
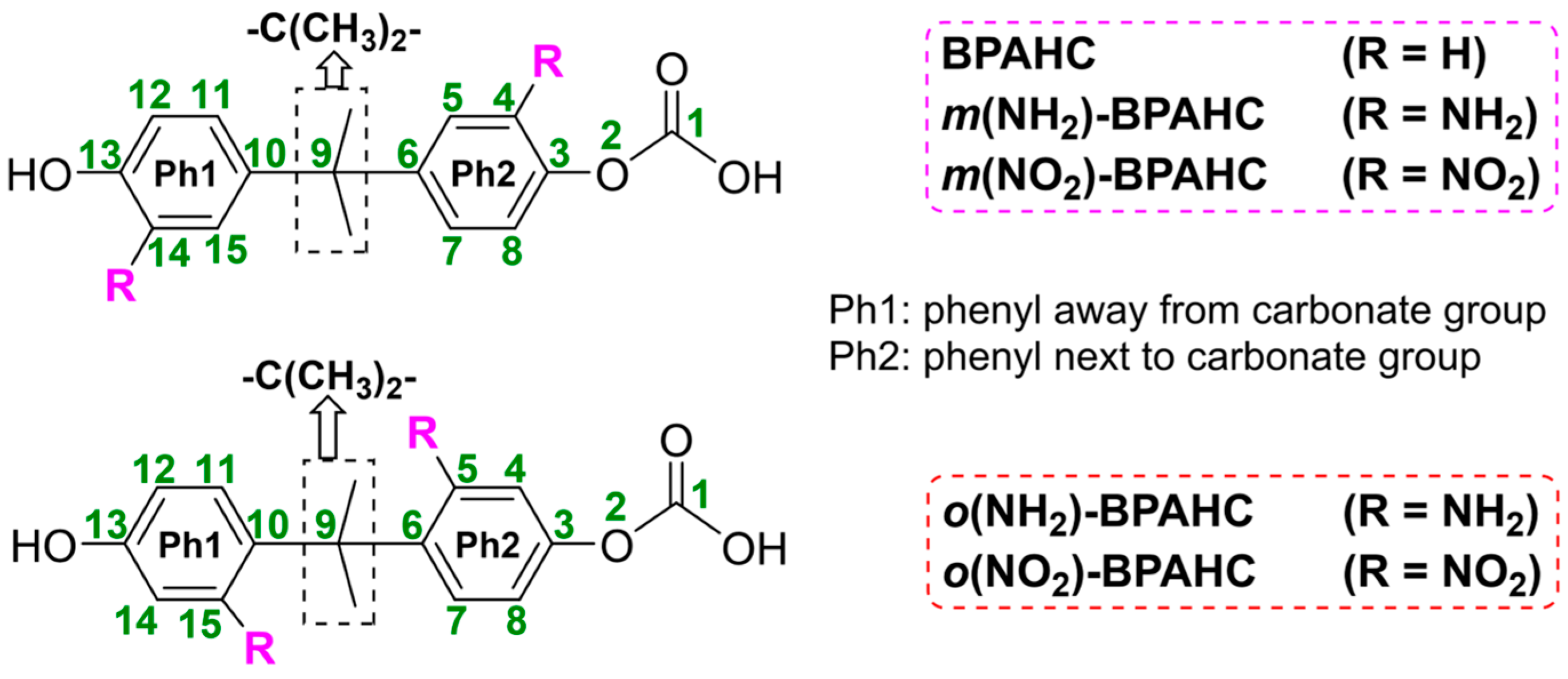
Mechanistic Study of Substituent Effect on Photoinduced O-C Bond Activation in Polycarbonate


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Effect of Electron-Donating -NH2 Group on O-C Bond Cleavage
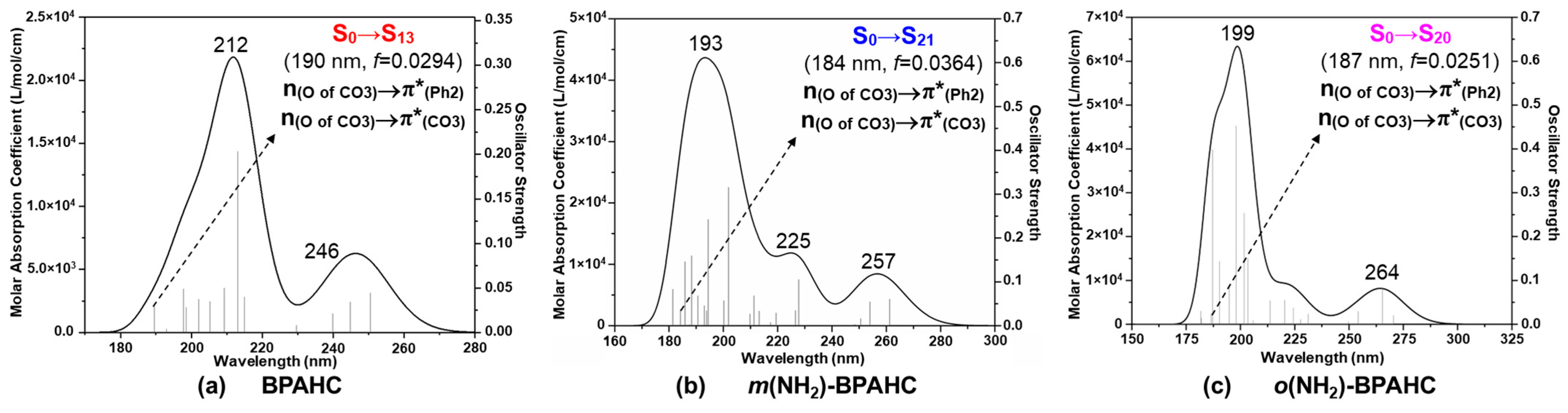
2.1.1. Absorption Spectra in the S0 Geometry Under -NH2 Effect
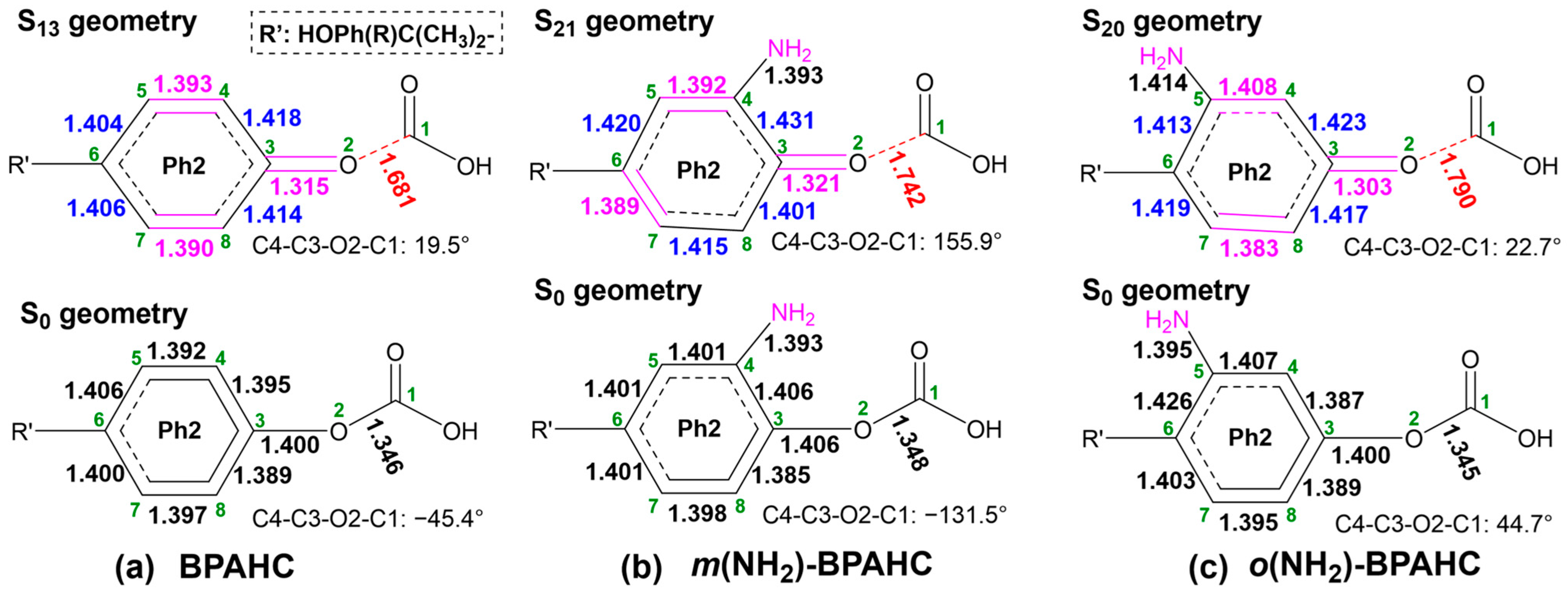
2.1.2. GS and ES Geometries Under -NH2 Effect
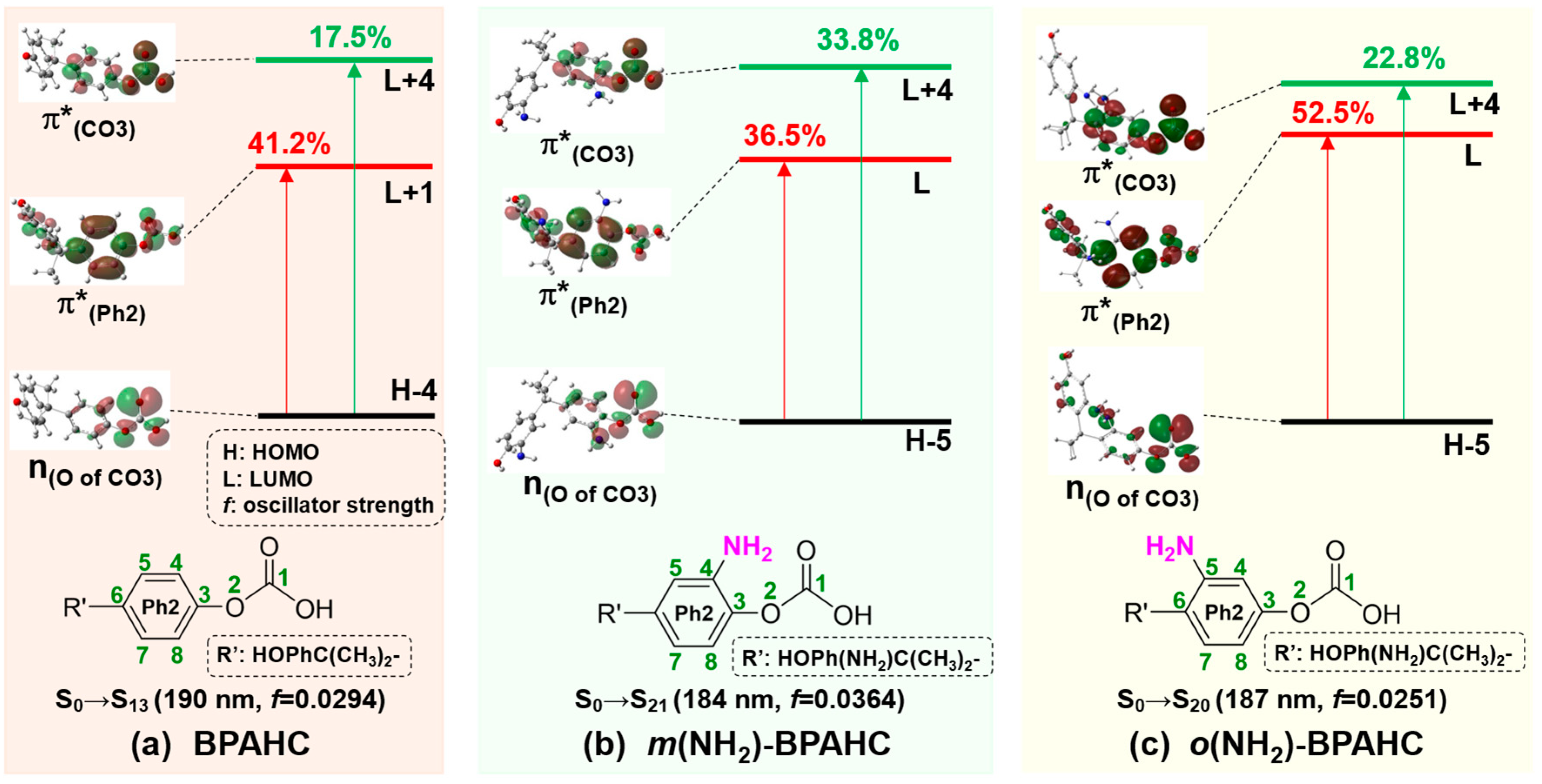
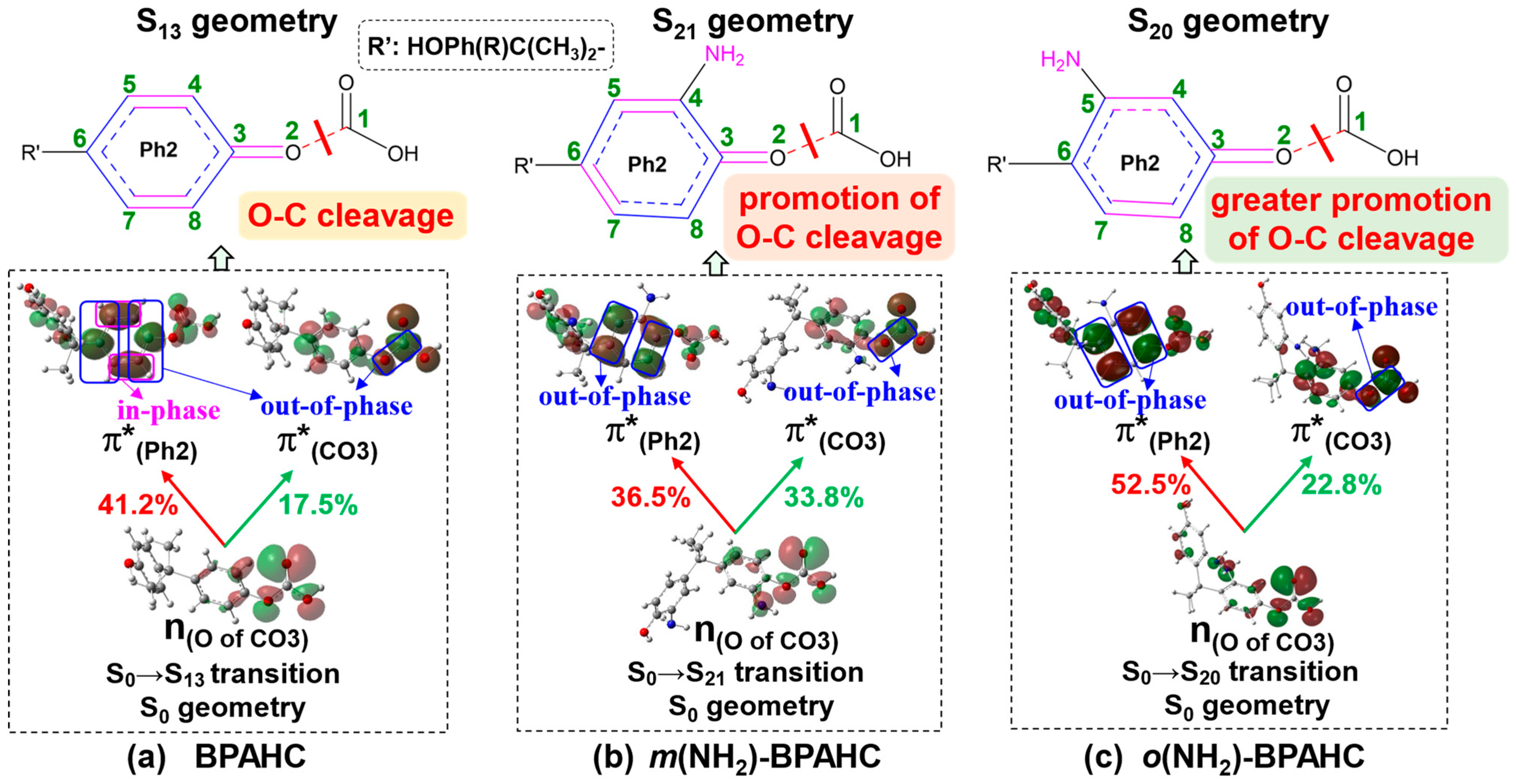
2.1.3. Characteristic MOs Under -NH2 Effect
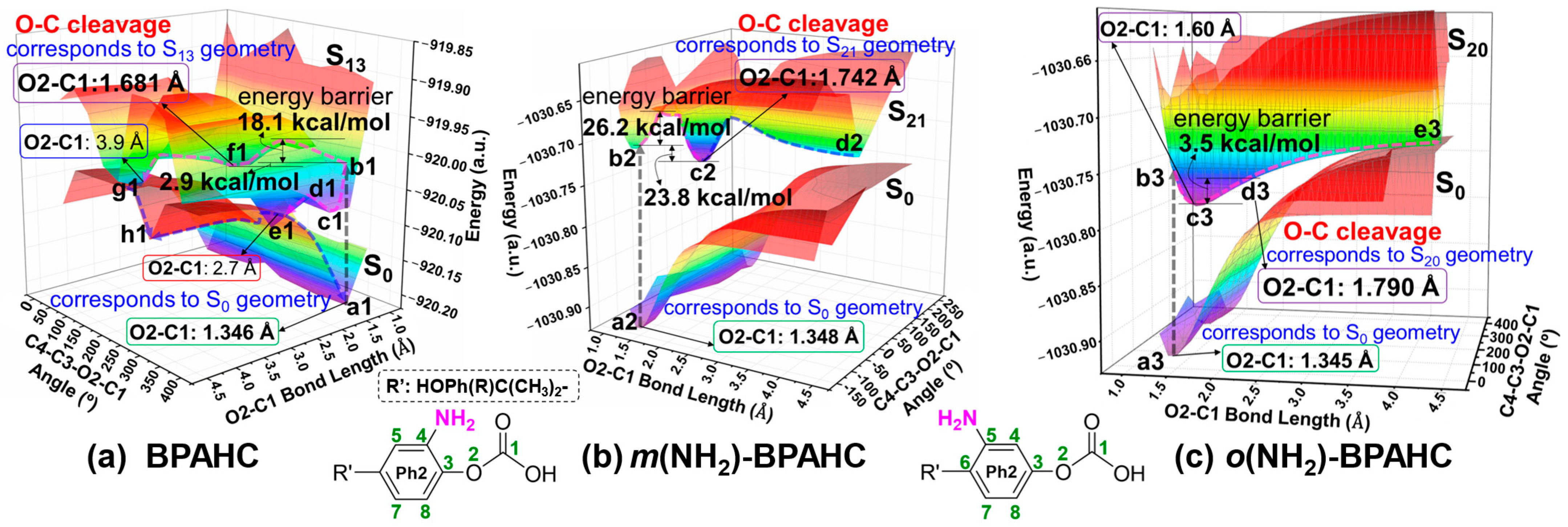
2.1.4. Potential Energy Surfaces (PESs) Under -NH2 Effect
2.2. Effect of Electron-Withdrawing -NO2 Group on O-C Bond Cleavage
2.2.1. Absorption Spectra in the S0 Geometry Under -NO2 Effect
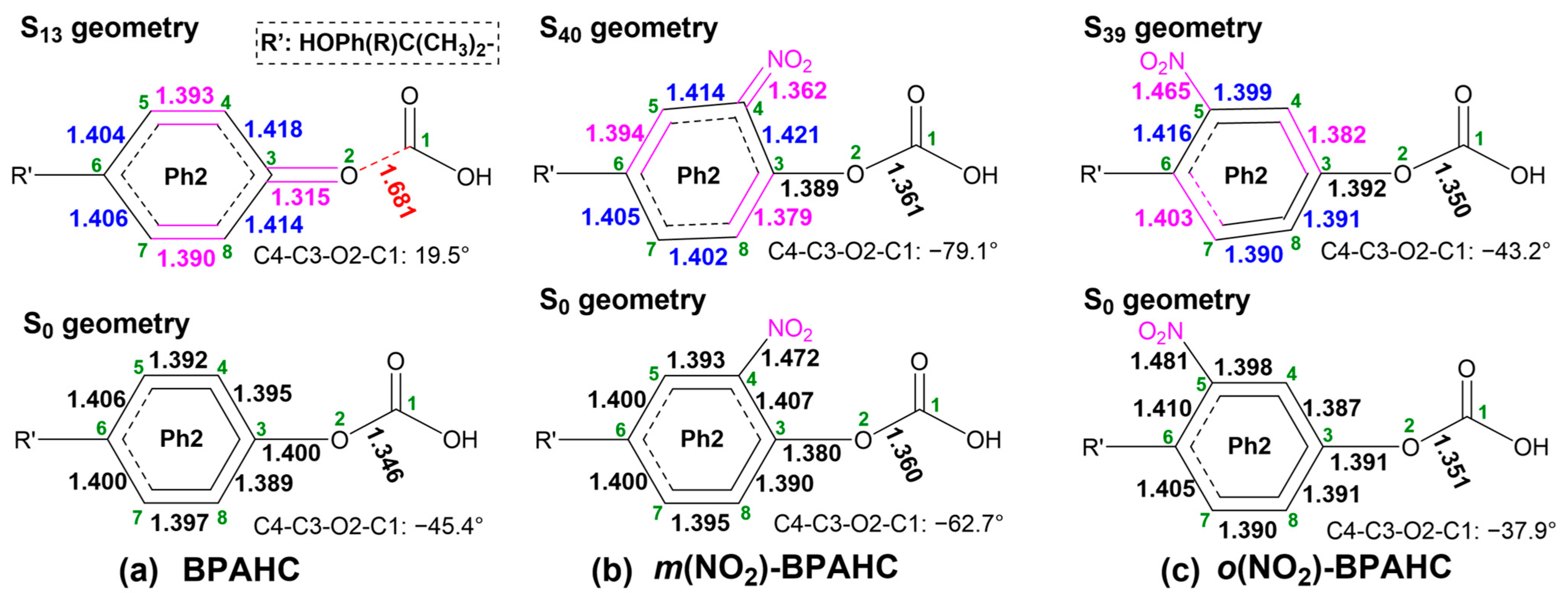
2.2.2. GS and ES Geometries Under -NO2 Effect
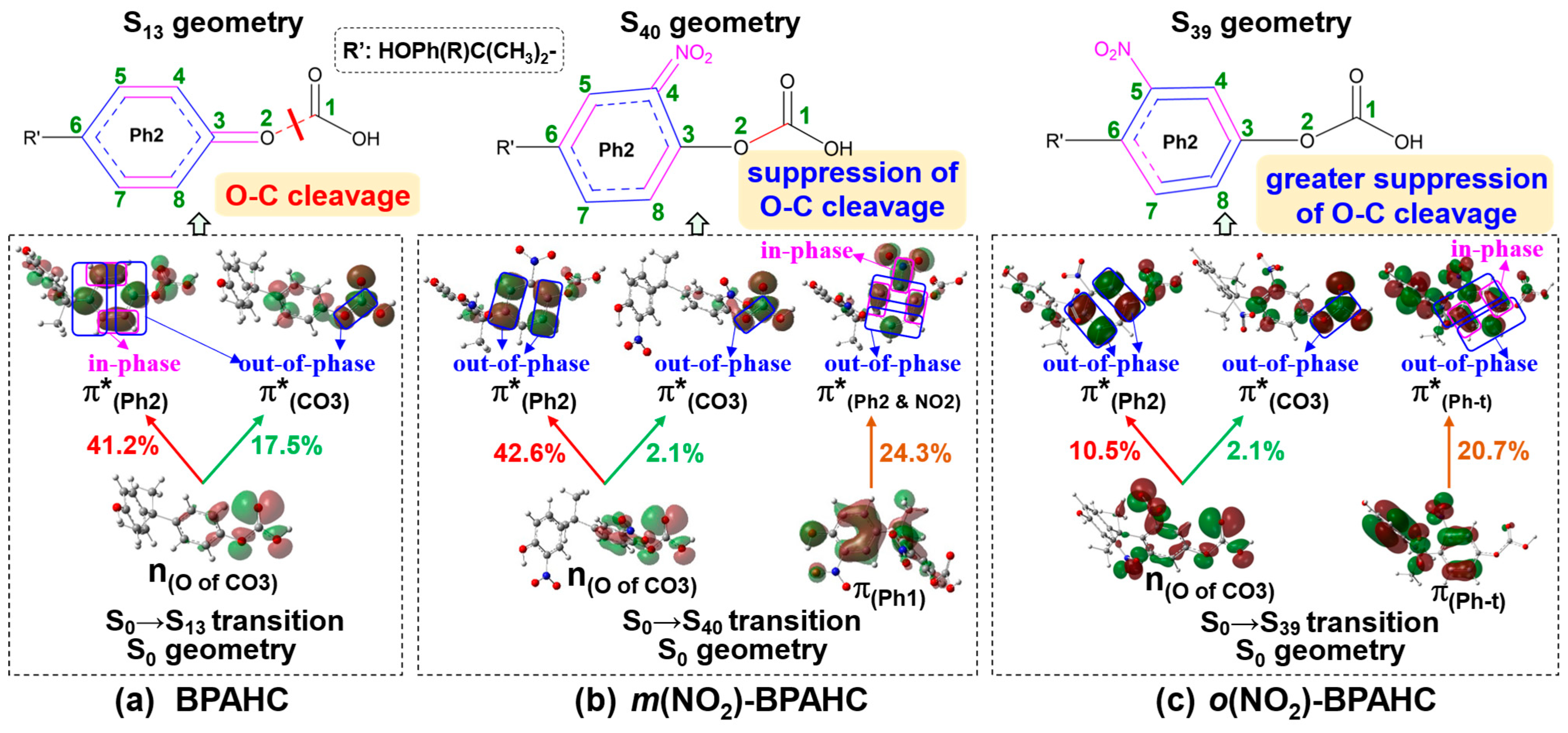
2.2.3. Characteristic MOs Under -NO2 Effect
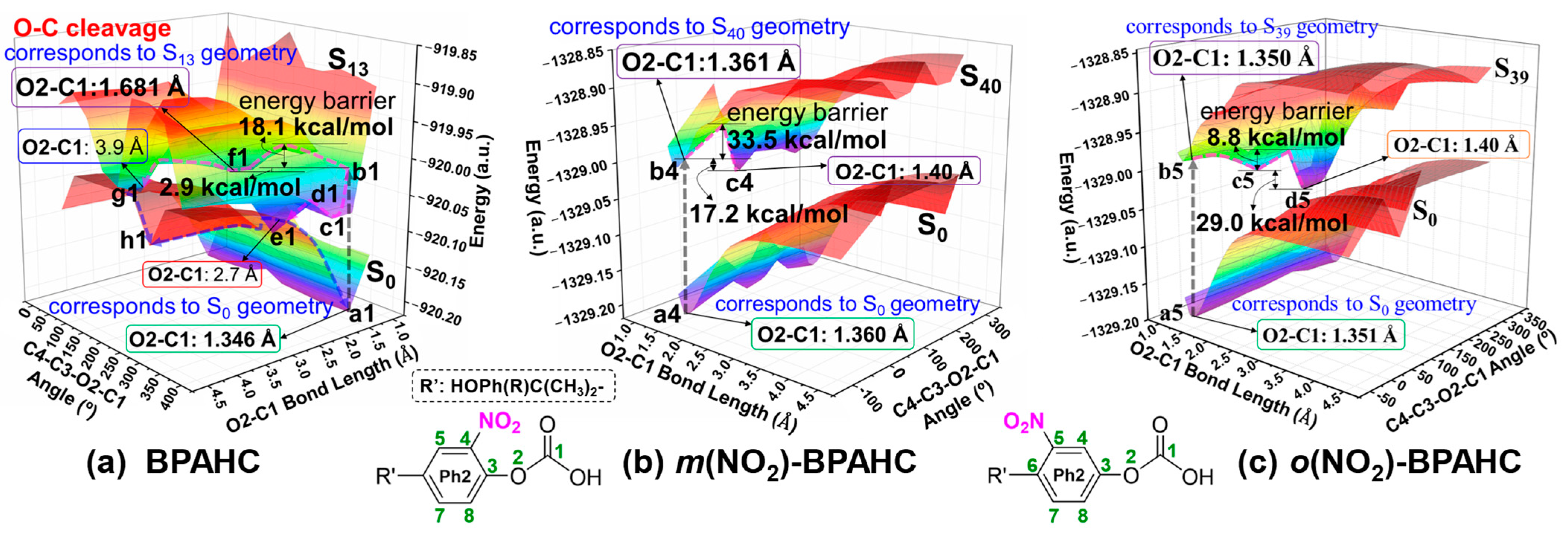
2.2.4. PESs Under -NO2 Effect
3. Computational Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ram, A.; Zilber, O.; Kenig, S. Life Expectation of Polycarbonate. Polym. Eng. Sci. 1985, 25, 535–540. [Google Scholar] [CrossRef]
- Rivaton, A. Recent Advances in Bisphenol-A Polycarbonate Photodegradation. Polym. Degrad. Stabil. 1995, 49, 163–179. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Li, X.; Pan, G.; Tong, H. Theoretical Studies on Thermal Degradation Reaction Mechanism of Model Compound of Bisphenol A Polycarbonate. Waste Manag. 2018, 71, 181–191. [Google Scholar] [CrossRef]
- Humphrey, J.S.; Shultz, A.R.; Jaquiss, D.B.G. Flash Photochemical Studies of Polycarbonate and Related Model Compounds, Photodegradation vs. Photo-Fries Rearrangement. Macromolecules 1973, 6, 305–314. [Google Scholar] [CrossRef]
- Rivaton, A.; Sallet, D.; Lemaire, J. The Photochemistry of Bisphenol-A Polycarbonate Reconsidered. Polym. Photochem. 1983, 3, 463–481. [Google Scholar] [CrossRef]
- Pickett, J.E. Influence of Photo-Fries Reaction Products on the Photodegradation of Bisphenol-A Polycarbonate. Polym. Degrad. Stabil. 2011, 96, 2253–2265. [Google Scholar] [CrossRef]
- Diepens, M.; Gijsman, P. Photostabilizing of Bisphenol A Polycarbonate by Using UV-Absorbers and Self Protective Block Copolymers Based on Resorcinol Polyarylate Blocks. Polym. Degrad. Stabil. 2009, 94, 1808–1813. [Google Scholar] [CrossRef]
- Diepens, M.; Gijsman, P. Photodegradation of Bisphenol A Polycarbonate with Different Types of Stabilizers. Polym. Degrad. Stabil. 2010, 95, 811–817. [Google Scholar] [CrossRef]
- Pankasem, S.; Kuczynski, J.; Thomas, J.K. Photochemistry and Photodegradation of Polycarbonate. Macromolecules 1994, 27, 3773–3781. [Google Scholar] [CrossRef]
- Liaw, D.-J.; Chang, P. Synthesis and Characterization of Polycarbonates Based on Bisphenol S by Melt Transesterification. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 2453–2460. [Google Scholar] [CrossRef]
- Rivaton, A.; Mailhot, B.; Soulestin, J.; Varghese, H.; Gardette, J.-L. Influence of the Chemical Structure of Polycarbonates on the Contribution of Crosslinking and Chain Scissions to the Photothermal Ageing. Eur. Polym. J. 2002, 38, 1349–1363. [Google Scholar] [CrossRef]
- Factor, A.; Engen, P.T. The Synthesis, Characterization, and Weathering Behavior of Polycarbonates Derived from Bis(p-hydroxyphenyl)dimethylsilane (BPSi). J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2231–2236. [Google Scholar] [CrossRef]
- Grimme, S. MO-Theoretical Investigation on the Photodissociation of Carbon-Oxygen Bonds in Aromatic Compounds. Chem. Phys. 1992, 163, 313–330. [Google Scholar] [CrossRef]
- Toldo, J.M.; Barbatti, M.; Gonçalves, P.F.B. A Three-State Model for the Photo-Fries Rearrangement. Phys. Chem. Chem. Phys. 2017, 19, 19103–19108. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, S.; Lv, Y.; Hu, P.; Huang, Y.; Kong, M.; Li, G. Atomic-Scale Insight into the Pyrolysis of Polycarbonate by ReaxFF-Based Reactive Molecular Dynamics Simulation. Fuel 2021, 287, 119484. [Google Scholar] [CrossRef]
- Huang, X.; Orimoto, Y.; Aoki, Y. Theoretical Analysis of Properties of Ground and Excited States for Photodissociation of the C-O Bond in Polycarbonates. J. Phys. Chem. A 2021, 125, 6662–6673. [Google Scholar] [CrossRef]
- Huang, X.; Orimoto, Y.; Aoki, Y. Theoretical Design of Durable and Strong Polycarbonates against Photodegradation. Phys. Chem. Chem. Phys. 2024, 26, 57–61. [Google Scholar] [CrossRef]
- Gross, E.K.U.; Kohn, W. Time-Dependent Density-Functional Theory. Adv. Quantum Chem. 1990, 21, 255–291. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained with the Correlation Energy Density Functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Fantacci, S.; Angelis, F.D.; Nazeeruddin, M.K.; Grätzel, M. Electronic and Optical Properties of the Spiro-MeOTAD Hole Conductor in Its Neutral and Oxidized Forms: A DFT/TDDFT Investigation. J. Phys. Chem. C 2011, 115, 23126–23133. [Google Scholar] [CrossRef]
- Orimoto, Y.; Ishimoto, K.; Aoki, Y. Role of Pyridinium Groups and Iodide Ions in Photoelectrochromism in Viologen-Based Ion-Pair Charge-Transfer Complexes: Molecular Orbital Analysis. J. Phys. Chem. C 2018, 122, 4546–4556. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yu, J.; Su, N.Q.; Yang, W. Describing Chemical Reactivity with Frontier Molecular Orbitalets. JACS Au 2022, 2, 1383–1394. [Google Scholar] [CrossRef]
- Mashkovtsev, D.; Orimoto, Y.; Aoki, Y. Fast and Accurate Calculation of the UV-Vis Spectrum with the Modified Local Excitation Approximation. J. Chem. Theory Comput. 2023, 19, 5548–5562. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electronic Transition | Energy (eV) | λ (nm) | f a | Contribution (%) | Transition b | Assignment | |
|---|---|---|---|---|---|---|---|
| BPAHC | S0→S13 | 6.54 | 190 | 0.0294 | 41.2 | H-4→L+1 | n(O of CO3)→π*(Ph2) |
| 17.5 | H-4→L+4 | n(O of CO3)→π*(CO3) | |||||
| 15.1 | H-3→L+1 | π(Ph2)→π*(Ph2) | |||||
| m(NH2)-BPAHC | S0→S21 | 6.73 | 184 | 0.0364 | 36.5 | H-5→L | n(O of CO3)→π*(Ph2) |
| 33.8 | H-5→L+4 | n(O of CO3)→π*(CO3) | |||||
| o(NH2)-BPAHC | S0→S20 | 6.65 | 187 | 0.0251 | 52.5 | H-5→L | n(O of CO3)→π*(Ph2) |
| 22.8 | H-5→L+4 | n(O of CO3)→π*(CO3) |
| Electronic Transition | Energy (eV) | λ (nm) | f a | Contribution (%) | Transition b | Assignment | |
|---|---|---|---|---|---|---|---|
| BPAHC | S0→S13 | 6.54 | 190 | 0.0294 | 41.2 | H-4→L+1 | n(O of CO3)→π*(Ph2) |
| 17.5 | H-4→L+4 | n(O of CO3)→π*(CO3) | |||||
| 15.1 | H-3→L+1 | π(Ph2)→π*(Ph2) | |||||
| m(NO2)-BPAHC | S0→S40 | 6.46 | 192 | 0.0272 | 42.6 | H-5→L+2 | n(O of CO3)→π*(Ph2) |
| 2.1 | H-5→L+6 | n(O of CO3)→π*(CO3) | |||||
| 24.3 | H-13→L+1 | π(Ph1)→π*(Ph2 & NO2) | |||||
| o(NO2)-BPAHC | S0→S39 | 6.42 | 193 | 0.0725 | 10.5 | H-7→L+2 | n(O of CO3)→π*(Ph2) |
| 2.1 | H-7→L+6 | n(O of CO3)→π*(CO3) | |||||
| 20.7 | H-3→L+4 | π(Ph-t)→π*(Ph-t) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Orimoto, Y.; Aoki, Y. Mechanistic Study of Substituent Effect on Photoinduced O-C Bond Activation in Polycarbonate. Molecules 2025, 30, 1839. https://doi.org/10.3390/molecules30081839
Huang X, Orimoto Y, Aoki Y. Mechanistic Study of Substituent Effect on Photoinduced O-C Bond Activation in Polycarbonate. Molecules. 2025; 30(8):1839. https://doi.org/10.3390/molecules30081839
Chicago/Turabian StyleHuang, Xiao, Yuuichi Orimoto, and Yuriko Aoki. 2025. "Mechanistic Study of Substituent Effect on Photoinduced O-C Bond Activation in Polycarbonate" Molecules 30, no. 8: 1839. https://doi.org/10.3390/molecules30081839
APA StyleHuang, X., Orimoto, Y., & Aoki, Y. (2025). Mechanistic Study of Substituent Effect on Photoinduced O-C Bond Activation in Polycarbonate. Molecules, 30(8), 1839. https://doi.org/10.3390/molecules30081839