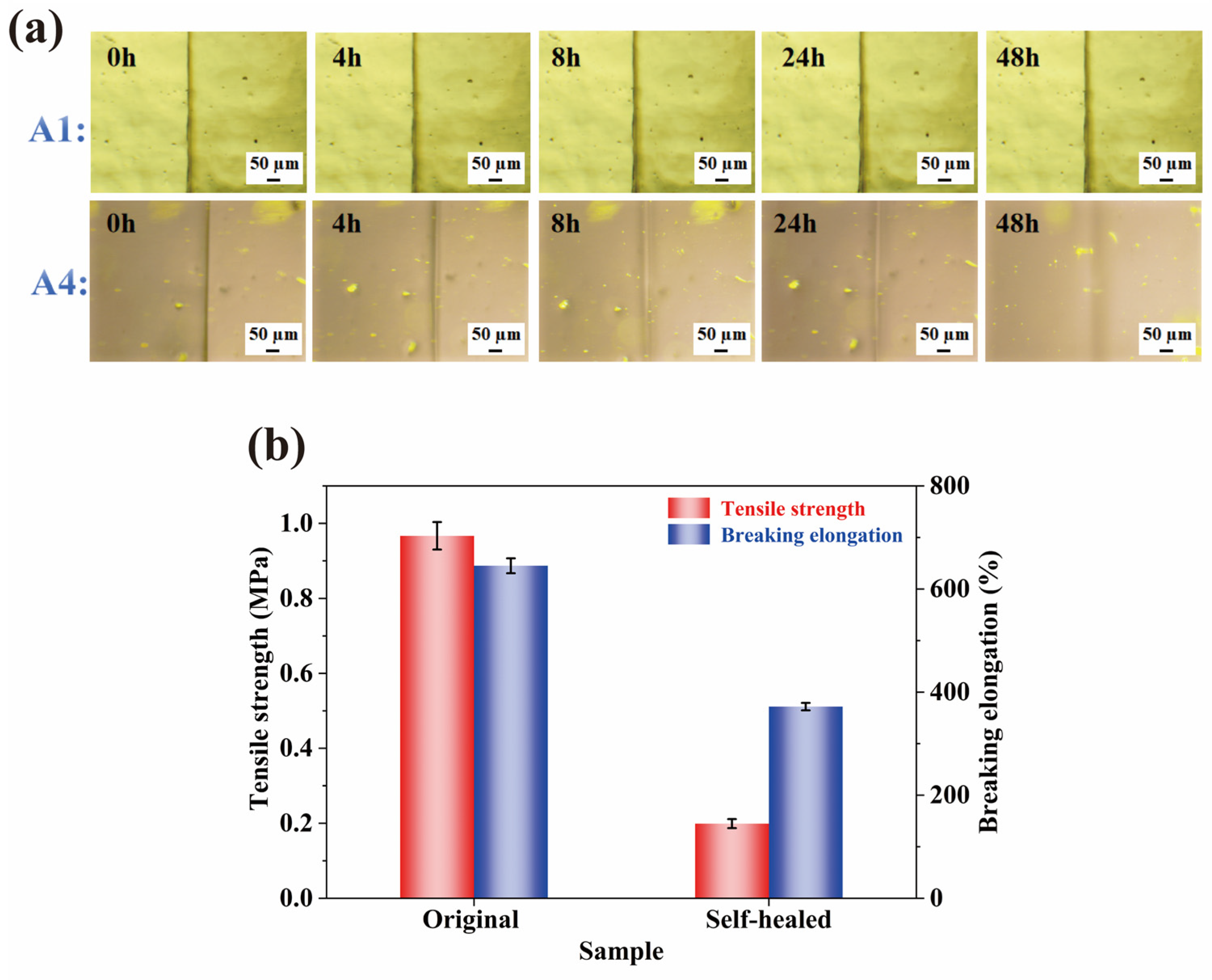
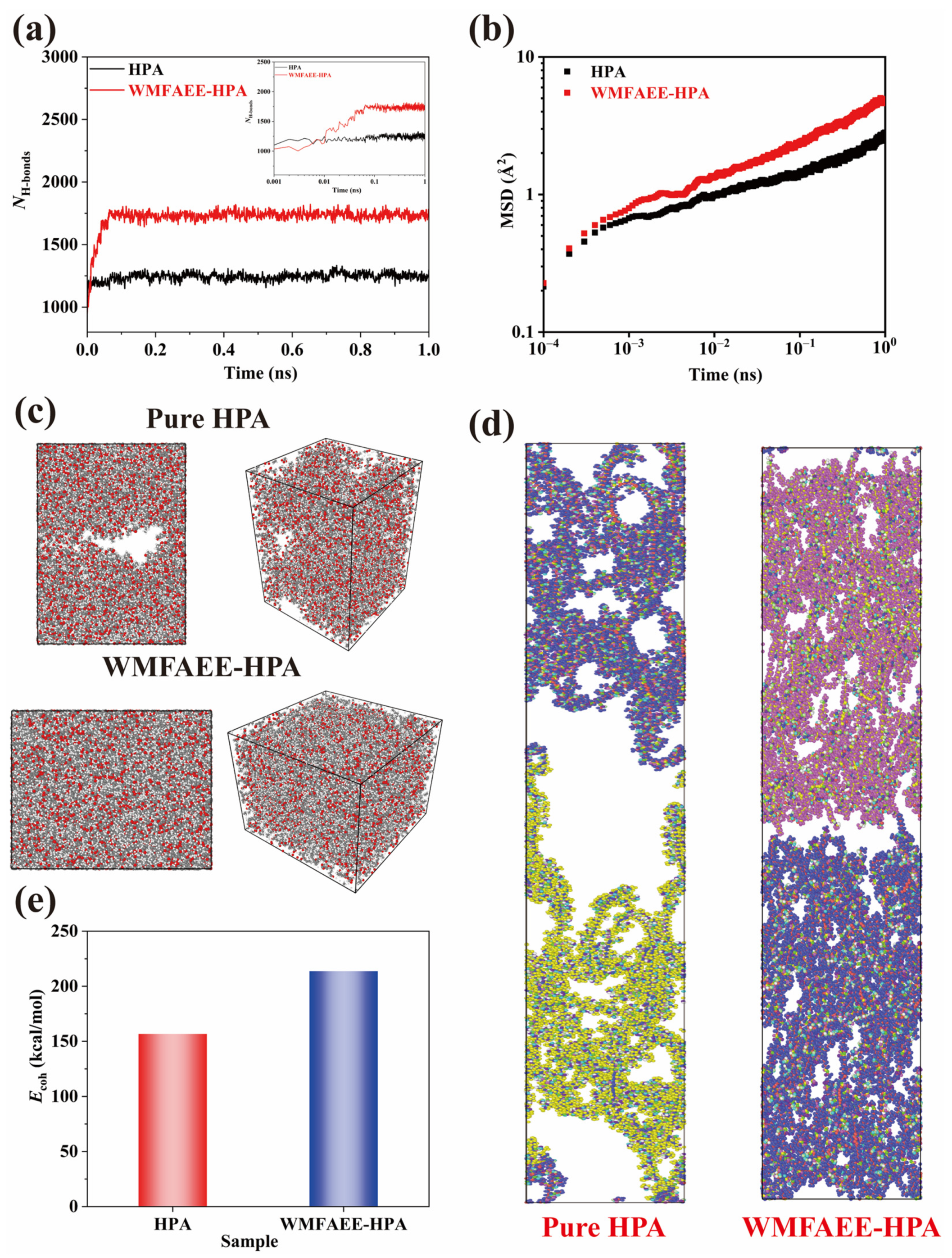
2.6. Self-Healing Properties
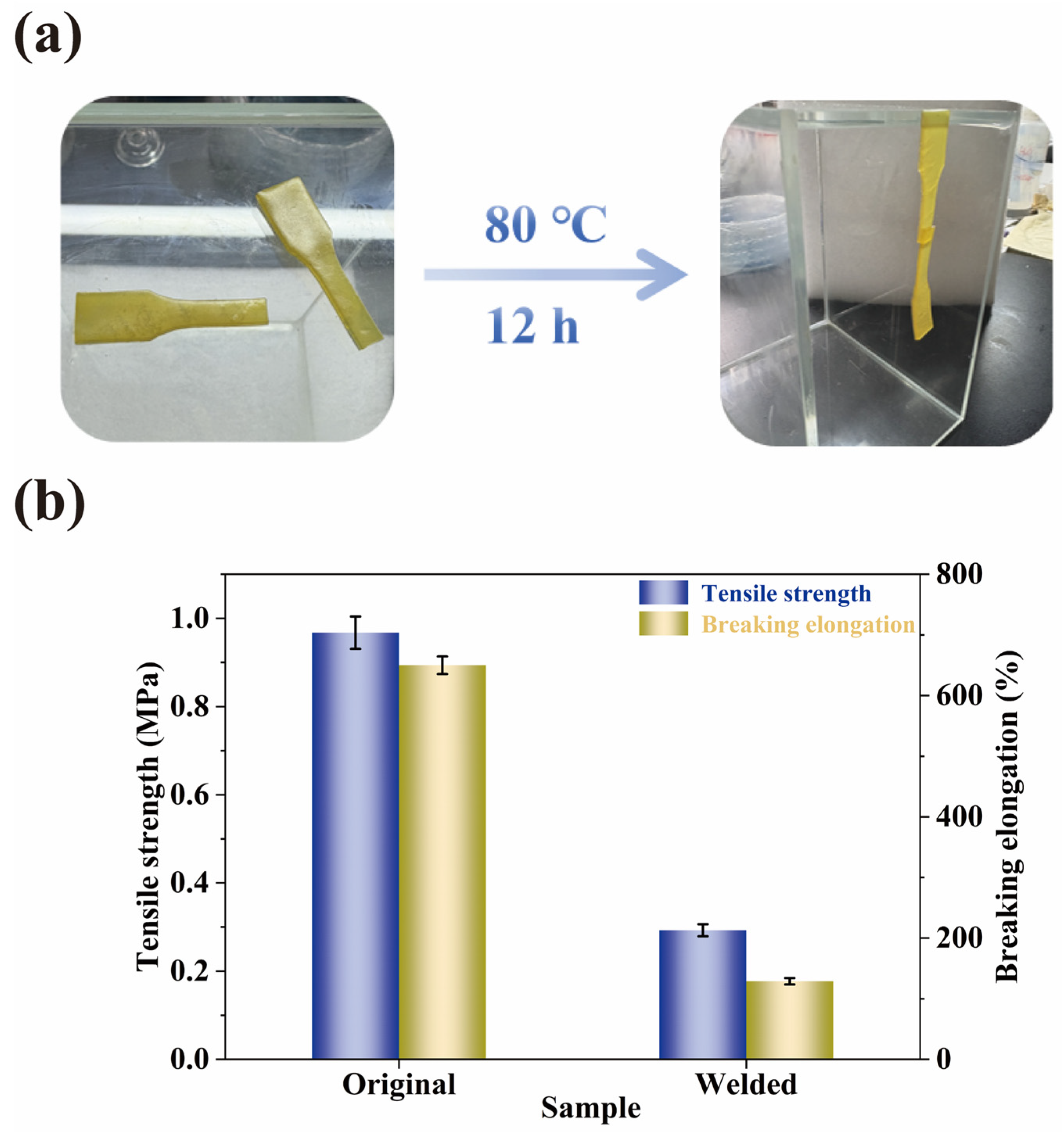
2.7. Welding and Physical Reprocessing Properties
2.8. Pressure-Sensitive Adhesive Properties
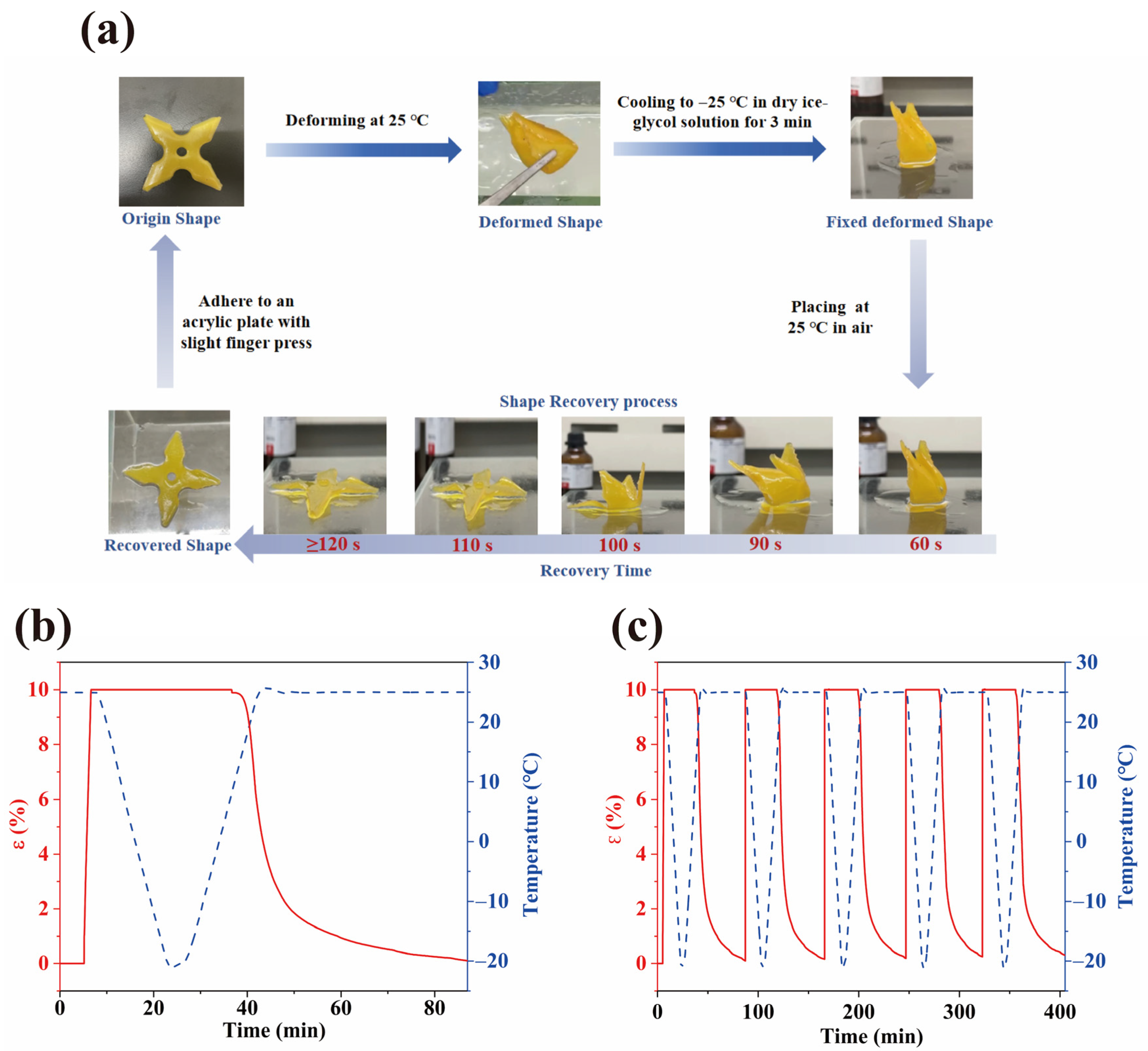
2.9. Shape Memory Properties
2.10. Biodegradability
3. Materials and Methods
3.1. Materials
3.2. Preparation of WMFAEE Monomer
3.3. Synthesis of Liquid WMFAEE-HPA Elastomer
3.4. Molding of WCO-Based Elastomer
3.5. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| WCO | Waste cooking oil |
| WFAEE | WCO-based fatty acid ethyl ester |
| WEFAEE | WCO-based epoxidized fatty acid ethyl ester |
| WMFAEE | WCO-based methacrylate fatty acid ethyl ester |
| HPA | Hydroxypropyl acrylate |
| MAA | Methacrylic acid |
| PPh3 | Triphenylphosphine |
| Irgacure 819 | phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide |
| DMAB | P-dimethylaminobenzaldehyde |
References
- Hosseinzadeh-Bandbafha, H.; Nizami, A.-S.; Kalogirou, S.A.; Gupta, V.K.; Park, Y.-K.; Fallahi, A.; Sulaiman, A.; Ranjbari, M.; Rahnama, H.; Aghbashlo, M.; et al. Environmental Life Cycle Assessment of Biodiesel Production from Waste Cooking Oil: A Systematic Review. Renew. Sustain. Energy Rev. 2022, 161, 112411. [Google Scholar] [CrossRef]
- Yang, J.; Shan, H. The Willingness of Submitting Waste Cooking Oil (WCO) to Biofuel Companies in China: An Evolutionary Analysis in Catering Networks. J. Clean. Prod. 2021, 282, 125331. [Google Scholar] [CrossRef]
- Lam, S.S.; Liew, R.K.; Jusoh, A.; Chong, C.T.; Ani, F.N.; Chase, H.A. Progress in Waste Oil to Sustainable Energy, with Emphasis on Pyrolysis Techniques. Review. Renew. Sustain. Energy Rev. 2016, 53, 741–753. [Google Scholar] [CrossRef]
- Farooq, M.; Ramli, A.; Subbarao, D. Biodiesel Production from Waste Cooking Oil Using Bifunctional Heterogeneous Solid Catalysts. J. Clean. Prod. 2013, 59, 131–140. [Google Scholar] [CrossRef]
- Juan, J.C.; Kartika, D.A.; Wu, T.Y.; Hin, T.-Y.Y. Biodiesel Production from Jatropha Oil by Catalytic and Non-Catalytic Approaches: An Overview. Bioresour. Technol. 2011, 102, 452–460. [Google Scholar] [CrossRef]
- Yaakob, Z.; Mohammad, M.; Alherbawi, M.; Alam, Z.; Sopian, K. Overview of the Production of Biodiesel from Waste Cooking Oil. Renew. Sustain. Energy Rev. 2013, 18, 184–193. [Google Scholar] [CrossRef]
- Paul, A.K.; Borugadda, V.B.; Goud, V.V. In-Situ Epoxidation of Waste Cooking Oil and Its Methyl Esters for Lubricant Applications: Characterization and Rheology. Lubricants 2021, 9, 27. [Google Scholar] [CrossRef]
- Appiah, G.; Tulashie, S.K.; Akpari, E.E.A.; Rene, E.R.; Dodoo, D. Biolubricant Production via Esterification and Transesterification Processes: Current Updates and Perspectives. Int. J. Energy Res. 2022, 46, 3860–3890. [Google Scholar] [CrossRef]
- Attia, N.K.; El-Mekkawi, S.A.; Elardy, O.A.; Abdelkader, E.A. Chemical and Rheological Assessment of Produced Biolubricants from Different Vegetable Oils. Fuel 2020, 271, 117578. [Google Scholar] [CrossRef]
- Azzena, U.; Montenero, A.; Carraro, M.; Crisafulli, R.; De Luca, L.; Gaspa, S.; Muzzu, A.; Nuvoli, L.; Polese, R.; Pisano, L.; et al. Recovery, Purification, Analysis and Chemical Modification of a Waste Cooking Oil. Waste Biomass Valorization 2023, 14, 145–157. [Google Scholar] [CrossRef]
- Mannu, A.; Ferro, M.; Pietro, M.E.D.; Mele, A. Innovative Applications of Waste Cooking Oil as Raw Material. Sci. Prog. 2019, 102, 153–160. [Google Scholar] [CrossRef]
- Orozco, L.M.; Cardona, S.; Gomez, C.; Inciarte, H.; Villada, Y.; Rios, L. Evaluation of KHSO4 as a Recyclable Catalyst in the Production of Dehydrated Castor Oil to Be Applied in Alkyd Resins. Prog. Org. Coat. 2021, 161, 106467. [Google Scholar] [CrossRef]
- Gaur, V.K.; Sharma, P.; Sirohi, R.; Varjani, S.; Taherzadeh, M.J.; Chang, J.-S.; Yong Ng, H.; Wong, J.W.C.; Kim, S.-H. Production of Biosurfactants from Agro-Industrial Waste and Waste Cooking Oil in a Circular Bioeconomy: An Overview. Bioresour. Technol. 2022, 343, 126059. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Toor, S.S.; Brandão, J.; Pedersen, T.H.; Rosendahl, L.A. Optimized Conversion of Waste Cooking Oil into Ecofriendly Bio-Based Polymeric Surfactant—A Solution for Enhanced Oil Recovery and Green Fuel Compatibility. J. Clean. Prod. 2021, 294, 126214. [Google Scholar] [CrossRef]
- Sharma, P.; Usman, M.; Salama, E.-S.; Redina, M.; Thakur, N.; Li, X. Evaluation of Various Waste Cooking Oils for Biodiesel Production: A Comprehensive Analysis of Feedstock. Waste Manag. 2021, 136, 219–229. [Google Scholar] [CrossRef]
- Frota De Albuquerque Landi, F.; Fabiani, C.; Castellani, B.; Cotana, F.; Pisello, A.L. Environmental Assessment of Four Waste Cooking Oil Valorization Pathways. Waste Manag. 2022, 138, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, H.; Wang, Z.; Liu, J. Tracing the Source of Cooking Oils with an Integrated Approach of Using Stable Carbon Isotope and Fatty Acid Abundance. J. Agric. Food Chem. 2012, 60, 8069–8073. [Google Scholar] [CrossRef]
- Awogbemi, O.; Kallon, D.V.V.; Aigbodion, V.S.; Panda, S. Advances in Biotechnological Applications of Waste Cooking Oil. Case Stud. Chem. Environ. Eng. 2021, 4, 100158. [Google Scholar] [CrossRef]
- Ibanez, J.; Martel Martín, S.; Baldino, S.; Prandi, C.; Mannu, A. European Union Legislation Overview about Used Vegetable Oils Recycling: The Spanish and Italian Case Studies. Processes 2020, 8, 798. [Google Scholar] [CrossRef]
- Can, Ö. Combustion Characteristics, Performance and Exhaust Emissions of a Diesel Engine Fueled with a Waste Cooking Oil Biodiesel Mixture. Energy Convers. Manag. 2014, 87, 676–686. [Google Scholar] [CrossRef]
- Xiong, Y.; Miao, W.; Wang, N.; Chen, H.; Wang, X.; Wang, J.; Tan, Q.; Chen, S. Solid Alcohol Based on Waste Cooking Oil: Synthesis, Properties, Micromorphology and Simultaneous Synthesis of Biodiesel. Waste Manag. 2019, 85, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, M.-Y.; Qi, Y.-X.; Jin, X.-Y.; Xu, H.-R.; Chen, Y.; Chen, S.; Su, H. Synthesis and Properties of Wax Based on Waste Cooking Oil. RSC Adv. 2022, 12, 3365–3371. [Google Scholar] [CrossRef]
- Liu, M.-Y.; Li, G.-M.; Wang, P.-Y.; Ying, W.-Y.; Yang, Y.; Tang, C.-Y.; Li, Y.-Y.; Chen, S.-P. Mechanically Enhanced 3D Printable Photocurable Resin Composed of Epoxy Waste Cooking Oil and Triethylene Glycol Dimethacrylate. J. Polym. Res. 2024, 31, 177. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M.-Y.; Fan, X.-G.; Wang, P.-Y.; Chen, S.-P. A 4D-Printable Photocurable Resin Derived from Waste Cooking Oil with Enhanced Tensile Strength. Molecules 2024, 29, 2162. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M.-Y.; Fan, X.-G.; Wang, L.; Liang, J.-Y.; Jin, X.-Y.; Che, R.-J.; Ying, W.-Y.; Chen, S.-P. Four-Dimensional Printing of Multifunctional Photocurable Resin Based on Waste Cooking Oil. ACS Sustain. Chem. Eng. 2022, 10, 16344–16358. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Y.; Wang, P.; Ying, W.; Liu, Q.; Ding, G.; Chen, S. Synthesis and Properties of a Photocurable Coating Based on Waste Cooking Oil. Coatings 2023, 13, 1553. [Google Scholar] [CrossRef]
- Mosadegh, B.; Polygerinos, P.; Keplinger, C.; Wennstedt, S.; Shepherd, R.F.; Gupta, U.; Shim, J.; Bertoldi, K.; Walsh, C.J.; Whitesides, G.M. Pneumatic Networks for Soft Robotics That Actuate Rapidly. Adv. Funct. Mater. 2014, 24, 2163–2170. [Google Scholar] [CrossRef]
- Ritere, A.; Jurinovs, M.; Platnieks, O.; Barkane, A.; Gaidukovs, S. A Super-Tough Plant Oil Based Elastomer for UV-Light Assisted 3D Printed Soft Robotics and Shape-Memory. J. Mater. Chem. A 2024, 12, 16569–16582. [Google Scholar] [CrossRef]
- Asbeck, A.T.; De Rossi, S.M.M.; Galiana, I.; Ding, Y.; Walsh, C.J. Stronger, Smarter, Softer: Next-Generation Wearable Robots. IEEE Robot. Autom. Mag. 2014, 21, 22–33. [Google Scholar] [CrossRef]
- White, S.R.; Caruso, M.M.; Moore, J.S. Autonomic Healing of Polymers. MRS Bull. 2008, 33, 766–769. [Google Scholar] [CrossRef]
- Kim, D.-M.; Song, I.-H.; Choi, J.-Y.; Jin, S.-W.; Nam, K.-N.; Chung, C.-M. Self-Healing Coatings Based on Linseed-Oil-Loaded Microcapsules for Protection of Cementitious Materials. Coatings 2018, 8, 404. [Google Scholar] [CrossRef]
- Norambuena-Contreras, J.; Concha, J.; Arteaga-Pérez, L.; Gonzalez-Torre, I. Synthesis and Characterisation of Alginate-Based Capsules Containing Waste Cooking Oil for Asphalt Self-Healing. Appl. Sci. 2022, 12, 2739. [Google Scholar] [CrossRef]
- Lee, J.K.; Hong, S.J.; Liu, X.; Yoon, S.H. Characterization of Dicyclopentadiene and 5-Ethylidene-2-Norbornene as Self-Healing Agents for Polymer Composite and Its Microcapsules. Macromol. Res. 2004, 12, 478–483. [Google Scholar] [CrossRef]
- Zeng, C.; Seino, H.; Ren, J.; Hatanaka, K.; Yoshie, N. Bio-Based Furan Polymers with Self-Healing Ability. Macromolecules 2013, 46, 1794–1802. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Yin, Q.-Y.; Dai, C.-H.; Chen, H.; Gou, K.; Guan, H.-Z.; Wang, P.-H.; Jiang, J.-T.; Weng, G.-S. Tough Double Metal-Ion Cross-Linked Elastomers with Temperature-Adaptable Self-Healing and Luminescence Properties. Chin. J. Polym. Sci. 2021, 39, 554–565. [Google Scholar] [CrossRef]
- Cao, J.; Lu, C.; Zhuang, J.; Liu, M.; Zhang, X.; Yu, Y.; Tao, Q. Multiple Hydrogen Bonding Enables the Self-Healing of Sensors for Human–Machine Interactions. Angew. Chem. Int. Ed. 2017, 56, 8795–8800. [Google Scholar] [CrossRef] [PubMed]
- Meurer, J.; Hniopek, J.; Ahner, J.; Schmitt, M.; Popp, J.; Zechel, S.; Peneva, K.; Hager, M.D. In-Depth Characterization of Self-Healing Polymers Based on π–π interactions. Beilstein J. Org. Chem. 2021, 17, 2496–2504. [Google Scholar] [CrossRef]
- Utrera-Barrios, S.; Verdejo, R.; López-Manchado, M.A.; Hernández Santana, M. Evolution of Self-Healing Elastomers, from Extrinsic to Combined Intrinsic Mechanisms: A Review. Mater. Horiz. 2020, 7, 2882–2902. [Google Scholar] [CrossRef]
- Rao, Y.-L.; Chortos, A.; Pfattner, R.; Lissel, F.; Chiu, Y.-C.; Feig, V.; Xu, J.; Kurosawa, T.; Gu, X.; Wang, C.; et al. Stretchable Self-Healing Polymeric Dielectrics Cross-Linked Through Metal–Ligand Coordination. J. Am. Chem. Soc. 2016, 138, 6020–6027. [Google Scholar] [CrossRef]
- Li, T.; Zheng, T.-Z.; Guo, Z.-X.; Xu, J.; Guo, B.-H. A Well-Defined Hierarchical Hydrogen Bonding Strategy to Polyureas with Simultaneously Improved Strength and Toughness. Chin. J. Polym. Sci. 2019, 37, 1257–1266. [Google Scholar] [CrossRef]
- Bodor, M.; Lasagabáster-Latorre, A.; Arias-Ferreiro, G.; Dopico-García, M.S.; Abad, M.-J. Improving the 3D Printability and Mechanical Performance of Biorenewable Soybean Oil-Based Photocurable Resins. Polymers 2024, 16, 977. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, X.; Hu, J.; Li, J.; Zhou, R.; Lin, S. Green Synthesis of Soybean Oil-Derived UV-Curable Resins for High-Resolution 3D Printing. Addit. Manuf. 2024, 95, 104543. [Google Scholar] [CrossRef]
- Peng, S.; Li, Y.; Wu, L.; Zhong, J.; Weng, Z.; Zheng, L.; Yang, Z.; Miao, J.-T. 3D Printing Mechanically Robust and Transparent Polyurethane Elastomers for Stretchable Electronic Sensors. ACS Appl. Mater. Interfaces 2020, 12, 6479–6488. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Zhang, J.; Huang, J.; Yu, X.; Cheng, J.; Shang, Q.; Hu, Y.; Liu, C.; Hu, L.; Zhou, Y. High-Performance 3D Printing UV-Curable Resins Derived from Soybean Oil and Gallic Acid. Green Chem. 2021, 23, 5911–5923. [Google Scholar] [CrossRef]
- Stouten, J.; Schnelting, G.H.M.; Hul, J.; Sijstermans, N.; Janssen, K.; Darikwa, T.; Ye, C.; Loos, K.; Voet, V.S.D.; Bernaerts, K.V. Biobased Photopolymer Resin for 3D Printing Containing Dynamic Imine Bonds for Fast Reprocessability. ACS Appl. Mater. Interfaces 2023, 15, 27110–27119. [Google Scholar] [CrossRef]
- Bassett, A.W.; Honnig, A.E.; Breyta, C.M.; Dunn, I.C.; La Scala, J.J.; Stanzione, J.F. Vanillin-Based Resin for Additive Manufacturing. ACS Sustain. Chem. Eng. 2020, 8, 5626–5635. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, J.; Huang, J.; Yu, X.; Cheng, J.; Shang, Q.; Hu, Y.; Liu, C.; Zhang, M.; Hu, L.; et al. Self-Healing, Antibacterial, and 3D-Printable Polymerizable Deep Eutectic Solvents Derived from Tannic Acid. ACS Sustain. Chem. Eng. 2022, 10, 7954–7964. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Parra-Cabrera, C.; Kim, Y.T.; Kuo, A.P.; Folch, A. Desktop-Stereolithography 3D-Printing of a Poly(Dimethylsiloxane)-Based Material with Sylgard-184 Properties. Adv. Mater. 2018, 30, 1800001. [Google Scholar] [CrossRef]
- Zhu, G.; Liu, M.; Weng, S.; Zhang, G.; Hu, Y.; Kou, Z.; Bo, C.; Hu, L.; Wu, S.; Zhou, Y. Dye-Free and Reprintable Multi-Color DLP 3D Printing Using ZnCl2-Based Polymerizable Deep Eutectic Solvents and Type I Photoinitiators. Chem. Eng. J. 2023, 472, 144987. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, J.; Huang, J.; Qiu, Y.; Liu, M.; Yu, J.; Liu, C.; Shang, Q.; Hu, Y.; Hu, L.; et al. Recyclable and Reprintable Biobased Photopolymers for Digital Light Processing 3D Printing. Chem. Eng. J. 2023, 452, 139401. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Han, H. Synthesis of UV Curable, Highly Stretchable, Transparent Poly(Urethane-Acrylate) Elastomer and Applications Toward Next Generation Technology. Macromol. Res. 2020, 28, 896–902. [Google Scholar] [CrossRef]
- Chen, J.-H.; An, X.-P.; Li, Y.-D.; Wang, M.; Zeng, J.-B. Reprocessible Epoxy Networks with Tunable Physical Properties: Synthesis, Stress Relaxation and Recyclability. Chin. J. Polym. Sci. 2018, 36, 641–648. [Google Scholar] [CrossRef]
- Cao, L.; Fan, J.; Huang, J.; Chen, Y. A Robust and Stretchable Cross-Linked Rubber Network with Recyclable and Self-Healable Capabilities Based on Dynamic Covalent Bonds. J. Mater. Chem. A 2019, 7, 4922–4933. [Google Scholar] [CrossRef]
- Debnath, S.; Kaushal, S.; Ojha, U. Catalyst-Free Partially Bio-Based Polyester Vitrimers. ACS Appl. Polym. Mater. 2020, 2, 1006–1013. [Google Scholar] [CrossRef]
- Wu, Y.; Fei, M.; Chen, T.; Li, C.; Fu, T.; Qiu, R.; Liu, W. H-Bonds and Metal-Ligand Coordination-Enabled Manufacture of Palm Oil-Based Thermoplastic Elastomers by Photocuring 3D Printing. Addit. Manuf. 2021, 47, 102268. [Google Scholar] [CrossRef]
- Zhao, T.; Yu, R.; Li, S.; Li, X.; Zhang, Y.; Yang, X.; Zhao, X.; Wang, C.; Liu, Z.; Dou, R.; et al. Superstretchable and Processable Silicone Elastomers by Digital Light Processing 3D Printing. ACS Appl. Mater. Interfaces 2019, 11, 14391–14398. [Google Scholar] [CrossRef] [PubMed]
- Peñas-Caballero, M.; Chemello, E.; Grande, A.M.; Hernández Santana, M.; Verdejo, R.; Lopez-Manchado, M.A. Poly(Methyl Methacrylate) as Healing Agent for Carbon Fibre Reinforced Epoxy Composites. Polymers 2023, 15, 1114. [Google Scholar] [CrossRef]
- Fan, F.; Szpunar, J. The Self-healing Mechanism of an Industrial Acrylic Elastomer. J. Appl. Polym. Sci. 2015, 132, 42135. [Google Scholar] [CrossRef]
- Li, X.; Zhai, Y.; Yang, K.; Bai, J.; Qiu, Y.; Wang, Y. Preparation and Characterization of a Novel Self-Healing Transparent Polyimide Film Based on Dynamic Disulfide Bonds. Polymers 2024, 16, 3461. [Google Scholar] [CrossRef]
- Lee, D.-I.; Kim, S.-H.; Lee, D.-S. Synthesis of Self-Healing Waterborne Polyurethane Systems Chain Extended with Chitosan. Polymers 2019, 11, 503. [Google Scholar] [CrossRef] [PubMed]
- GB/T 5532-2008; Animal and Vegetable Fats and Oils—Determination of Iodine Value. Standards Press of China: Beijing, China, 2008.
- GB/T 1677-2008; Determinating the Epoxy Value of Plasticizers. Standards Press of China: Beijing, China, 2008.
- GB 5009.168-2016; National Food Safety Standard—Determination of Fatty Acids in Foods. Standards Press of China: Beijing, China, 2016.
- Wu, Y.; Li, R.; Wang, J.; Situ, Y.; Huang, H. A New Carbazolyl-basedacylphosphine Oxide Photoinitiator with High Performance and Low Migration. J. Polym. Sci. 2022, 60, 52–61. [Google Scholar] [CrossRef]
- GB/T 1040.2-2006; Plastics—Determination of Tensile Properties—Part 2: Test Conditions for Moulding and Extrusion Plastics. Standards Press of China CN-GB: Beijing, China, 2006.
- GB/T 4852-2002; Test Method for Tack of Pressure Sensitive Adhesive Tapes by Rolling Ball. Standards Press of China: Beijing, China, 2002.
- GB/T 4851-2014; Measurement of Static Shear Adhesion for Adhesive Types. Standards Press of China: Beijing, China, 2014.
- GB/T 2792-2014; Measurement of Peel Adhesion Properties for Adhesive Tapes. Standards Press of China: Beijing, China, 2014.
- Wu, B.; Sufi, A.; Ghosh Biswas, R.; Hisatsune, A.; Moxley-Paquette, V.; Ning, P.; Soong, R.; Dicks, A.; Simpson, A. Direct Conversion of McDonald’s Waste Cooking Oil into a Biodegradable High-Resolution 3D-Printing Resin. ACS Sustain. Chem. Eng. 2020, 8, 1171–1177. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; In’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun. 2022, 271, 108171. [Google Scholar] [CrossRef]
- Cui, X.; Zhang, L.; Yang, Y.; Tang, P. Understanding the Application of Covalent Adaptable Networks in Self-Repair Materials Based on Molecular Simulation. Soft Matter 2024, 20, 1486–1498. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO—The Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Major Compositions | Dp (mm) | Ec (mJ/cm2) | Reference |
|---|---|---|---|
| WCO-based methacrylate fatty acid ethyl ester (WMFAEE) Hydroxypropyl acrylate (HPA) | 0.224 | 55.44 | This work |
| Acrylated epoxidized soybean oil (AESO) | 1.318 | 77 | [42] |
| Commercial resin | 0.314 | 16.32 | [45] |
| Bifunctional vanillin-vased vuilding vlock (It contains Priamine 1075 and MEV) Trifunctional crosslinker (It contains TREN and MEV) Isobornyl methacrylate (IBOMA) Phenylbis(2,4,6-trimethylbenzoyl) phosphine oxide (BAPO) | 0.404 | 17.5 | [46] |
| Methacrylated vanillin (MV) Glycerol dimethacrylate (GDM) | 0.340 | 187 | [47] |
| Choline chloride (ChCl) Hydroxyethyl methacrylate (HEMA) Tannic acid (TA) | 0.192 | 8.70 | [48] |
| Poly(dimethylsiloxane) (PDMS) PDMS macromolecular chains with methacryloxypopyl functional groups at the terminal ends (3DP-PDMS-E) PDMS copolymers containing methacryloxypropyl functional groups in the side chains (3DP-PDMS-S) | 0.213 | - | [49] |
| Acrylic acid (AA) ZnCl2 H2O Citric acid (CA) | 0.24 | - | [50] |
| Castor oil (CO) Isophorone diisocyanate (IPDI) 2-(tert-Butylamino) ethyl methacrylate (TBEM) | 0.176 | - | [51] |
| Major Compositions | Elongation at Break (%) | Tensile Strength (MPa) | Reference |
|---|---|---|---|
| WCO-based methacrylate fatty acid ethyl ester (WMFAEE) Hydroxypropyl acrylate (HPA) | 645.09 | 0.967 | This work |
| Epoxy waste oil methacrylate (EWOMA) 2-phenoxyethyl acrylate (PHEA) Methacrylic acid (MAA) | 230.1 | 0.48 | [25] |
| Poly(tetrahydrofuran-based) polyurethane acrylate oligomer (PPTMGA) Isobornyl acrylate (IBOA) | 414.3 ± 7.6 | 15.7 ± 0.7 | [44] |
| Methyl Acrylate (MA) n-Butyl Acrylate (BA) | 575.15 | 16.97 | [52] |
| Diglycidyl ether of bisphenol A (DGEBA) 1,4-Cyclohexanedicarboxylic acid (CHDA) Sebacic acid (SA) 1,5,7-Triazabicyclo [4.4.0] dec -5-ene (TBD) | 280 | 22 | [53] |
| Epoxidized derivatives of natural rubber (ENR) TEMPO-oxidized cellulose nanocrystals (TOCNs) | 750 | 5.8 | [54] |
| Poly(2-hydroxyethyl methacrylate) (PHEMA) | 130 | 25.4 | [55] |
| Palm oil fatty acid—ethylacrylamid (POFA-EA) Acrylic acid (AA) ZnCl2 | 867 | 2.1 | [56] |
| Vinyl-terminated polydimethylsiloxane (VPS-22000) Branched mercapto-functionalized polysiloxane (MPS) Precipitated silica (PSi) | 1400 | - | [57] |
| Major Compositions | Self-Healing Efficiency Based on Elongation at Break | Self-Healing Efficiency Based on Tensile Strength | Possible Self-Healing Mechanism | Reference |
|---|---|---|---|---|
| WCO-based methacrylate fatty acid ethyl ester (WMFAEE) Hydroxypropyl acrylate (HPA) | 57.82% | 20.68% | Molecular chain diffusion and entanglement + hydrogen bonding | This work |
| Epoxy resin (Bisphenol F diglycidyl ether (DGEBF), Bisphenol A diglycidyl ether (DGEBA) and1,6-Hexanediol diglycidyl ether) Carbon fibre | 53% | - | Thermally driven physical flow filling for crack healing | [58] |
| Commercial acrylic elastome | 85% | Molecular chain diffusion + hydrogen bonding | [59] | |
| Cystamine 2,2-Bis [4-(3,4-dicarboxyphenoxy)phenyl]propane dianhydride (BPADA) 2,2-Bis [4-(4-aminophenoxy)phenyl]hexafluoropropane (HFBAPP) Methyl-2-pyrrolidinone (NMP) N,N-dimethylacetamide (DMAc) Triethylamine (TEA) | 91.8% | - | Dynamic disulfide bonds | [60] |
| Poly(tetramethylene ether glycol) (PTMEG) Isophorone diisocyanate (IPDI) 2,2-Bis(hydroxymethyl) propionic acid (DMPA) Chitosan | - | 35% | Hydroxyl-driven dynamic exchange reaction | [61] |
| Sample | WMFAEE (g) | EWOMA (g) | HPA (g) | Mass Ratio of WMFAEE (or EWOMA) to HPA | 819 (g) | DMAB (g) |
|---|---|---|---|---|---|---|
| A1 (pure HPA, control sample) | 0 | - | 100 | 0:1 | 2 | 2 |
| A2 | 50 | - | 150 | 1:3 | 4 | 4 |
| A3 | 50 | - | 100 | 1:2 | 3 | 3 |
| A4 | 100 | - | 150 | 2:3 | 5 | 5 |
| A5 | 100 | - | 100 | 1:1 | 4 | 4 |
| A6 | 150 | - | 100 | 3:2 | 5 | 5 |
| A7 (EWOMA-HPA, control sample) | - | 100 | 150 | 2:3 | 5 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Sun, J.; Liu, M.; Tang, C.; Yang, Y.; Ding, G.; Liu, Q.; Chen, S. Multifunctional 3D-Printable Photocurable Elastomer with Self-Healing Capability Derived from Waste Cooking Oil. Molecules 2025, 30, 1824. https://doi.org/10.3390/molecules30081824
Wang P, Sun J, Liu M, Tang C, Yang Y, Ding G, Liu Q, Chen S. Multifunctional 3D-Printable Photocurable Elastomer with Self-Healing Capability Derived from Waste Cooking Oil. Molecules. 2025; 30(8):1824. https://doi.org/10.3390/molecules30081824
Chicago/Turabian StyleWang, Pengyu, Jiahui Sun, Mengyu Liu, Chuanyang Tang, Yang Yang, Guanzhi Ding, Qing Liu, and Shuoping Chen. 2025. "Multifunctional 3D-Printable Photocurable Elastomer with Self-Healing Capability Derived from Waste Cooking Oil" Molecules 30, no. 8: 1824. https://doi.org/10.3390/molecules30081824
APA StyleWang, P., Sun, J., Liu, M., Tang, C., Yang, Y., Ding, G., Liu, Q., & Chen, S. (2025). Multifunctional 3D-Printable Photocurable Elastomer with Self-Healing Capability Derived from Waste Cooking Oil. Molecules, 30(8), 1824. https://doi.org/10.3390/molecules30081824