Abstract
Moyamoya angiopathy (MA) is a cerebrovascular disease determining a progressive stenosis of the terminal part of the internal carotid arteries (ICAs) and their proximal branches and the compensatory development of abnormal “moyamoya” vessels. MA occurs as an isolated cerebral angiopathy (so-called moyamoya disease) or in association with various conditions (moyamoya syndromes) including several heritable conditions such as Down syndrome, neurofibromatosis type 1 and other genomic defects. Although the mechanism that links MA to these genetic syndromes is still unclear, it is believed that the involved genes may contribute to the disease susceptibility. Herein, we describe the case of a 43 years old woman with bilateral MA and peculiar facial characteristics, having a 484-kb microduplication of the chromosomal region 15q13.3 and a previously unreported 786 kb microdeletion in 18q21.32. This patient may have a newly-recognized genetic syndrome associated with MA. Although the relationship between these genetic variants and MA is unclear, our report would contribute to widening the genetic scenario of MA, in which not only genic mutation, but also genome unbalances are possible candidate susceptibility factors.
1. Introduction
Moyamoya angiopathy (MA) is a rare (prevalence close to 6/100,000 in Japan, around ten-times less in Europe) cerebrovascular disease characterized by a progressive stenosis of the terminal part of the supraclinoid internal carotid arteries (ICAs) and their proximal branches, resulting in a compensatory development of fragile basal collateral vessels (moyamoya vessels) [1,2]. This condition leads to a severe hemodynamic impairment, conditioning ischemic strokes and cerebral hemorrhages, which are the most important disease clinical features. MA is commonly classified in moyamoya disease (MMD) when MA is bilateral, which is the idiopathic disease form, or moyamoya syndrome (MMS), or pseudo-moyamoya, or quasi-moyamoya when MA is associated with a series of well-known acquired (i.e., head irradiation, autoimmune disorders) or constitutional (genomic unbalances or mutations) conditions. Several genetic syndromes such as sickle cell disease, neurofibromatosis type 1, and Down, Noonan, Costello, and Alagille syndromes have been associated with MA [3,4].
The etiology of MA is still unknown. Although the detection of increased cytokine and growth factor concentration would support the role of impaired angiogenic and arteriogenic mechanism, genetic factors have long been considered, given the high familial rate (10–15%) and the ethnic differences [4,5,6]. The inheritance pattern and the involved genes are mostly unknown. RNF213 gene encoding for an enzyme with a Really Interesting New Gene (RING) finger and AAAA+ Adenosine TriPhosphate (ATP)ase domain is the only gene identified as conferring susceptibility to MA in Asian countries [7,8]. However, although missense mutations in this gene have been recently significantly associated with MA European patients, particularly in childhood-onset and familial cases [7,8], the causative role of this gene in Caucasians is still debated [9]. Recently, familial cases of MA associated with peculiar facial characteristics have been identified by copy number analysis, detecting microdeletion in Xq28 involving the BRCC3 gene that encodes a 36-kDa ubiquitous deubiquitinating enzyme [10]. Risk of MA disease has been also attributed to mutations in GUCY1A3 gene, encoding the major nitric oxide receptor in vascular smooth muscle cells (vSMCs) in achalasia cases [11]. Other sporadic syndromic cases of MA have been reported, as resumed in Table 1 [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33]. However, these observations are not able to fully explain the pathogenesis of MA, which is believed to be much more complex. A mechanism including the role of multiple genes and angiogenic abnormalities, in which unknown triggering environmental factors (including infections, immune failure, and hemodynamic stress to specific vascular loci), has been postulated for starting the first pathological changes of the disease [5,6].

Table 1.
Sporadic syndromic cases of moyamoya angiopathy (MA).
Herein, we describe the case of a 43 years old woman with bilateral MA and peculiar facial characteristics, belonging to the population of the GENetics of mOyaMoyA (GENOMA) project, screened for copy number imbalances as suggested by the syndromic phenotype, resulting in a 484-kb microduplication of the chromosomal region 15q13.3, previously associated with clinical phenotype having incomplete penetrance [34] and a previously unreported 786-kb microdeletion in 18q21.32. This patient may have a newly-recognized genetic syndrome associated with MA.
2. Case Report
A 43 years old woman, born from non-consanguineous parents and with no documented developmental structural or mental defects, presented in February 2014 a progressive dysarthria, motor slowness, and changes of behavior, becoming listless and aggressive. In April 2014 she presented a sudden aphasia and right hemiparesis for which she was hospitalized elsewhere. The acute phase cerebral Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) scan showed acute cortico-subcortical left fronto-parietal and left internal capsule acute lesions. Cerebral Computed Tomography Angiography (CTA) scan detected a bilateral stenosis of internal carotid arteries (ICAs), middle cerebral arteries (MCAs), and anterior cerebral arteries (ACAs). Coagulation and autoimmunity screening were normal, as well as electrocardiography (ECG) monitoring and echocardiography. Aspirin therapy was introduced, and the patient was addressed to a rehabilitation course. After an initial improvement of the right hemiparesis, her clinical status worsened, and she developed a vertical ophthalmoparesis and dystonic movements in the left arm. For these symptoms, a biochemistry examination was performed, which resulted in being normal, as well as an abdominal ultrasound examination showing liver steatosis. A new cerebral MRI did not show any changes and detected the previously observed left frontal and insular lesions, as well as new right frontal and parietal lesions. Cerebral MRA confirmed the severe stenosis of the ICAs, MCAs, and ACAs, mostly in the left side (Figure 1). Thus, a suspicion of moyamoya angiopathy was posed, and in June 2015, she was addressed to our center. The neurological evaluation, performed at that time, showed the presence of severe attention deficit, orofacial apraxia, dysarthria, slight left hemianopia, and right hemiparesis with bilateral coreoatetosic movements and Babinski sign. Peculiar characteristics were a long narrowing face with bifrontal prominence, a low frontal hairline, arched eyebrows, downslanting palpebral fissures, hypertelorism, broad nasal tip, upturned nares, and large and low-set ears. Digital subtraction angiography (DSA) revealed stenosis of the ICAs with apex occlusion in the right one and hypertrophic aspects of lenticulostriate arteries with leptomeningeal anastomosis from ophthalmic artery and frontal artery to provide blood flow to the frontal regions. Blood flow in the temporal, parietal, and occipital regions was supported by posterior cerebral artery. Electroencephalography revealed left temporal epileptic activity. Perfusion MRI showed a decreased relative cerebral blood flow (rCBF) and cerebral blood volume (rCBV) into ischemic areas, as well as a bilateral increased time to peak (rTTP) and mean transit time (rMTT). A diagnosis of MA was confirmed, and given the somatic peculiar characteristics and the cognitive impairment associated with the MA, the patient underwent an array-comparative genomic hybridization (aCGH), after informed consent.
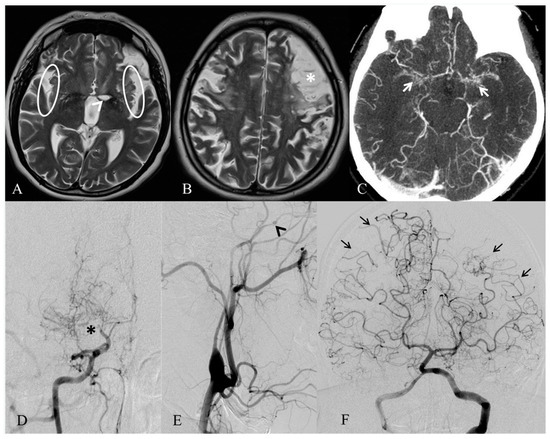
Figure 1.
MRI and angiographic images of MA of our case. Axial T2-weighted MRI (A,B) exhibits only a few vascular voids in the Sylvian fissures (white circles), due to a reduced flow in both the middle cerebral arteries. A well-demarcated ischemic stroke in the left nucleo-capsular region (white arrowhead) and an infarct involving the left frontal lobe (white asterisk) are also noticeable. Axial angio-CT (C) shows proliferation and enlargement of the lenticulostriate arteries (white arrows) in the basal ganglia. Right and left common carotid arteries’ angiograms (D–E) demonstrate a distal stenosis of the right internal carotid artery (black asterisk), with development of prominent collateral vessels, giving the characteristic puff of smoke appearance of moyamoya disease, and an occlusion of the left internal carotid artery, distally to the ophthalmic artery (black arrowhead). Left vertebral artery angiogram (F) indicates leptomeningeal cortical anastomosis from both the posterior cerebral arteries to the parietal and temporal lobes (black arrows).
In September 2016 and in May 2017, the patient underwent a direct (STA-MCA) and indirect revascularization (encephalo-duro-arterio-synangiosis (EDAS)) surgery in the left and right hemispheres, respectively. The post-surgical clinical course was regular, and the post-surgery DSA confirmed the by-pass perviety. A few months later, she presented focal seizures for which she performed a new electroencephalogram showing left parietotemporal sharp waves and started treatment with levetiracetam, with complete resolution of seizures. At the two-year follow-up evaluation, she did not refer to any other symptoms, and we did not detect any other signs of cerebral or systemic involvement.
3. Results
The aCGH analysis of the patient revealed a 484-kb microduplication of the chromosomal region 15q13.3 (from the 32065077–32539670 chromosome coordinate), involving the genes OTUD7A, coding for Ovarian Tumor Domain (OTU) deubiquitinase 7A, and CHRNA7, coding for neuronal nicotinic cholinergic receptor alpha polypeptide 7, and a 786-kb microdeletion in the 18q21.32 (from 58014483–58816361), involving the gene MC4R, coding for melanocortin 4 receptor, and a regulatory element lincRNA MIR122 (Figure 2).
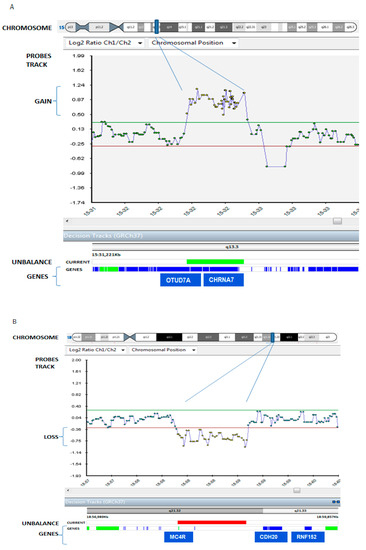
Figure 2.
Patient’s array comparative genomic hybridization (aCGH) data result. The patient carried: (panel A) a 484 Kb microduplication in the chromosomal region 15q13.3 (the blue bar in the chromosome track), from 32065077 to 32539670 chromosome coordinate, zoomed in the probes track as indicated by purple lines, and (panel B) a 786 Kb microdeletion in the 18q21.32 region (the blue bar in the chromosome), from 58014483 to 58816361 chromosome coordinate, zoomed in the probes track as indicated by purple lines. Duplication and deletion are revealed by a disclosure of at least 3 probes from the +0.4 (duplication)/−0.6 (deletion range) of Log2ratio value in X axis (normal values are included in the range from +0.30, green line, to −0.30, red line; probed duplicated and deleted in the patient are yellow in the figure, while green dots are balanced probes. In the track “unbalance”, the patient’s duplication in panel A and the patient’s deletion in panel B are indicated with a green and red bars respectively. Genes are represented as blue strips in the genes truck, and genes associated with pathology are colored in green.
Array-CGH data of the patient showed a 484-kb microduplication of the chromosomal region 15q13.3, from the 32065077–32539670 chromosome coordinate (Panel A), and a 786-kb microdeletion in the 18q21.32 region, from 58014483–58816361 (Panel B). Duplication and deletion were revealed by a disclosure of at least three probes from the −0.6 (deletion)/+0.4 (duplication) range of the Log2 ratio value in the X axis (probed duplicated, and deleted in the patient are yellow in the figure).
Both unbalances resulted by being inherited from the mother, who did not have neurovascular signs, but looked similar to the proband (Figure 3) and presenting less dysmorphic features than her daughter (low frontal hairline and hypertelorism were not present in the mother). The 15q13.3 microduplication was detected also in the healthy patient’s brother and sister. We did not perform the aCGH analysis on the 13 y.o patient’s son since he was underage. This unbalance is a common variant with a described low penetrance in neuropsychiatric disorders [34,35,36,37], and it has never been previously reported to be associated with vascular diseases. The microdeletion 18q21.32 was rarely observed and not reported in the databases of normal variants. The patient’s siblings did not have this unbalance and were not phenotypically similar to the patient. We analyzed the mRNA expression of MC4R and mir-122, located in the deleted region, and RNF152, which is located distal to the deletion, in position (59475296_59561480) of chromosome 18. mRNA expression of these three genes was detectable, but not significantly different in the patient compared to her relatives (parents, one brother, and one sister) and to the pool of healthy controls (data not shown).
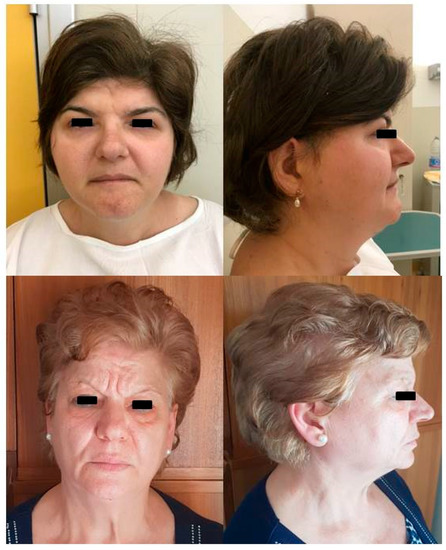
Figure 3.
Proband and proband’s mother’s photos. Front and profile photos of the proband (upper panels) and her mother (lower panels). The proband shows peculiar characteristics such as a long narrowing face with bifrontal prominence, low frontal hairline, arched eyebrows, downslanting palpebral fissures, hypertelorism, broad nasal tip, upturned nares, and large and low-set ears. The mother is similar, but not having low frontal hairline and hypertelorism.
4. Discussion
Herein, we describe the first MA patient having a microduplication of the chromosomal region 15q13.3 and a microdeletion in the 18q21.32. Our patient, other than the typical moyamoya neuroradiological features, has some peculiar facial characteristics such as a long narrowing face with bifrontal prominence, low frontal hairline, arched eyebrows, downslanting palpebral fissures, hypertelorism, broad nasal tip, upturned nares, and large and low-set ears. Facial dysmorphism, which commonly includes a combination of different facial features, is a well-known characteristic of many genetic syndromes and has been described in moyamoya syndromic cases [4,38]. The phenotypic presentation and the genomic unbalances observed in our patient supported the diagnosis of moyamoya syndrome, rather than moyamoya disease. Microduplications of 15q13.3 ranging from 350 kb–1.6 Mb involving OTUD7A and CHRNA7 have been previously described, with various breakpoints within the 15q13.3 region. OTUD7A encodes for out deubiquitinase 7A, a protein expressed mainly in the central nervous system and having a supposed tumor suppressor role. CHRNA7 encodes the α7 subunit of the neuronal nicotinic acetylcholine receptor (nAChR), which is composed of five subunits, arranged in a barrel-like configuration in the cell membrane. The subunits exist as 16 isoforms (α1–α10, β1–β4, δ, γ, and ε), which may be arranged in homomeric or heteromeric configurations. Each subunit contains four domains (M1–M4) in the cell membrane, with the M2 transmembrane domain of each subunit contributing to the channel of the receptor. Combinatorial association of various α- and β-subunits result in functionally-diverse nAChR subtypes that vary in ion permeability, open time, ligand affinity, and other functions [36,37,39]. Given the low estimation of penetrance, the pathogenicity of 15q13.3 duplication encompassing OTUD7A and CHRNA7 has been associated with a highly variable intra- and inter-familial phenotype. Patients with such microduplication exhibit various clinical symptoms varying from asymptomatic or mild developmental delay and learning problems during childhood to mental retardation, autism, muscular hypotonia, and a variety of neuropsychiatric disorders, including seizures [34,35,37]. Although MA has never been described as an associated finding, we cannot completely exclude a possible role of such a microduplication in endothelial dysfunction, since in the endothelial cells, the α7 homomeric nAChR has been observed to act as a mediator of cholinergic angiogenesis and to be upregulated in in vitro and in vivo ischemic experimental conditions [39]. However, since this variant was present also in the proband’s healthy relatives, it is probably benign or has incomplete penetrance.
More interestingly, the patient carried also a microdeletion of 18q21.32, in the site of the MC4R (melanocortin-4 receptor) gene. Haploinsufficiency of the MC4R has been described as the most common heritable obesity syndrome in man and is responsible for 0.5–2.5% of all early onset morbid obesity [40]. Moreover, MC4R has been found to be expressed most heavily in the central nervous system, where it plays a critical role in energy balance [41]. MC4R-expressing neurons within the hypothalamus receive orexigenic and anorexigenic inputs from arcuate nucleus pro-opiomelanocortin POMC and agouti-related protein-expressing neurons AgRP projections and act to maintain energy homeostasis through modulation of both food intake and energy expenditure [42,43].
However, MC4R has been reported to be involved also in angiogenic balance, endothelial function, and blood pressure control. MC4R+/− obese pregnant rats were observed to have a pro-angiogenic phenotype with circulating VEGF greater that MC4R+/+ pregnant rats [42,43].
Notably, the deleted region included a non-coding microRNA (miRNA), mir-122, which is considered as a potential biomarker in acute liver failure. Because miRNAs are regulator of mRNA expression, mir-122 could influence the expression of genes located nearby: we noticed a gene of the ring finger protein (RNF) family, RNF152, close to the above-mentioned microdeletion region. RNF152 is an E3 ligase and is self-polyubiquitinated. It is localized in lysosomes and displays a pro-apoptosis activity when overexpressed [44]. Given the well-known association between RNF213 and MA, we speculated on a possible role of this ring finger protein, RNF152, in the development of MA in our patient. Since this variant was not included in the deleted region, we hypothesized that deletion of a regulatory element, such as mir-122, may influence the expression of the flanking gene in a tissue-specific manner and with variable penetrance and expressivity. In fact, 18q21.32 microdeletion was carried also by the mother of our patient, who did not present MA. For this reason, we analyzed the expression of mir-122 and RNF152 to evaluate a supposed mutual modulation. Unfortunately, the expression data did not show any differences in RNF152 mRNA expression comparing our patient with relatives or controls.
Although we cannot exclude a different way of regulation or a tissue-dependent expression variability of mir-122, MC4R and RNF152, which could have a different behavior in the central nervous system, we believe that the microdeletion or microduplication might be primarily responsible for the disease susceptibility,. Other modifiers, genetic and environmental factors could ultimately trigger the phenotypic presentation, explaining the variable phenotypes within the patient and her mother. An alternative hypothesis is that these loci are the modifiers for other yet-to-be-identified mutations in the genome. Variability in these other factors, among individuals with the same microdeletion, may explain their varying phenotypes.
Although our results should be confirmed in other families carrying the same genomic microdeletions or duplications, we hope that this report would open a new genetic scenario leading to the search and possibly identification of genetic factors predisposing to MA disease, not only genic mutation, but also genome unbalances being candidate susceptibility factors.
5. Materials and Methods
Array CGH (aCGH) was performed on DNA from peripheral blood. Blood samples for DNA extraction were collected from the patient and her family, including parents, one brother, and one sister, after informed consent. Array-CGH analyses were performed using the oligo ISCA180K platform (BlueGnome array by Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer’s protocol. Data were analyzed using Bluefuse software (BlueGnome, Cambridge, U.K.). Clinical interpretation of array CGH results were based on published literature and public databases such as ENSEMBL (www.ensambl.org), UCSC (genome.uscs.edu), Database for Genetic Variants (dgv.tcag.ca), DECIPHER (decipher.sanger.ac.uk) and the Italian database of Troina, (gvarianti.homelinux.net) following Cytogenetic European and International Guidelines [45,46]. Total RNA was isolated from lymphocytes of peripheral blood (from 3 independent blood collections) of the patient’s family (parents, one brother, and one sister) and 3 unrelated healthy controls (3 age- and sex-matched to the proband and with normal CGH analysis), using the RNeasy® Mini Kit (Qiagen, Hilden, Germany, UE). mRNA expressions of MC4R, MIR122, and RNF152 were studied by real-time PCR with SYBR green standard procedures (Applied Biosystem, Foster City, CA, USA), using the CFX96 system (Bio-Rad, Hercules, CA, USA) and analyzed by Bio-Rad CFX Manager software (www.bio-rad.com/en-eh/product/cfx-manager-software?ID=aed9803-cb4d-4ecb-9263-3efc9e650edb). Specific target sequences were selected for real-time PCR using Primer 3 software (http://frodo.wi.mit.edu/primer3/), analyzing two regions for each gene. The sequences of primers used (Metabion, International AG) were as follows: MC4R (forward 5′-CCAGTGAGTCCCTTGGAAAA, reverse 5′-CTTGGCTATTGCCACAATCA; forward 5′-CCCCATTCTTCCTCCACTTA, reverse 5′-TCCGGAGTGCATAAATCAGA), mir122 (forward 5′-CTGAAAGGCCATAGCAGGAG, reverse 5′-CTGCCACCACTGTCAACAAG; forward 5′-GTAGGTGCCCTTCGGTACAA, reverse 5′-CTTTCTGCCCAGGAACTGTC) and RNF152 (forward 5′-TCTCCAGCCCTAGAAGTTGC, reverse 5′-CCTTCACAGCGCTCTCTAATC; forward 5′, AGCACACCTGCTGTTCAGTG, reverse 5′-CTTCTGGCTGGTCCTCATCT). A control amplicon was selected with the same parameters in the MAPK1 gene (forward 5′-CTAACGTTCTGCACCGTGAC, reverse 5′-GTGGTGTTGAGCAGCAGGTT) on 22q11.2; size (approximately 60 bp) and the melting temperature (60°C) were the same for all amplicons. Genomic coordinates herein are based on the February 2009 Human Genome Build (GRCh37/hg19).
The patient belongs to the MA cohort patients of the GENetics of mOyaMoyA (GENOMA) project, for which Ethical Committee approval was obtained (Regione Lombardia Ethical Committee, Fondazione Istituto di Ricovero e Cura a Carattere Scientifico IRCCS Neurologico Carlo Besta, n.12, October 1th, 2014). Specific consent was obtained from the patient and her relatives for clinical data, pictures, and DNA obtainment.
Author Contributions
F.L.S. performed the genetic analysis and drafted the manuscript. A.R. contributed to genetic analysis. J.V.G., E.A.P., G.B. and S.N. contributed to the paper drafting. F.C. and V.D.L. managed the patient and revised the paper. F.A. and P.F. managed the patient and performed the revascularization surgery. D.S.R. and S.B. performed the neurophysiological evaluations. G.F., A.G., M.G. and M.G.B. performed and evaluated the neuroimaging. C.P. and L.C. contributed to the patient management. A.B. conceived of the paper, managed the patient, and drafted the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors wish to thank the patient and her family for their cooperation. This patient belongs to the GENetics of mOyaMoyA (GEN-O-MA) project, which is a multicenter cohort study implemented in Italy for moyamoya disease study.
Conflicts of Interest
The authors have no conflict of interest.
References
- Kudo, T. Spontaneous occlusion of the circle of Willis. A disease apparently confined to Japanese. Neurology 1968, 18, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Takaku, A. Cerebrovascular ‘moyamoya’ disease. Disease showing abnormal net-like vessels in base of brain. Arch. Neurol. 1969, 20, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Phi, J.H.; Wang, K.C.; Lee, J.Y.; Kim, S.K. Moyamoya syndrome: A window of moyamoya disease. J. Korean Neurosurg. Soc. 2015, 57, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Guey, S.; Tournier-Lasserve, E.; Hervé, D.; Kossorotoff, M. Moyamoya disease and syndromes: From genetics to clinical management. Appl. Clin. Genet. 2015, 16, 49–68. [Google Scholar]
- Bersano, A.; Guey, S.; Bedini, G.; Nava, S.; Hervé, D.; Vajkoczy, P.; Tatlisumak, T.; Sareela, M.; van der Zwan, A.; Klijn, C.J.; et al. European Moyamoya Disease Initiative. Research Progresses in Understanding the Pathophysiology of Moyamoya Disease. Cerebrovas. Dis. 2016, 41, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Bedini, G.; Blecharz, K.G.; Nava, S.; Vajkoczy, P.; Alessandri, G.; Ranieri, M.; Acerbi, F.; Ferroli, P.; Riva, D.; Esposito, S.; et al. Vasculogenic and Angiogenic Pathways in Moyamoya Disease. Curr. Med. Chem. 2016, 23, 315–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Touho, H.; Miyake, N.; Ohba, C.; Doi, H.; Saitsu, H.; Taguri, M.; Morita, S.; Matsumoto, N. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G>A variant in RNF213 showed varying clinical course and severity. J. Hum. Genet. 2012, 57, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Guey, S.; Kraemer, M.; Hervé, D.; Ludwig, T.; Kossorotoff, M.; Bergametti, F.; Schwitalla, J.C.; Choi, S.; Broseus, L.; Callebaut, I.; et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur. J. Hum. Genet. 2017, 25, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Miskinyte, S.; Butler, M.G.; Hervé, D.; Sarret, C.; Nicolino, M.; Petralia, J.D.; Bergametti, F.; Arnould, M.; Pham, V.N.; Gore, A.V.; et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am. J. Hum. Genet. 2011, 88, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Philippi, A.; Belbouab, R.; Zerah, M.; Chabrier, S.; Collardeau-Frachon, S.; Bergametti, F.; Essongue, A.; Berrou, E.; Krivosic, V.; et al. Loss of α1β1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am. J. Hum. Genet. 2014, 94, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Santoro, C.; Di Rocco, F.; Kossorotoff, M.; Zerah, M.; Boddaert, N.; Calmon, R.; Vidaud, D.; Cirillo, M.; Cinalli, G.; Mirone, G.; et al. Moyamoya syndrome in children with neurofibromatosis type 1: Italian-French experience. Am. J. Med. Genet. 2017, 173, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Ebisudani, Y.; Aoi, M.; Fukuhara, T. Adult-Onset Hemorrhagic Quasi-Moyamoya Disease with Unilateral Steno-occlusive Lesion in a Patient with Neurofibromatosis Type 1. J. Stroke Cerebrovas. Dis. 2018, 27, 1423–1424. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Kirkham, F.J. Noonan syndrome and moyamoya. Pediatr. Neurol. 1997, 16, 256–258. [Google Scholar] [CrossRef]
- Gupta, M.; Choudhri, O.A.; Feroze, A.H.; Do, H.M.; Grant, G.A.; Steinberg, G.K. Management of moyamoya syndrome in patients with Noonan syndrome. J. Clin. Neurosci. 2016, 28, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shiihara, T.; Kato, M.; Mitsuhashi, Y.; Hayasaka, K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr. Neurol. 2005, 32, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Soro, I.; Leitão, A.; Silva, M.L.; Leão, M. Moyamoya vascular pattern in Alagille syndrome. Pediatr. Neurol. 2012, 47, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Yokote, H.; Tsuura, M.; Nakai, K.; Ohshima, A.; Itakura, T. Marfan syndrome associated with moyamoya phenomenon and aortic dissection. Acta Neurochir. (Wien) 1999, 141, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Vinícius Ribeiro, E.; Silva, M.; Tavares Mendonça, F.; Garcia Dusi, R. Anesthetic management in a child with moya-moya disease and sickle cell anemia: Case report. Rev. Esp. Anestesiol. Reanim. 2016, 63, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Alamri, A.; Hever, P.; Cheserem, J.; Gradil, C.; Bassi, S.; Tolias, C.M. Encephaloduroateriosynangiosis (EDAS) in the management of Moyamoya syndrome in children with sickle cell disease. Br. J. Neurosurg. 2017, 15, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xin, B.; Jones, S.; Puffenberger, E.G.; Hinze, C.; Bright, A.; Tan, H.; Zhou, A.; Wu, G.; Vargus-Adams, J.; Agamanolis, D.; et al. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc. Natl. Acad. Sci. USA 2011, 108, 5372–5377. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Bernardi, B.; Stafa, A.; Garone, C.; Franzoni, E.; Abinun, M.; Mitchell, P.; Mitra, D.; Friswell, M.; Nelson, J.; et al. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev. Med. Child Neurol. 2010, 52, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Bober, M.B.; Khan, N.; Kaplan, J.; Lewis, K.; Feinstein, J.A.; Scott, C.I., Jr.; Steinberg, G.K. Majewski osteodysplastic primordial dwarfism type II (MOPD II): Expanding the vascular phenotype. Am. J. Med. Genet. A 2010, 152A, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Moftakhar, P.; Smith, E.R.; Choulakian, A.; Scott, R.M.; Pour, D. Moyamoya disease in children with congenital dwarfing conditions. Pediatr. Neurosurg. 2010, 46, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Codd, P.J.; Scott, R.M.; Smith, E.R. Seckel syndrome and moyamoya. J. Neurosurg. Pediatr. 2009, 3, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.E.; Egan, M.; Rodgers, S.; Harter, D.; Burnside, R.D.; Milla, S.; Pappas, J. Complex chromosome rearrangement of 6p25.3->p23 and 12q24.32->qter in a child with Moyamoya. Pediatrics 2013, 131, e1996–e2001. [Google Scholar] [CrossRef] [PubMed]
- Janczar, S.; Fogtman, A.; Koblowska, M.; Baranska, D.; Pastorczak, A.; Wegner, O.; Kostrzewska, M.; Laguna, P.; Borowiec, M.; Mlynarski, W. Novel severe hemophilia A and moyamoya (SHAM) syndrome caused by Xq28 deletions encompassing F8 and BRCC3 genes. Blood 2014, 123, 4002–4004. [Google Scholar] [CrossRef] [PubMed]
- Girirajan, S.; Mendoza-Londono, R.; Vlangos, C.N.; Dupuis, L.; Nowak, N.J.; Bunyan, D.J.; Hatchwell, E.; Elsea, S.H. Smith-Magenis syndrome and Moyamoya disease in a patient with del(17)(p11.2p13.1). Am. J. Med. Genet. A 2007, 143, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Deleyiannis, F.; Bernard, T.J.; Fenton, L.Z.; Somme, S.; Wilkinson, C.C. Moyamoya in a Patient with Smith-Magenis Syndrome. Pediatr. Neurosurg. 2017, 52, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.O.; Baek, H.J.; Woo, Y.J.; Choi, Y.Y.; Chung, T.W. Moyamoya syndrome in a child with trisomy 12p syndrome. Pediatr. Neurol. 2006, 35, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Rogaia, D.; Mencarelli, A.; Ottaviani, V.; Sallicandro, E.; Guercini, G.; Esposito, S.; Bersano, A.; Merla, G.; Stangoni, G. Juvenile Moyamoya and Craniosynostosis in a Child with Deletion 1p32p31: Expanding the Clinical Spectrum of 1p32p31 Deletion Syndrome and a Review of the Literature. Int. J. Mol. Sci. 2017, 18, 1998. [Google Scholar] [CrossRef] [PubMed]
- Kainth, D.S.; Chaudhry, S.A.; Kainth, H.S.; Suri, F.K.; Qureshi, A.I. Prevalence and characteristics of concurrent down syndrome in patients with moyamoya disease. Neurosurgery 2013, 72, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Spengos, K.; Kosmaidou-Aravidou, Z.; Tsivgoulis, G.; Vassilopoulou, S.; Grigori-Kostaraki, P.; Zis, V. Moyamoya syndrome in a Caucasian woman with Turner’s syndrome. Eur. J. Neurol. 2006, 13, e7–e8. [Google Scholar] [CrossRef] [PubMed]
- Szafranski, P.; Schaaf, C.P.; Person, R.E.; Gibson, I.B.; Xia, Z.; Mahadevan, S.; Wiszniewska, J.; Bacino, C.A.; Lalani, S.; Potocki, L.; et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum. Mutat. 2010, 31, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Gillentine, M.A.; Schaaf, C.P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Conti-Tronconi, B.M.; McLane, K.E.; Raftery, M.A.; Grando, S.A.; Protti, M.P. The nicotinic acetylcholine receptor: Structure and autoimmune pathology. Crit. Rev. Biochem. Mol. Biol. 1994, 29, 69–123. [Google Scholar] [CrossRef] [PubMed]
- Van Bon, B.W.; Mefford, H.C.; Menten, B.; Koolen, D.A.; Sharp, A.J.; Nillesen, W.M.; Innis, J.W.; de Ravel, T.J.; Mercer, C.L.; Fichera, M.; et al. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J. Med. Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, M.; Huynh, Q.B.; Wieczorek, D.; Balliu, B.; Mikat, B.; Boehringer, S. Distinctive facial features in idiopathic Moyamoya disease in Caucasians: A first systematic analysis. PeerJ 2018, 6, e4740. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Weis, M.; Aicher, A.; Dimmeler, S.; Cooke, J.P. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Investig. 2002, 110, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Farooq, I.S.; Yeo, G.S.; O’Rahilly, S. Binge eating as a phenotype of melanocortin 4 receptor gene mutations. N. Engl. J. Med. 2003, 349, 606–609. [Google Scholar]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Huszar, D.; Lynch, C.A.; Fairchild-Huntress, V.; Dunmore, J.H.; Fang, Q.; Berkemeier, L.R.; Gu, W.; Kesterson, R.A.; Boston, B.A.; Cone, R.D.; et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 1997, 88, 131–141. [Google Scholar] [CrossRef]
- Ste Marie, L.; Miura, G.I.; Marsh, D.J.; Yagaloff, K.; Palmiter, R.D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 12339–12344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wu, W.; Wu, Y.; Zheng, J.; Suo, T.; Tang, H.; Tang, J. RNF152, a novel lysosome localized E3 ligase with pro-apoptotic activities. Protein Cell 2010, 1, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Hastings, R.; Howell, R.; Dagna Bricarelli, F.; Kristoffersson, U.; Cavani, S. Specific Constitutional Cytogenetic Guidelines of European Cytogeneticist Association E.C.A. Newsletter 2012, 30, 11–19. [Google Scholar]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T.; Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standard and Guidelines for interpretation and reporting of postnatal constitutional of copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).