Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
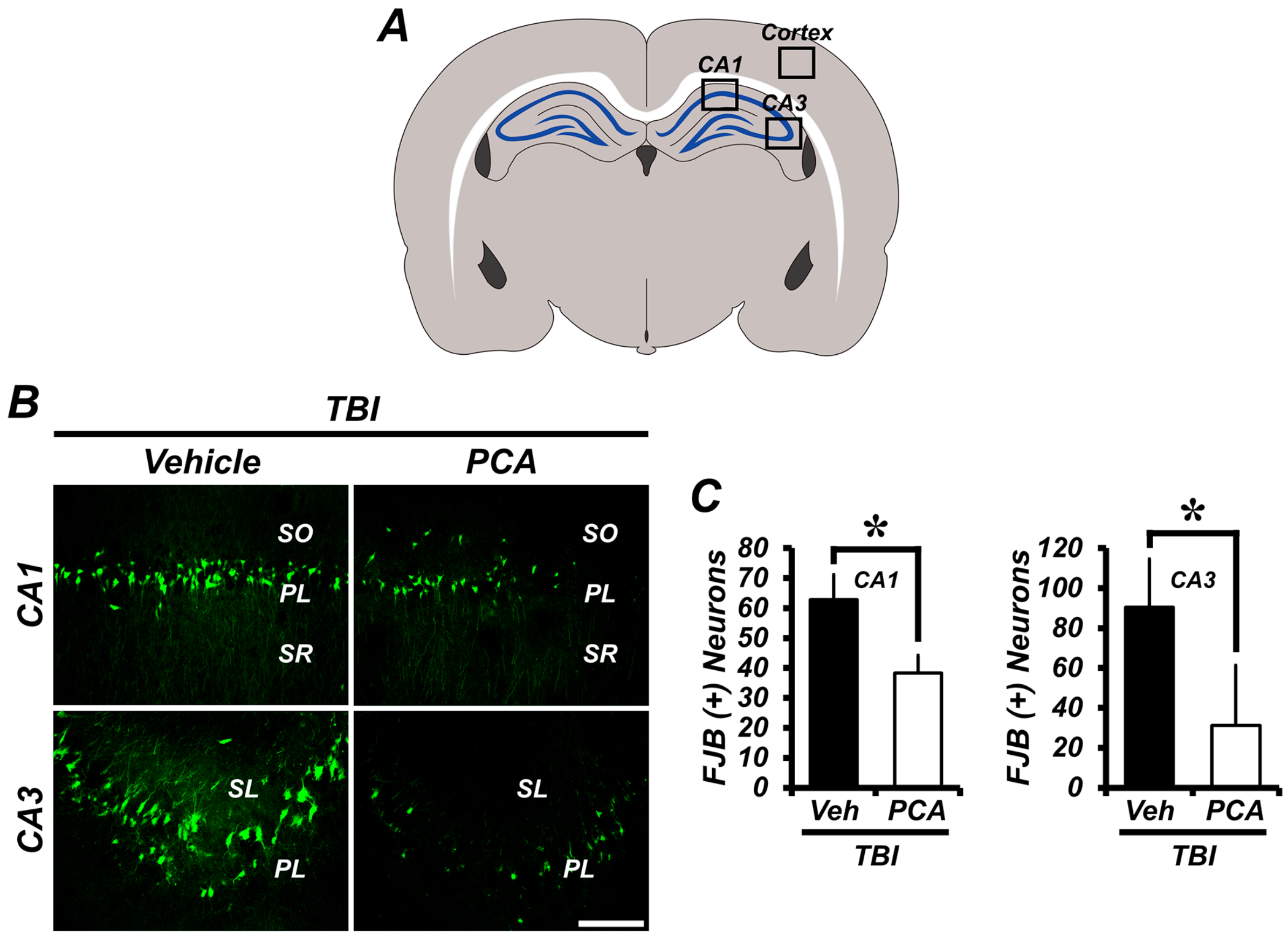
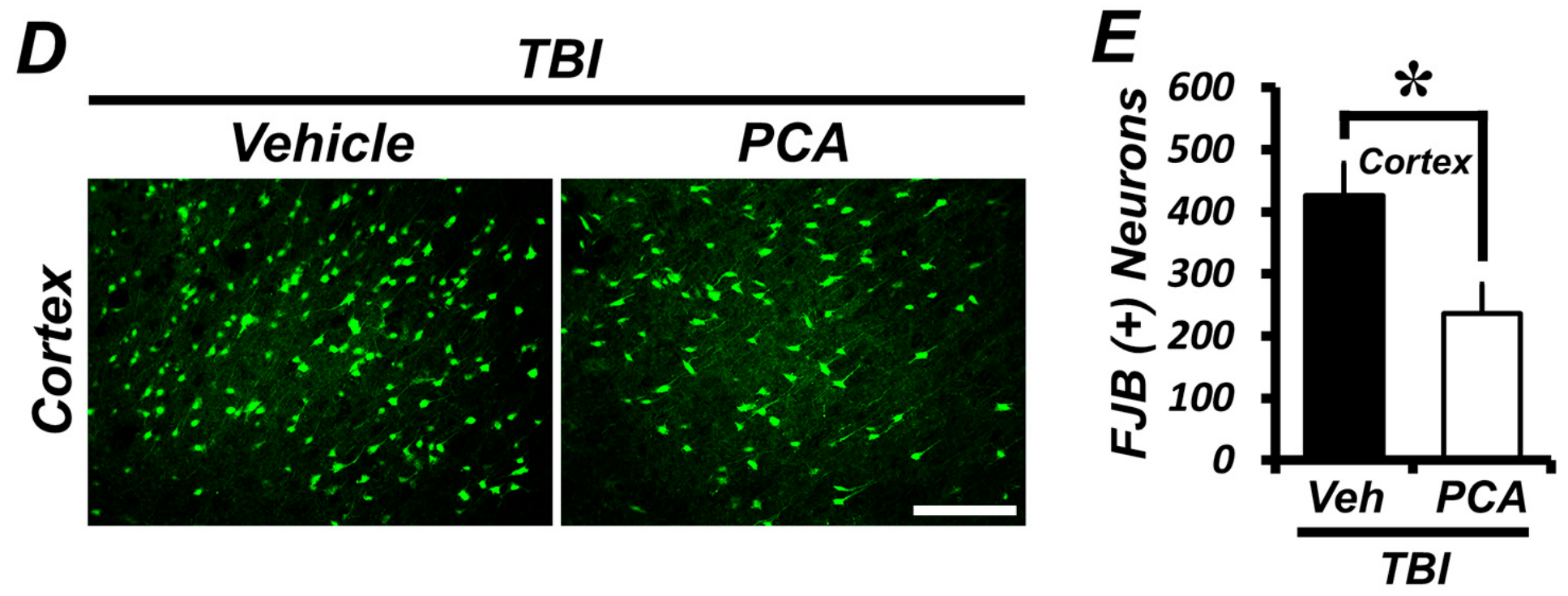
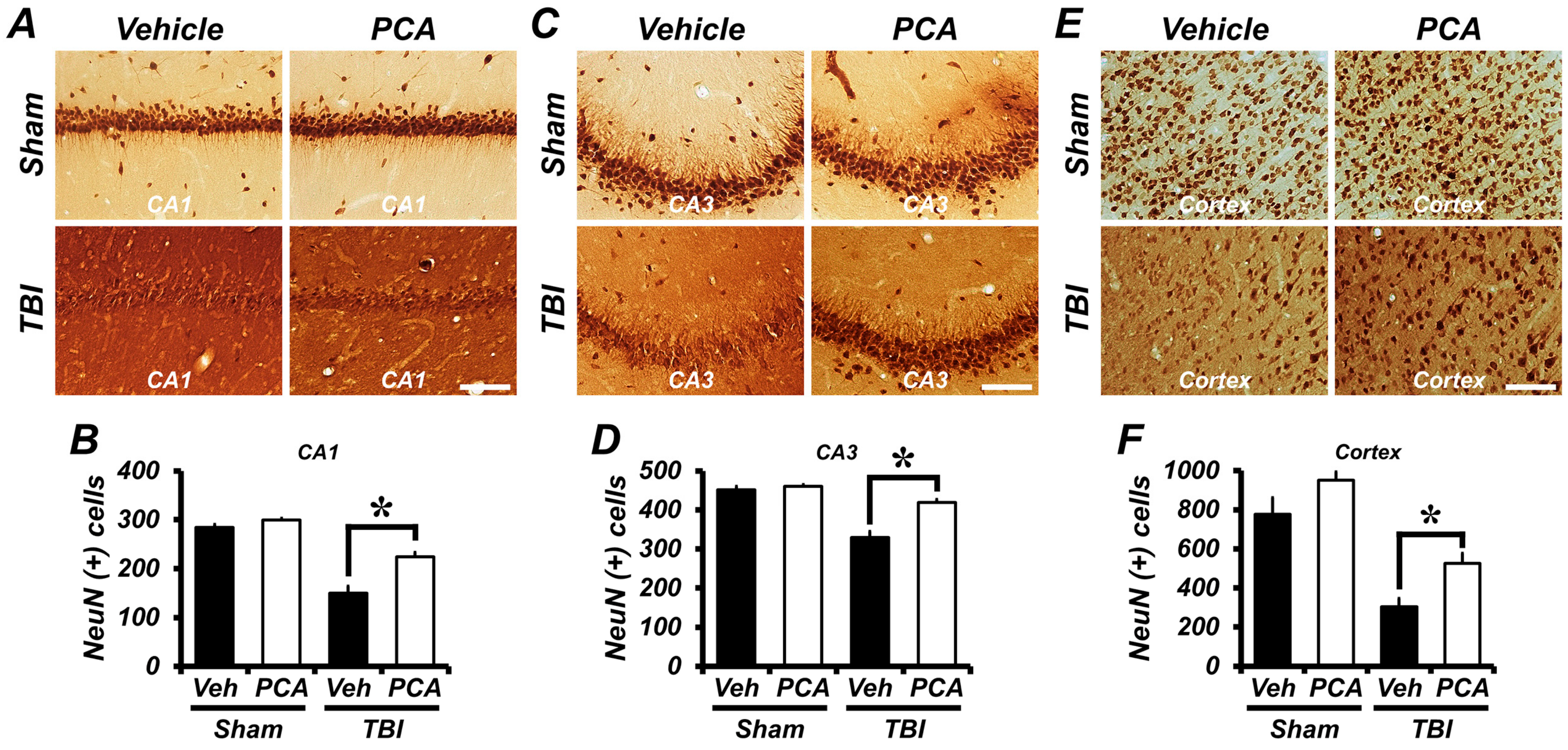
2.1. Protocatechuic Acid (PCA) Decreases the Number of Degenerating Neurons after Traumatic Brain Injury (TBI)
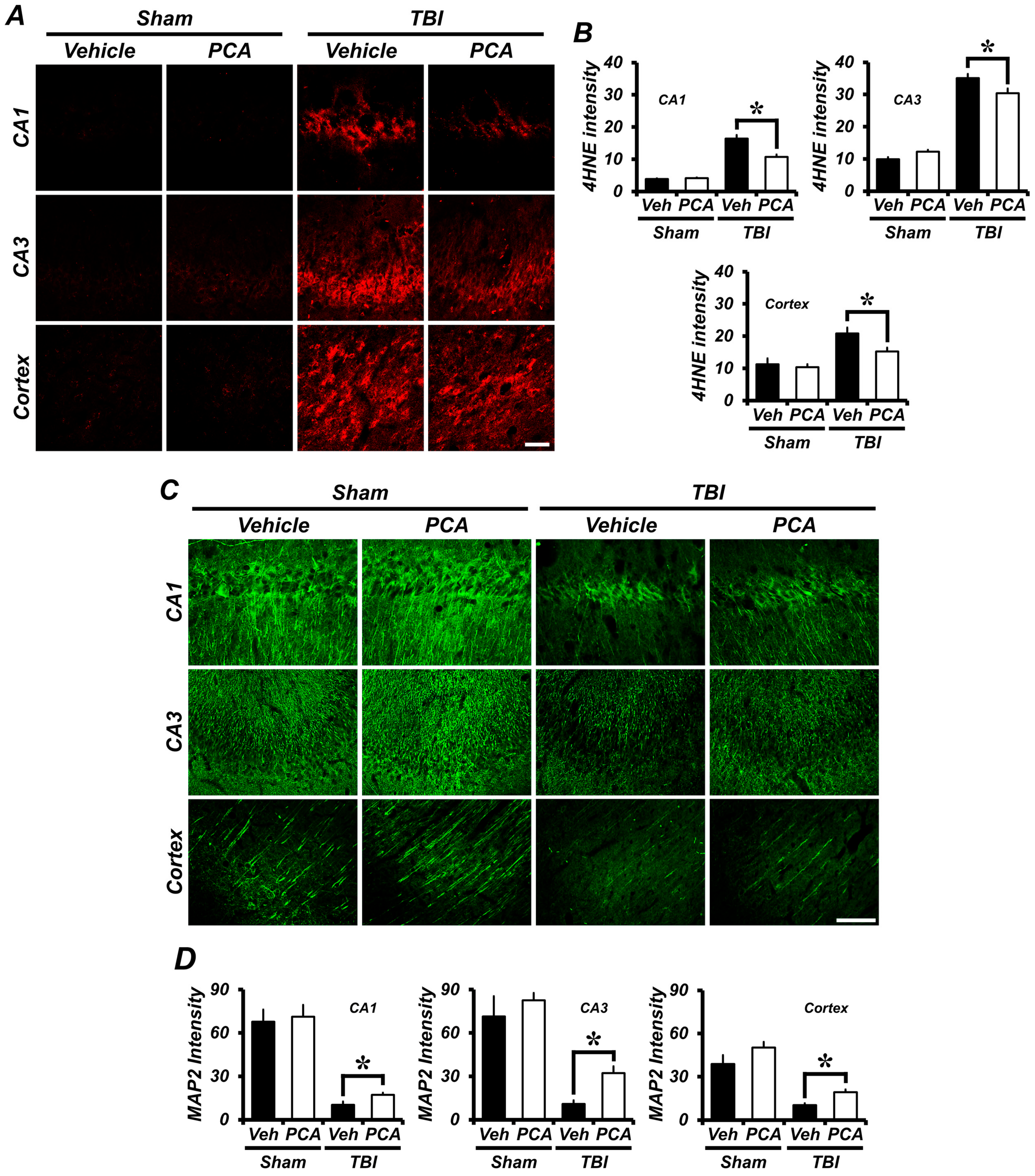
2.2. PCA Decreases TBI-Induced Oxidative Injury in the Hippocampus and Cortex
2.3. PCA Decreases TBI-Induced Dendritic Damage in the Hippocampus and Cortex
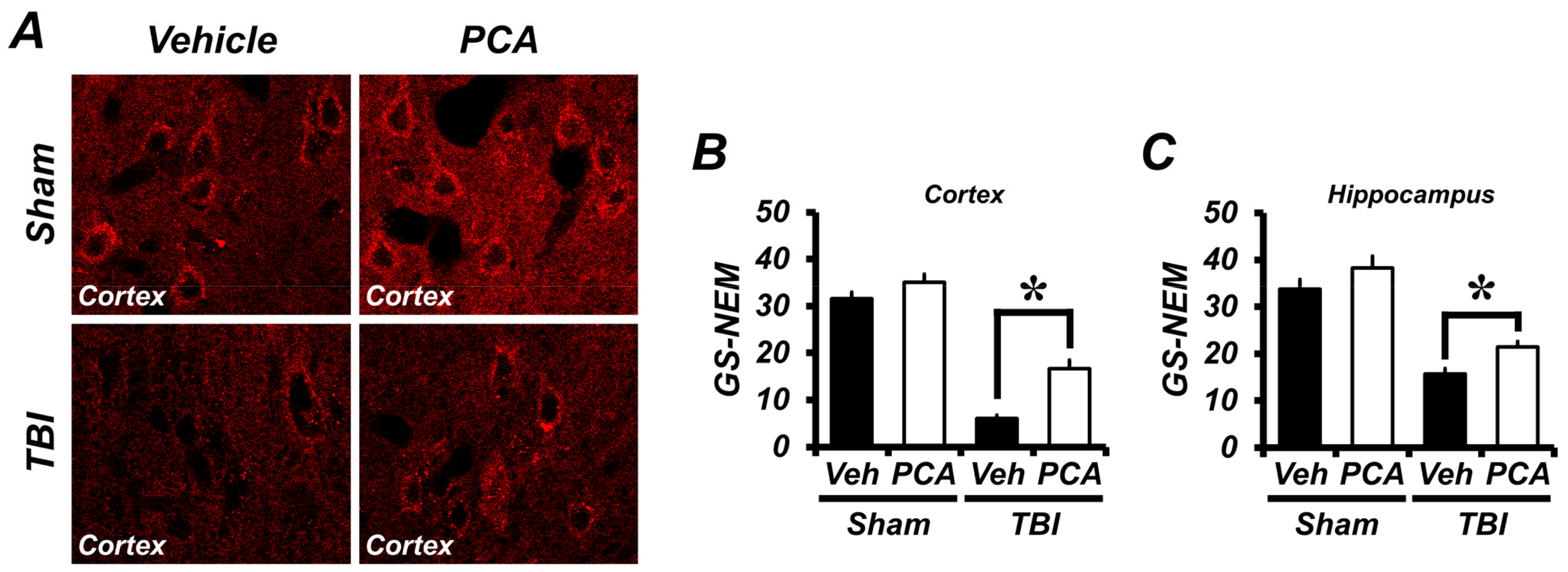
2.4. PCA Decreases TBI-Induced Glutathione (GSH) Depletion in the Hippocampus and Cortex
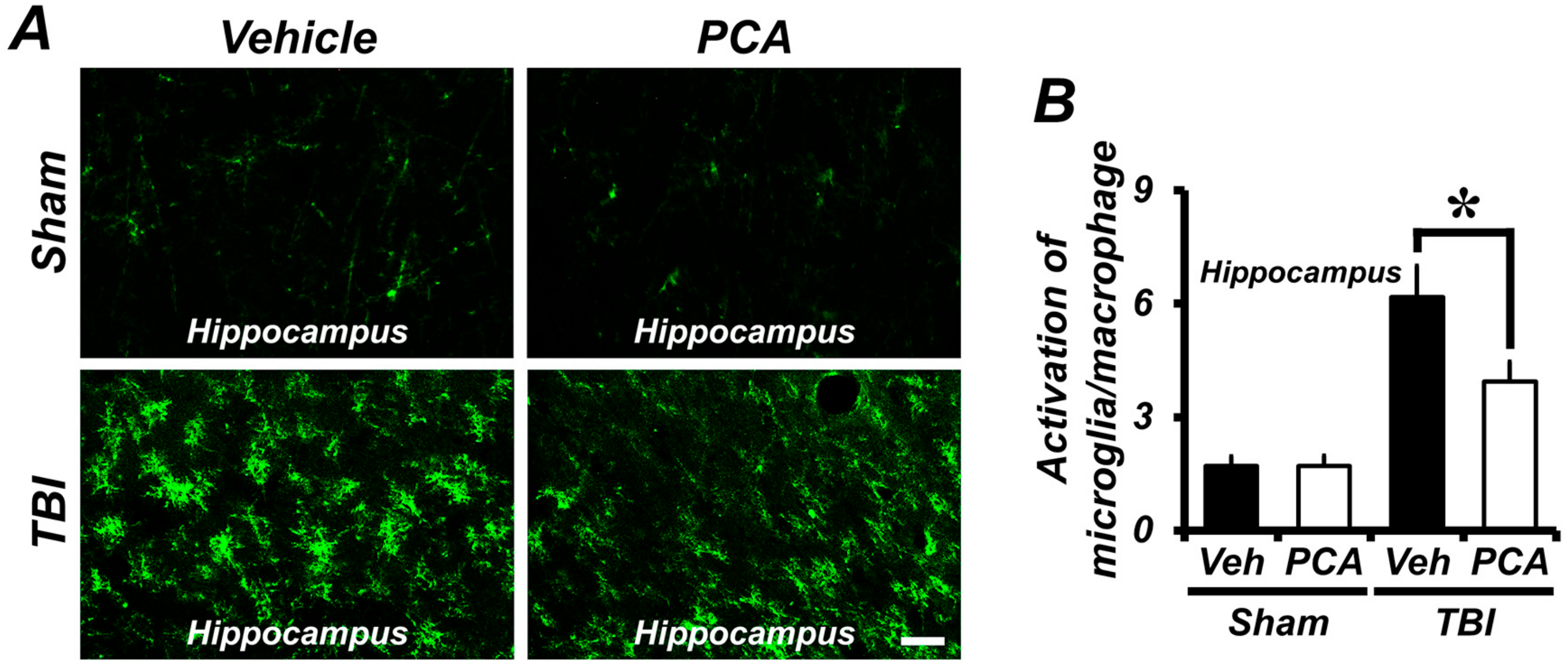
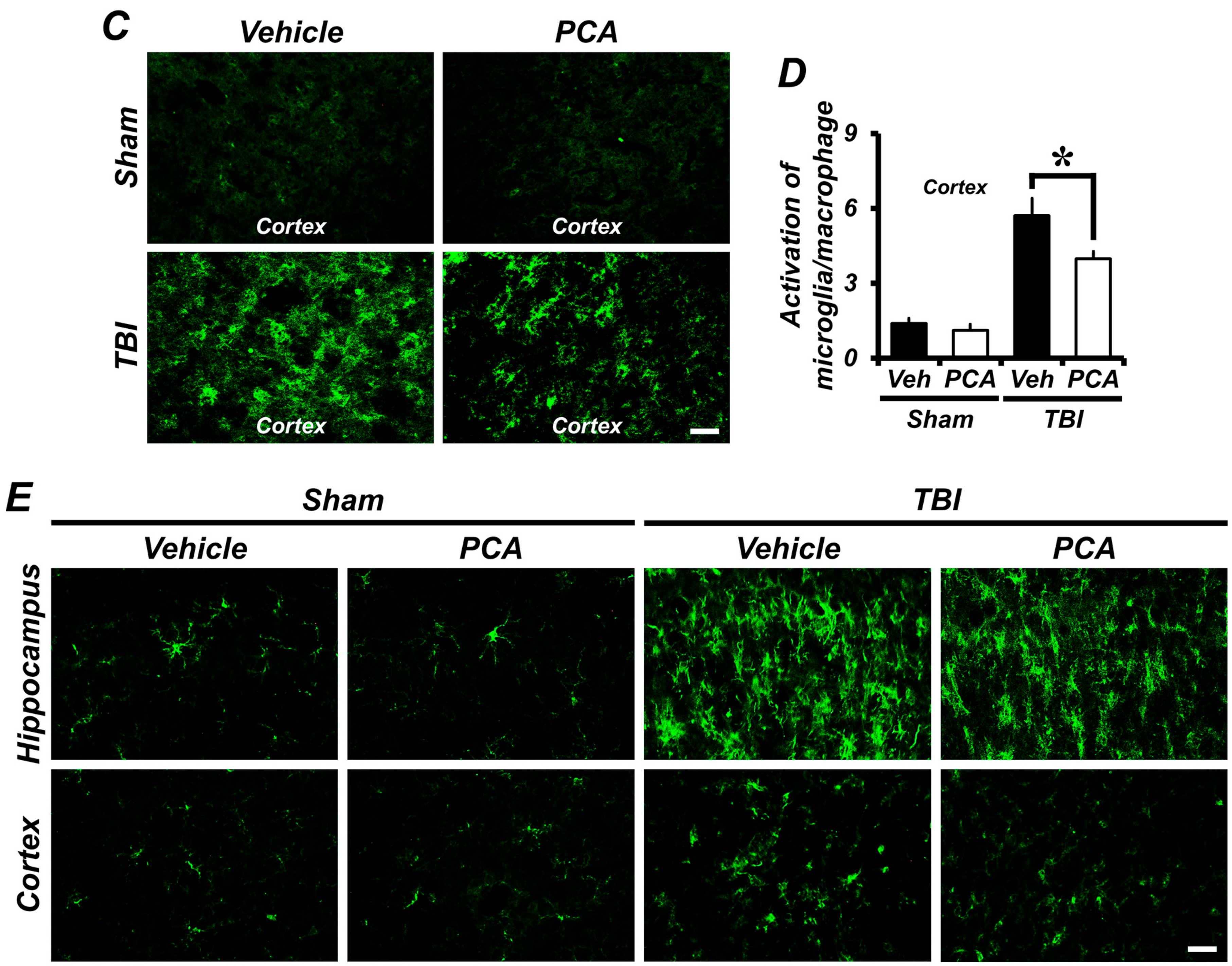
2.5. PCA Decreases TBI-Induced Microglial and Macrophage Activation
2.6. PCA Prevents Delayed Neuronal Death after TBI
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Controlled Cortical Impact Model for TBI
4.3. PCA Administration
4.4. Brain Section Groundwork
4.5. Evaluation of Neuronal Death
4.6. Detection of Oxidative Injury and Dendritic Damage
4.7. Determination of Glutathione Concentration
4.8. Evaluation of Microglia and Macrophage Activation
4.9. Detection of Live Neurons
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parikh, S.; Koch, M.; Narayan, R.K. Traumatic brain injury. Int. Anesthesiol. Clin. 2007, 45, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Singh, P. Missile injuries of the brain: Results of less aggressive surgery. Neurol. India 2003, 51, 215–219. [Google Scholar] [PubMed]
- Hall, E.D. Inhibition of lipid peroxidation in central nervous system trauma and ischemia. J. Neurol. Sci. 1995, 134 (Suppl.), 79–83. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Role of free radicals and catalytic metal ions in human disease: An overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar] [PubMed]
- Love, S. Oxidative stress in brain ischemia. Brain Pathol. 1999, 9, 119–131. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, T.K.; Juhler, M.; Wieloch, T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury: 1998. J. Neurotrauma 1998, 15, 731–769. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Agardh, C.D.; Bengtsson, F. Free radicals and brain damage. Cerebrovasc. Brain Metab. Rev. 1989, 1, 165–211. [Google Scholar] [PubMed]
- Smith, S.L.; Andrus, P.K.; Zhang, J.R.; Hall, E.D. Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 1994, 11, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Lewen, A.; Hillered, L. Involvement of reactive oxygen species in membrane phospholipid breakdown and energy perturbation after traumatic brain injury in the rat. J. Neurotrauma 1998, 15, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Marklund, N.; Clausen, F.; McIntosh, T.K.; Hillered, L. Free radical scavenger posttreatment improves functional and morphological outcome after fluid percussion injury in the rat. J. Neurotrauma 2001, 18, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Althaus, J.S.; Andrus, P.K.; Williams, C.M.; VonVoigtlander, P.F.; Cazers, A.R.; Hall, E.D. The use of salicylate hydroxylation to detect hydroxyl radical generation in ischemic and traumatic brain injury. Reversal by tirilazad mesylate (U-74006F). Mol. Chem. Neuropathol. 1993, 20, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Sakata, S.; Ikeda, M. Direct detection of ascorbyl radical in experimental brain injury: Microdialysis and an electron spin resonance spectroscopic study. J. Neurochem. 1995, 65, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Kontos, H.A.; Wei, E.P. Superoxide production in experimental brain injury. J. Neurosurg. 1986, 64, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Sun, A.Y. Oxidative mechanisms involved in kainate-induced cytotoxicity in cortical neurons. Neurochem. Res. 1994, 19, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Giampietro, D.E.; Cherici, G.; Alesiani, M.; Carla, V.; Moroni, F. Excitatory amino acid release and free radical formation may cooperate in the genesis of ischemia-induced neuronal damage. J. Neurosci. 1990, 10, 1035–1041. [Google Scholar] [PubMed]
- Chao, C.Y.; Yin, M.C. Antibacterial effects of roselle calyx extracts and protocatechuic acid in ground beef and apple juice. Foodborne Pathog. Dis. 2009, 6, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, J.; Han, W.; Mai, W.; Wang, L.; Li, Q.; Lin, M.; Bai, M.; Zhang, L.; Chen, D. Antioxidant ability and mechanism of rhizoma Atractylodes macrocephala. Molecules 2012, 17, 13457–13472. [Google Scholar] [CrossRef] [PubMed]
- Scazzocchio, B.; Vari, R.; Filesi, C.; D’Archivio, M.; Santangelo, C.; Giovannini, C.; Iacovelli, A.; Silecchia, G.; Li Volti, G.; Galvano, F.; et al. Cyanidin-3-O-β-glucoside and protocatechuic acid exert insulin-like effects by upregulating PPARgamma activity in human omental adipocytes. Diabetes 2011, 60, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.F.; An, L.J.; Jiang, B.; Guan, S.; Bao, Y.M. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jiang, W.; Zhu, H.; Hou, J. Antifibrotic effects of protocatechuic aldehyde on experimental liver fibrosis. Pharm. Biol. 2012, 50, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Lende, A.B.; Kshirsagar, A.D.; Deshpande, A.D.; Muley, M.M.; Patil, R.R.; Bafna, P.A.; Naik, S.R. Anti-inflammatory and analgesic activity of protocatechuic acid in rats and mice. Inflammopharmacology 2011, 19, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, O.; Disli, O.M.; Timurkaan, N. Protective effects of protocatechuic acid on TCDD-induced oxidative and histopathological damage in the heart tissue of rats. Toxicol. Ind. Health 2013, 29, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pan, X.; Wang, D.; Sun, H.; Han, F.; Lv, C.; Zhang, X. Protective effects of protocatechuic acid on retinal ganglion cells from oxidative damage induced by H2O2. Neurol. Res. 2015, 37, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.N.; Brenner, M.C.; Punessen, N.; Snodgrass, M.; Byars, C.; Arora, Y.; Linseman, D.A. Comparison of the neuroprotective and anti-inflammatory effects of the anthocyanin metabolites, protocatechuic acid and 4-hydroxybenzoic acid. Oxid. Med. Cell. Longev. 2017, 2017, 6297080. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zhang, X.; Lv, C.; Li, C.; Yu, Y.; Wang, X.; Han, F. Protocatechuic acid ameliorates neurocognitive functions impairment induced by chronic intermittent hypoxia. Sci. Rep. 2015, 5, 14507. [Google Scholar] [CrossRef] [PubMed]
- Krzysztoforska, K.; Mirowska-Guzel, D.; Widy-Tyszkiewicz, E. Pharmacological effects of protocatechuic acid and its therapeutic potential in neurodegenerative diseases: Review on the basis of in vitro and in vivo studies in rodents and humans. Nutr. Neurosci. 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Ge, D.; Liu, T.Q.; Ma, X.H.; Cui, Z.F. Protocatechuic acid promotes cell proliferation and reduces basal apoptosis in cultured neural stem cells. Toxicol. In Vitro 2009, 23, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Chu, X.; Guan, M.; Yang, X.; Xie, X.; Liu, F.; Chen, C.; Deng, X. Protocatechuic acid suppresses ovalbumin-induced airway inflammation in a mouse allergic asthma model. Int. Immunopharmacol. 2013, 15, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; An, C.N.; Xu, M.; Guo, D.A.; Li, M.; Pu, X.P. Protocatechuic acid inhibits rat pheochromocytoma cell damage induced by a dopaminergic neurotoxin. Biol. Pharm. Bull. 2009, 32, 1866–1869. [Google Scholar] [CrossRef] [PubMed]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Klein, J.A.; Ackerman, S.L. Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Investig. 2003, 111, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, A.; Li, L.; Huang, Y.; Xue, P.; Hao, A. Oxidative stress mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Seizure 2010, 19, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef]
- Lee, H.M.; Seo, J.H.; Kwak, M.K.; Kang, S.O. Methylglyoxal upregulates Dictyostelium discoideum slug migration by triggering glutathione reductase and methylglyoxal reductase activity. Int. J. Biochem. Cell Biol. 2017, 90, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Wang, H.; Wang, J.H.; Wang, Q.; Ma, Q.F.; Chen, Y.Y. Protocatechuic acid inhibits inflammatory responses in LPS-stimulated BV2 microglia via NF-κB and MAPKs signaling pathways. Neurochem. Res. 2015, 40, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.S.; Schiding, J.K.; Kaczorowski, S.L.; Marion, D.W.; Kochanek, P.M. Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight drop and controlled cortical impact models. J. Neurotrauma 1994, 11, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Suh, S.W.; Frederickson, C.J.; Danscher, G. Neurotoxic zinc translocation into hippocampal neurons is inhibited by hypothermia and is aggravated by hyperthermia after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2006, 26, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Mouzon, B.; Chaytow, H.; Crynen, G.; Bachmeier, C.; Stewart, J.; Mullan, M.; Stewart, W.; Crawford, F. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J. Neurotrauma 2012, 29, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Suh, S.W.; Aoyama, K.; Chen, Y.; Garnier, P.; Matsumori, Y.; Gum, E.; Liu, J.; Swanson, R.A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003, 23, 10681–10690. [Google Scholar] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.N.; Berman, A.E.; Swanson, R.A.; Yenari, M.A. Digitally quantifying cerebral hemorrhage using Photoshop and Image J. J. Neurosci. Methods 2010, 190, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Hong, D.K.; Suh, S.W. ZnT3 Gene deletion reduces colchicine-induced dentate granule cell degeneration. Int. J. Mol. Sci. 2017, 18, 2189. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Yoo, J.H.; Song, H.K.; Sohn, M.; Won, S.J.; Suh, S.W. Pyruvate administration reduces recurrent/moderate hypoglycemia-induced cortical neuron death in diabetic rats. PLoS ONE 2013, 8, e81523. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. EAAC1 gene deletion increases neuronal death and blood brain barrier disruption after transient cerebral ischemia in female mice. Int. J. Mol. Sci. 2014, 15, 19444–19457. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. https://doi.org/10.3390/ijms18122510
Lee SH, Choi BY, Lee SH, Kho AR, Jeong JH, Hong DK, Suh SW. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. International Journal of Molecular Sciences. 2017; 18(12):2510. https://doi.org/10.3390/ijms18122510
Chicago/Turabian StyleLee, Sang Hwon, Bo Young Choi, Song Hee Lee, A. Ra Kho, Jeong Hyun Jeong, Dae Ki Hong, and Sang Won Suh. 2017. "Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death" International Journal of Molecular Sciences 18, no. 12: 2510. https://doi.org/10.3390/ijms18122510
APA StyleLee, S. H., Choi, B. Y., Lee, S. H., Kho, A. R., Jeong, J. H., Hong, D. K., & Suh, S. W. (2017). Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. International Journal of Molecular Sciences, 18(12), 2510. https://doi.org/10.3390/ijms18122510