Targeting Angiogenesis in Pancreatic Neuroendocrine Tumors: Resistance Mechanisms

 ,
,
Abstract
:1. Introduction
2. Neuroendocrine Tumors Classification
3. Treatment
4. Molecular Biology
5. Angiogenesis in P-NETs and Drug Development: Sunitinib
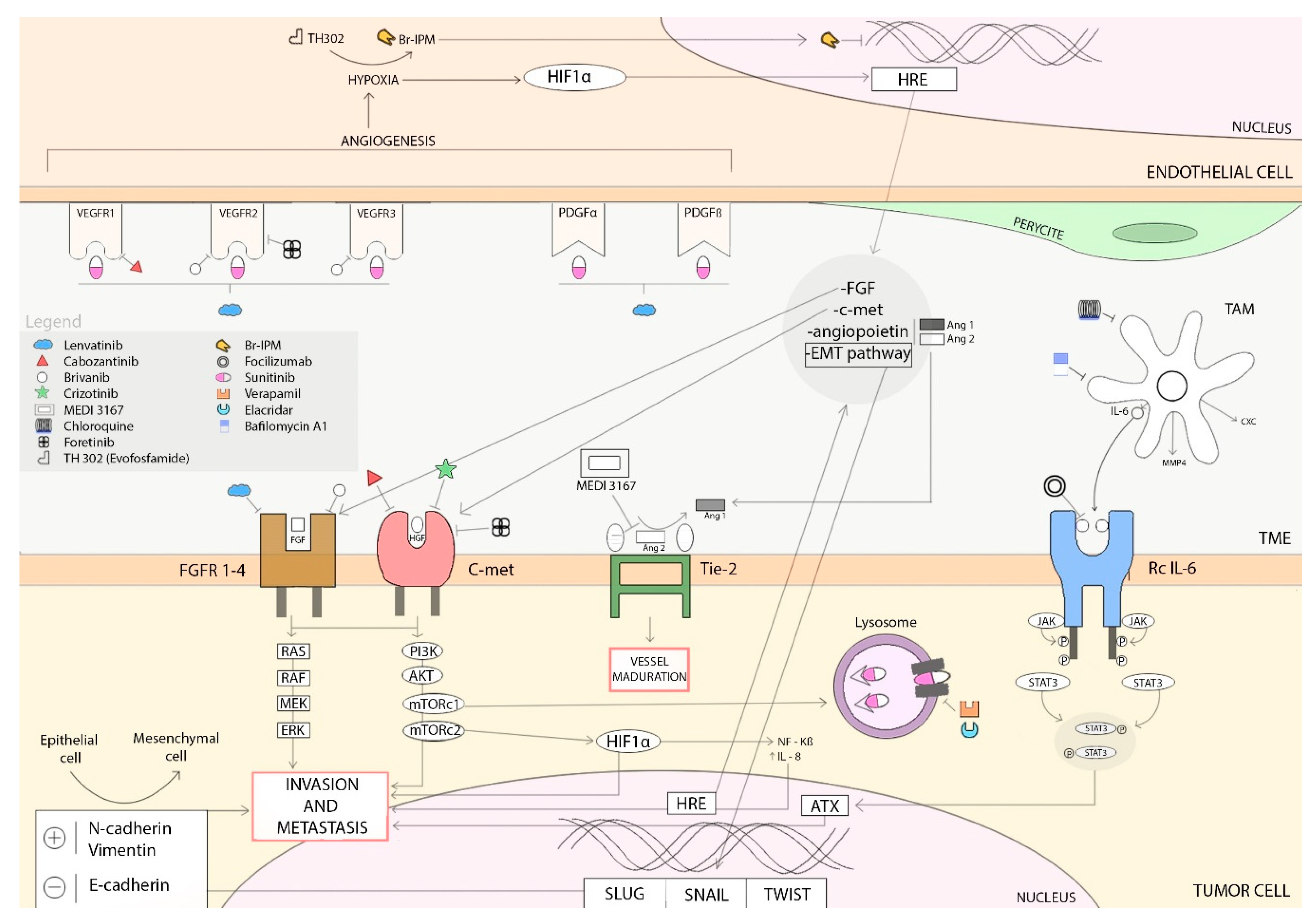
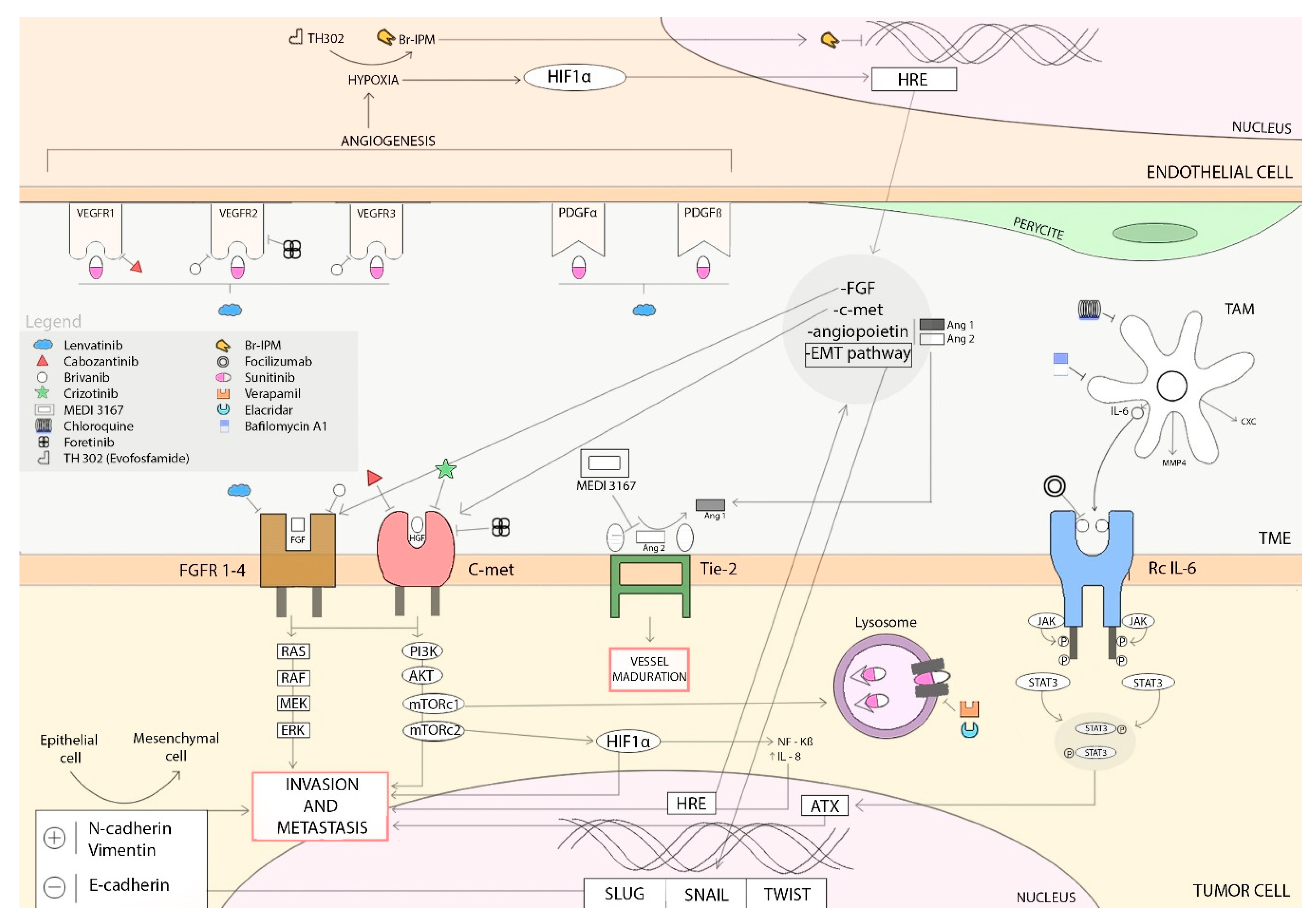
6. Resistance Mechanisms to Antiangiogenic Treatments
6.1. Hypoxia-Induced Activation of Alternative Proangiogenic Pathways and Metastasic Dissemination
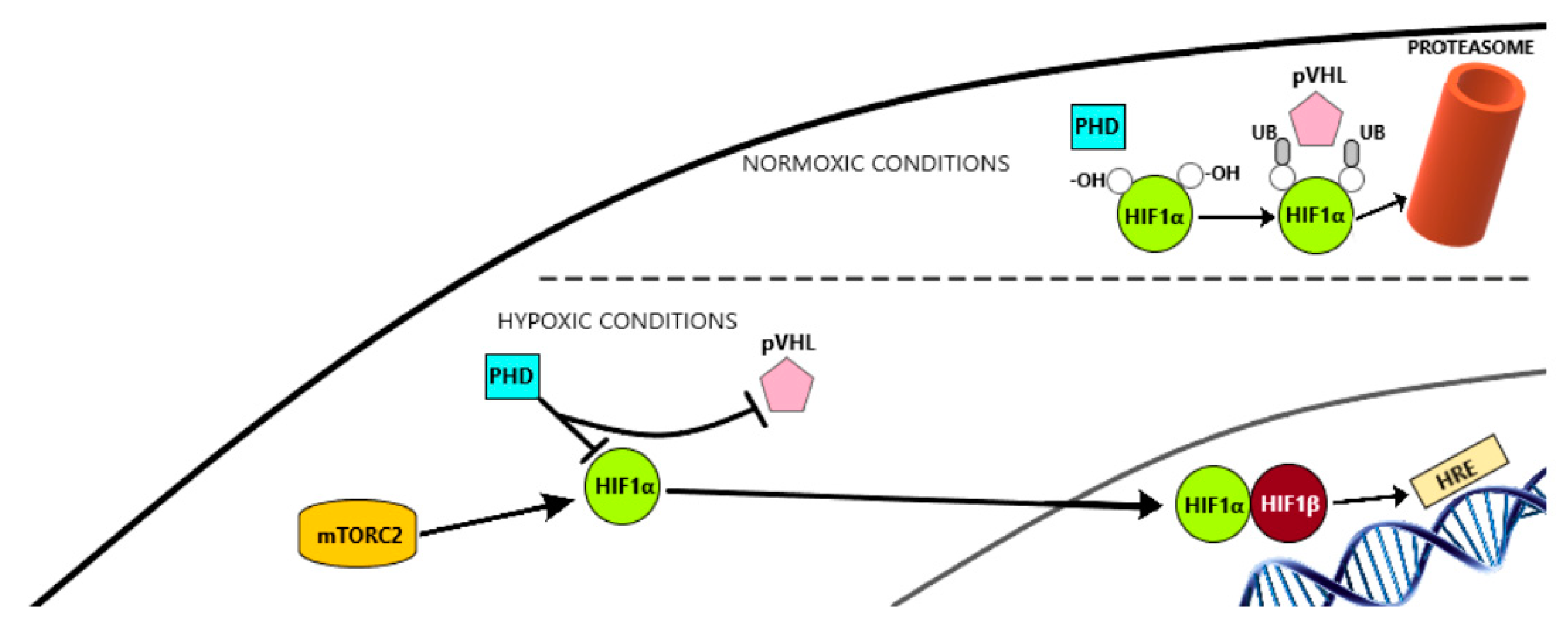
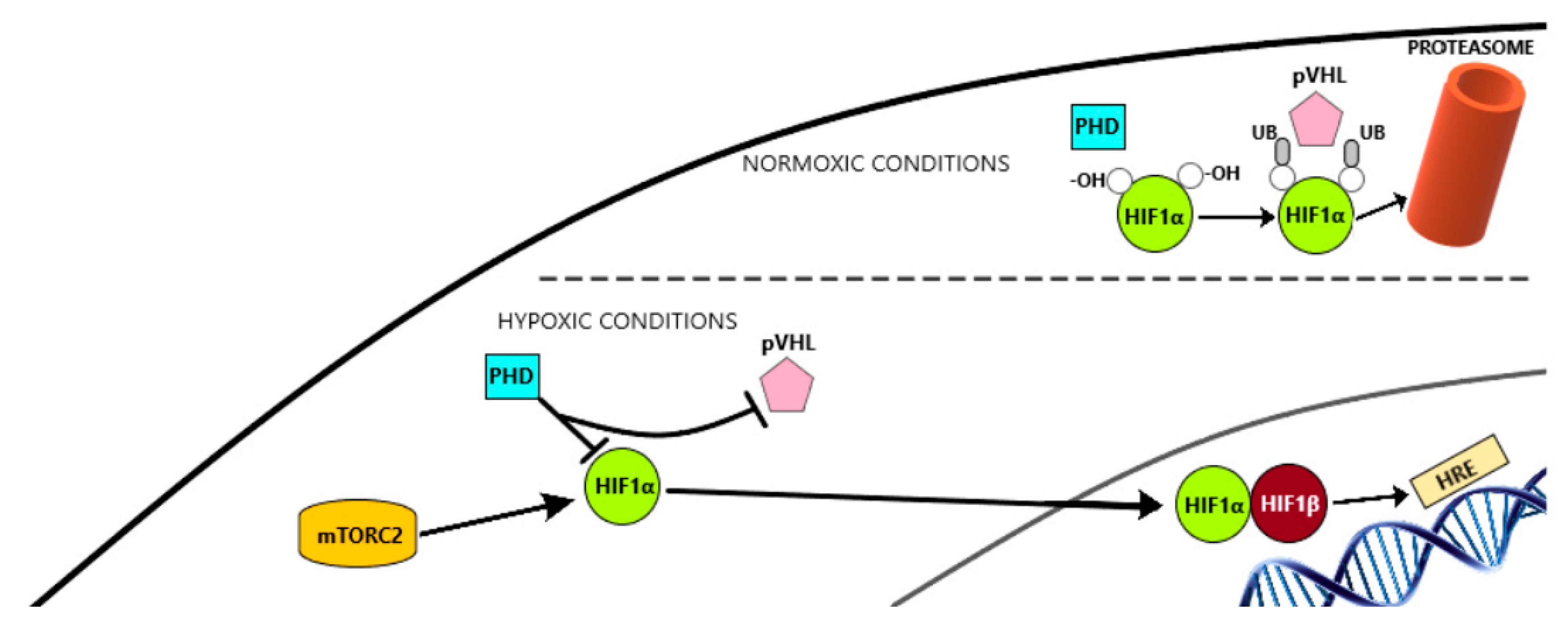
6.1.1. HIF-1α Pathway
6.1.2. Epithelial-Mesenchymal Transition (EMT)
6.1.3. c-Met Activation
6.2. Alternatives Modes of Vascularization
6.3. Hypoxia-Induced Recruitment of Bone-Marrow Derived Cells
6.4. Inflammatory Cells Infiltration: Tumor-Associated Macrophages
6.5. Increase of Pericyte Coverage
6.6. Autotaxin Upregulation
6.7. Sunitinib-induced Autophagy
6.8. Overexpression of EZH2
6.9. IL8 Serum Levels
6.10. Placental Growth Factor
7. Overview
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cives, M.; Strosberg, J. An update on gastroenteropancreatic neuroendocrine tumors. Oncology 2014, 28, 749–756. [Google Scholar] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Man, D.; Wu, J.; Shen, Z.; Zhu, X. Prognosis of patients with neuroendocrine tumor: A SEER database analysis. Cancer Manag. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Gordoa, T.; Díez, J.J.; Molina, J.; Reguera, P.; Martínez-Sáez, O.; Grande, E. An Overview on the Sequential Treatment of Pancreatic Neuroendocrine Tumors (pNETs). Rare Cancers Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Öberg, K.; Knigge, U.; Kwekkeboom, D.; Perren, A. Neuroendocrine gastro-entero-pancreatic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, Y.R.; Kloppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs; WHO Press: Geneva, Switzerland, 2017. [Google Scholar]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology 2016. [Google Scholar] [CrossRef]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID study group. J. Clin. Oncol. 2009. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 trial of 177lu-dotatate for midgut neuroendocrine tumors. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef]
- Raj, N.; Fazio, N.; Strosberg, J. Biology and Systemic Treatment of Advanced Gastroenteropancreatic Neuroendocrine Tumors. Am. Soc. Clin. Oncol. Educ. B. 2018. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.A.; Noonan, K.L.; Pavlovich, P.; Kennecke, H.F.; Lim, H.J. Outcomes of patients treated with capecitabine and temozolamide for advanced pancreatic neuroendocrine tumors (PNETs) and non-PNETs. J. Gastrointest Oncol. 2014, 5, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Fine, R.L.; Gulati, A.P.; Tsushima, D.; Mowatt, K.B.; Oprescu, A.; Bruce, J.N.; Chabot, J.A. Prospective phase II study of capecitabine and temozolomide (CAPTEM) for progressive, moderately, and well-differentiated metastatic neuroendocrine tumors. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Kunz, P.L.; Catalano, P.J.; Nimeiri, H.; Fisher, G.A.; Longacre, T.A.; Suarez, C.J.; Yao, C.J.; Kulke, M.H.; Hendifar, A.E.; Shanks, J.C.; et al. A randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors: A trial of the ECOG-ACRIN Cancer Research Group (E2211). J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- de Mestier, L.; Walter, T.; Evrard, C.; de Boissieu, P.; Hentic, O.; Cros, J.; Tougeron, D.; Lombard-Bohas, C.; Rebours, V.; Hammel, P.; et al. Temozolomide alone or combined to capecitabine for the treatment of advanced pancreatic NET. Neuroendocrinology 2019. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Lenz, H.J.; Meropol, N.J.; Posey, J.; Ryan, D.P.; Picus, J.; Bergsland, E.; Stuart, K.; Tye, L.; Huang, X.; et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dahan, L.; Raoul, J.L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Faris, J.E.; Murphy, J.E.; Blaszkowsky, L.S.; Kwak, E.L.; McCleary, N.J.; Fuchs, C.S.; Meyerhardt, J.A.; Ng, K.; Zhu, A.X.; et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Capdevila, J.; Fazio, N.; Lopez Lopez, C.; Teule, A.; Valle, J.W.; Tafuto, S.; Custodio, A.B.; Reed, N.; Raderer, M.; Grande, E.; et al. Final results of the TALENT trial (GETNE1509): A prospective multicohort phase II study of lenvatinib in patients (pts) with G1/G2 advanced pancreatic (panNETs) and gastrointestinal (giNETs) neuroendocrine tumors (NETs). J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Grande, E.; Capdevila, J.; Castellano, D.; Teulé, A.; Durán, I.; Fuster, J.; Sevilla, I.; Escudero, P.; Sastre, J.; García-Donas, J.; et al. Pazopanib in pretreated advanced neuroendocrine tumors: A phase II, open-label trial of the Spanish Task Force Group for Neuroendocrine Tumors (GETNE). Ann. Onc. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bergsland, E.K.; Mahoney, M.R.; Asmis, T.R.; Hall, N.; Kumthekar, P.; Maitland, M.L.; Niedzwiecki, D.; Nixon, A.B.; O’Reilly, E.M.; Schwartz, L.H.; et al. Prospective randomized phase II trial of pazopanib versus placebo in patients with progressive carcinoid tumors (CARC) (Alliance A021202). J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Bosman, F.; Carneiro, F.; Hruban, R.; Theise, N. WHO Classification of Tumors of Digestive System; WHO Press: Geneva, Switzerland, 2010. [Google Scholar]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of Angiogenesis by Hypoxia: Role of the HIF System. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef]
- Couvelard, A.; Deschamps, L.; Rebours, V.; Sauvanet, A.; Gatter, K.; Pezzella, F.; Ruszniewski, P.; Bedossa, P. Overexpression of the oxygen sensors PHD-1, PHD-2, PHD-3, and FIH is associated with tumor aggressiveness in pancreatic endocrine tumors. Clin. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Couvelard, A.; O′Toole, D.; Turley, H.; Leek, R.; Sauvanet, A.; Degott, C.; Ruszniewski, P.; Belghiti, J.; Harris, A.L.; Gatter, K.; et al. Microvascular density and hypoxia-inducible factor pathway in pancreatic endocrine tumours: Negative correlation of microvascular density and VEGF expression with tumour progression. Br. J. Cancer 2005. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Demetri, G.; Sargent, W.; Raymond, E. Molecular basis for sunitinib efficacy and future clinical development. Nat. Rev. Drug Discov. 2007. [Google Scholar] [CrossRef]
- Pietras, K.; Hanahan, D. A multitargeted, metronomic, and maximum-tolerated dose ′chemo-switch′ regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J. Clin. Oncol. 2005. [Google Scholar] [CrossRef]
- Faivre, S.; Delbaldo, C.; Vera, K.; Robert, C.; Lozahic, S.; Lassau, N.; Bello, C.; Deprimo, S.; Brega, N.; Massimini, G. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Tijeras-Raballand, A.; Neuzillet, C.; Couvelard, A.; Serova, M.; de Gramont, A.; Hammel, P.; Raymond, E.; Faivre, S. Resistance to targeted therapies in pancreatic neuroendocrine tumors (PNETs): Molecular basis, preclinical data, and counteracting strategies. Target Oncol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Beyens, M.; Vandamme, T.; Peeters, M.; Van Camp, G.; De Beeck, K.O. Resistance to targeted treatment of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2019, 26, R109–R130. [Google Scholar] [CrossRef] [Green Version]
- Grande, E.; Lopez, C.; Alonso-Gordoa, T.; Benavent, M.; Capdevila, J.; Teulé, A.; Custodio, A.; Sevilla, I.; Gajate, P.; Molina-Cerrillo, J.; et al. The SUNEVO (GETNE-1408) trial to evaluate the activity and safety of thecombination of sunitinib with evofosfamide (TH-302) in patients with G1/G2 metastatic pancreatic neuroendocrine tumours (pNETs) naïve forsystemic treatment: A phase II study of the Spanish Task Force Group for Neuroendocrine and Endocrine Tumors (GETNE). J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005. [Google Scholar] [CrossRef] [PubMed]
- Maione, F.; Capano, S.; Regano, D.; Zentilin, L.; Giacca, M.; Casanovas, O.; Bussolino, F.; Serini, G.; Giraudo, E. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J. Clin. Investig. 2012. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Bullock, A.J.; Zhang, L.; O’Neill, A.M.; Percy, A.; Sukhatme, V.; Mier, J.W.; Atkins, M.B.; Bhatt, R.S. Plasma angiopoietin-2 (ANG2) as an angiogenic biomarker in renal cell carcinoma (RCC). J. Clin. Oncol. 2010. [Google Scholar] [CrossRef]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Rmili, C.W.; Leow, C.C.; de Palma, M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003. [Google Scholar] [CrossRef]
- Yonemori, K.; Kurahara, H.; Maemura, K.; Mataki, Y.; Sakoda, M.; Iino, S.; Ueno, S.; Shinchi, H.; Natsugoe, S. Impact of snail and E-cadherin expression in pancreatic neuroendocrine tumors. Oncol. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ikezono, Y.; Koga, H.; Akiba, J.; Abe, M.; Yoshida, T.; Wada, F.; Nakamura, T.; Iwamoto, H.; Masuda, A.; Sakaue, T.; et al. Pancreatic Neuroendocrine Tumors and EMT Behavior Are Driven by the CSC Marker DCLK1. Mol. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003. [Google Scholar] [CrossRef]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012. [Google Scholar] [CrossRef]
- You, W.K.; Sennino, B.; Williamson, C.W.; Falcón, B.; Hashizume, H.; Yao, L.C.; Aftab, D.T.; McDonald, D.M. VEGF and c-Met blockade amplify angiogenesis inhibition in Pancreatic Islet Cancer. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, X.D.; Sun, M.; Falcón, B.; Hashizume, H.; Yao, L.C.; Aftab, D.T.; McDonald, D.M. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007. [Google Scholar] [CrossRef]
- Hendrix, M.J.C.; Seftor, E.A.; Seftor, R.E.B.; Chao, J.T.; Chien, D.S.; Chu, Y.W. Tumor cell vascular mimicry: Novel targeting opportunity in melanoma. Pharmacol. Ther. 2016. [Google Scholar] [CrossRef]
- Chu, X.; Gao, X.; Jansson, L.; Quach, M.; Skogseid, B.; Barbu, A. Multiple microvascular alterations in pancreatic islets and neuroendocrine tumors of a Men1 mouse model. Am. J. Pathol. 2013. [Google Scholar] [CrossRef]
- Rivera, L.B.; Bergers, G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol. 2015. [Google Scholar] [CrossRef]
- Krug, S.; Abbassi, R.; Griesmann, H.; Sipos, B.; Wiese, D.; Rexin, P.; Blank, A.; Perren, A.; Haybaeck, J.; Hüttelmaier, S. Therapeutic targeting of tumor-associated macrophages in pancreatic neuroendocrine tumors. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Wei, I.H.; Harmon, C.M.; Arcerito, M.; Cheng, D.F.; Minter, R.M.; Simeone, D.M. Tumor-associated macrophages are a useful biomarker to predict recurrence after surgical resection of nonfunctional pancreatic neuroendocrine tumors. Ann. Surg. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.E. The pericyte—A review. Tissue Cell 1986. [Google Scholar] [CrossRef]
- Taylor, M.; Coleman, R.L.; Sood, A.K. The Role of Angiogenesis in Cancer. Target. Ther. Transl. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Heldin, C.H.; Hellberg, C.; Ostman, A. PDGF and vessel maturation. Recent Results Cancer Res. 2010. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Pàez-Ribes, M.; Cortez, E.; Casanovas, O.; Pietras, K. Use of a Mouse Model of Pancreatic Neuroendocrine Tumors to Find Pericyte Biomarkers of Resistance to Anti-angiogenic Therapy. Horm. Metab. Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to systemic therapies in clear cell renal cell carcinoma: Mechanisms and management strategies. Mol. Cancer Ther. 2018, 17, 1355–1364. [Google Scholar] [CrossRef]
- Ravishankaran, P.; Karunanithi, R. Clinical significance of preoperative serum interleukin-6 and C-reactive protein level in breast cancer patients. World J. Surg. Oncol. 2011. [Google Scholar] [CrossRef]
- Kim, D.K.; Oh, S.Y.; Kwon, H.C.; Lee, S.; Kwon, K.A.; Kim, B.G.; Kim, S.G.; Kim, S.H.; Jang, J.S.; Kim, M.C. Clinical significances of preoperative serum interleukin-6 and C-reactive protein level in operable gastric cancer. BMC Cancer 2009. [Google Scholar] [CrossRef]
- Groblewska, M.; Mroczko, B.; Wereszczyńska-Siemia̧tkowska, U.; Kedra, B.; Lukaszewicz, M.; Baniukiewicz, A.; Szmitkowski, M. Serum interleukin 6 (IL-6) and C-reactive protein (CRP) levels in colorectal adenoma and cancer patients. Clin. Chem. Lab. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, X.; Yang, Y. Autotaxin upregulated by STAT3 activation contributes to invasion in pancreatic neuroendocrine neoplasms. Endocr. Connect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Aguiar, A.G.; Postlewait, L.M.; Ethun, C.G.; Zaidi, M.Y.; Zhelnin, K.; Krasinskas, A.; Russell, M.C.; Kooby, D.A.; Cardona, K.; El-Rayes, B.F. STAT3 Inhibition for Gastroenteropancreatic Neuroendocrine Tumors: Potential for a New Therapeutic Target? J. Gastrointest. Surg. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Haber, T.; Breuksch, I.; Gebhard, S.; Sugino, T.; Kubo, H.; Hata, J.; Koguchi, Y.; Yabe, M.; Kataoka, M.; et al. Overriding TKI resistance of renal cell carcinoma by combination therapy with IL-6 receptor blockade. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [Google Scholar] [CrossRef]
- Azijli, K.; Gotink, K.J.; Verheul, H.M. The Potential Role of Lysosomal Sequestration in Sunitinib Resistance of Renal Cell Cancer. J. Kidney Cancer VHL. 2016. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grépin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Miranoni, I.; Perren, A. Autophagy inhibition improves sunitinib efficacy in pancreatic neuroendocrine tumors via a lysosome-dependent mechanism. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Adelaiye-Ogala, R.; Budka, J.; Damayanti, N.P.; Arrington, J.; Ferris, M.; Hsu, C.C.; Chintala, S.; Orillion, A.; Miles, K.M.; Shen, L.; et al. EZH2 modifies sunitinib resistance in renal cell carcinoma by kinome reprogramming. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Marconcini, R.; Faviana, P.; Campani, D.; Galli, L.; Antonuzzo, A.; Orlandini, C.; Falcone, A.; Ricci, S. Enhancer of zest homolog 2 (EZH2) expression in well and moderately differentiated pancreatic neuroendocrine tumor (pNET). Ann. Oncol. 2016. [Google Scholar] [CrossRef]
- Mateo, J.; Heymach, J.V.; Zurita, A.J. Circulating biomarkers of response to sunitinib in gastroenteropancreatic neuroendocrine tumors: Current data and clinical outlook. Mol. Diagnosis. Ther. 2012. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Hilfenhaus, G.; Göhrig, A.; Pape, U.F.; Neumann, T.; Jann, H.; Zdunek, D.; Hess, G.; Stassen, J.M.; Wiedenmann, B.; Detjen, K.; et al. Placental growth factor supports neuroendocrine tumor growth and predicts disease prognosis in patients. Endocr. Relat. Cancer 2013. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| WHO 2010 | Mitotic Count | Ki67 Index | Previous |
|---|---|---|---|
| WD NETs G1 WD NETs G2 | <2 × 10 HPF | ≤2% | G1 |
| 2–20 × 10 HPF | 3–20% | G2 | |
| PD NEC G3 | >20 × 10 HPF | >20% | G3 |
| MANEC | |||
| WHO 2017 | Mitotic Count | Ki67 index | |
| WD NETs G1 WD NETs G2 | <2 × 10 HPF | <3% | |
| 2–20 × 10 HPF | 3–20% | ||
| WD NETs G3 | >20 × 10 HPF | >20% | Difference is made upon molecular and histological features |
| PD NEC G3 | >20 × 10 HPF | >20% | |
| MINEN | To qualify as MENEN each component (endocrine and non-endocrine) must have at least 30% | ||
| Localization | Midgut | Pancreas | Liver Tumor Burden High (>25%) | |||
|---|---|---|---|---|---|---|
| Grade of Differentiation | G1 | G2 | G1 | G2 | ||
| Ki 67 | <2% | 2–10% | <2% | 2–10% | ||
| First line SSA treatment | Octreotide LAR |  | ||||
| Lanreotide Autogel | | | | | | |
| Sunitinib | Cabozantinib | Lenvatinib | Pazopanib | |||||
|---|---|---|---|---|---|---|---|---|
| Trial design | Phase II_NR | Phase III_R | Phase II_NR | Phase II_NR | Phase II_NR | Phase II_NR | Phase II_NR | Phase II_R |
| Primary tumor origin | Carcionid 41 | pNET 171 | GI NET 41 | pNET 20 | GI NET 56 | pNET 55 | GEPNET | Carcinoid 97 + 74 |
| Follow-Up (m) | 15.1 | 60 | 23.3 | 17 | 44 | |||
| Previous treatment (SSA + others) (%) | 53.7 + 44 | 35 + 66 | 98 + NA 1 (0–6) | 75% + NA 3 (0–8) | 98 + 0 | 84 + 100 | 82 + 100 | 94 + 26 |
| ORR (%) | 2.4 | 9 | 15 | 15 | 16.3 | 42.3 | 9.5 | 2.1 vs. 0 |
| SD (%) | 82.9 | 63 | 75 (10% UK) | 63 (17% UK) | 74 | 50 | 50.0 | 72.2 vs. 73.0 |
| PD (%) | 2.4 | 14 | 0 | 5 | 0 | 0.02 | 40.5 | 4.1 vs. 18.9 |
| mPFS (m) | 10.2 | 11.4 vs. 5.5 | 31.4 | 21.8 | 15.4 | 15.53 | 9.5 | 11.6 vs. 8.5 |
| mOS (m) | 25.3 | 38.6 vs. 29.1 | NA | NA | NA | NA | NA | 41.3 vs. 42.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozas, J.; San Román, M.; Alonso-Gordoa, T.; Pozas, M.; Caracuel, L.; Carrato, A.; Molina-Cerrillo, J. Targeting Angiogenesis in Pancreatic Neuroendocrine Tumors: Resistance Mechanisms. Int. J. Mol. Sci. 2019, 20, 4949. https://doi.org/10.3390/ijms20194949
Pozas J, San Román M, Alonso-Gordoa T, Pozas M, Caracuel L, Carrato A, Molina-Cerrillo J. Targeting Angiogenesis in Pancreatic Neuroendocrine Tumors: Resistance Mechanisms. International Journal of Molecular Sciences. 2019; 20(19):4949. https://doi.org/10.3390/ijms20194949
Chicago/Turabian StylePozas, Javier, María San Román, Teresa Alonso-Gordoa, Miguel Pozas, Laura Caracuel, Alfredo Carrato, and Javier Molina-Cerrillo. 2019. "Targeting Angiogenesis in Pancreatic Neuroendocrine Tumors: Resistance Mechanisms" International Journal of Molecular Sciences 20, no. 19: 4949. https://doi.org/10.3390/ijms20194949
APA StylePozas, J., San Román, M., Alonso-Gordoa, T., Pozas, M., Caracuel, L., Carrato, A., & Molina-Cerrillo, J. (2019). Targeting Angiogenesis in Pancreatic Neuroendocrine Tumors: Resistance Mechanisms. International Journal of Molecular Sciences, 20(19), 4949. https://doi.org/10.3390/ijms20194949