Intraspecific Variation within the Utricularia amethystina Species Morphotypes Based on Chloroplast Genomes

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
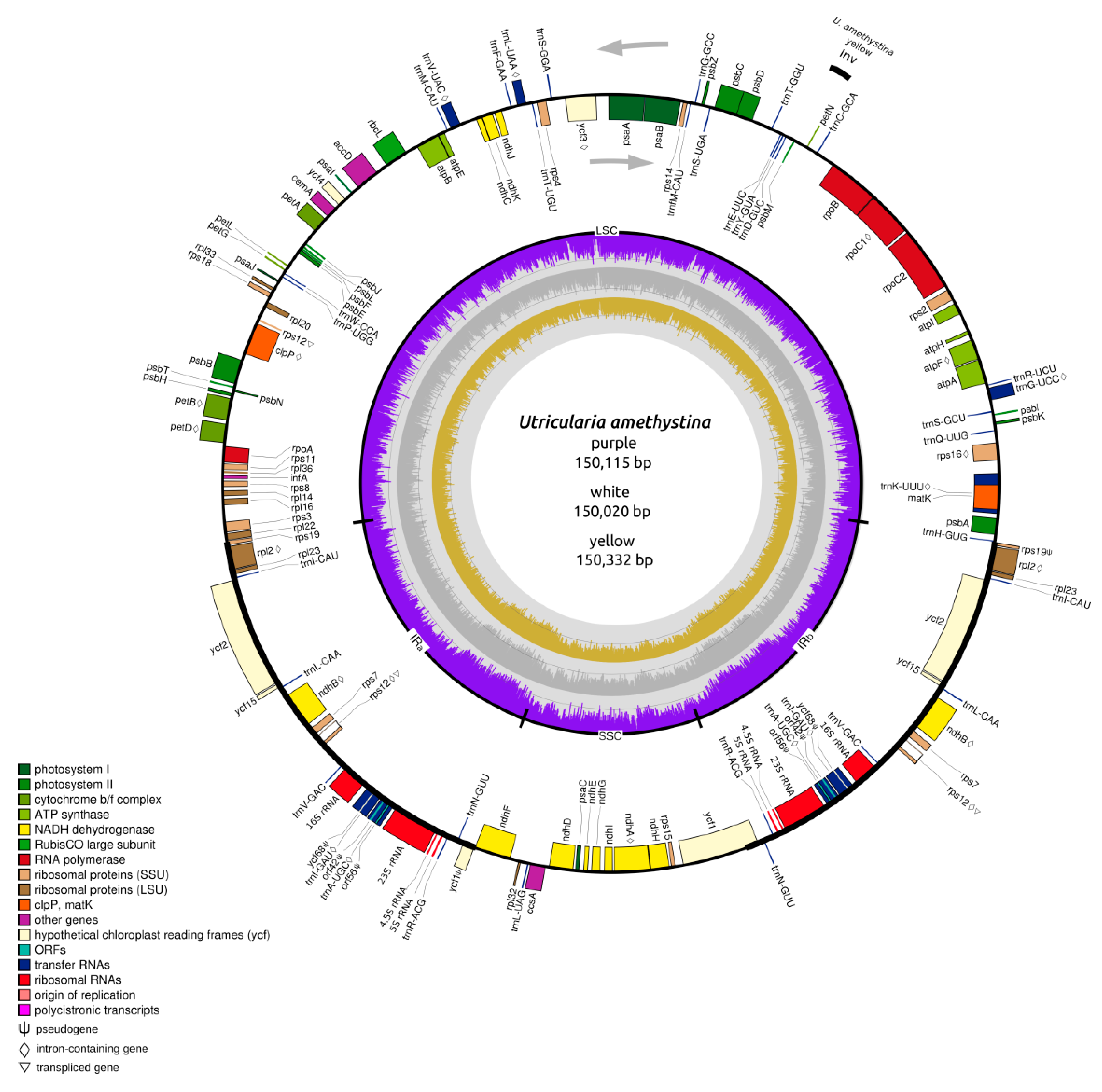
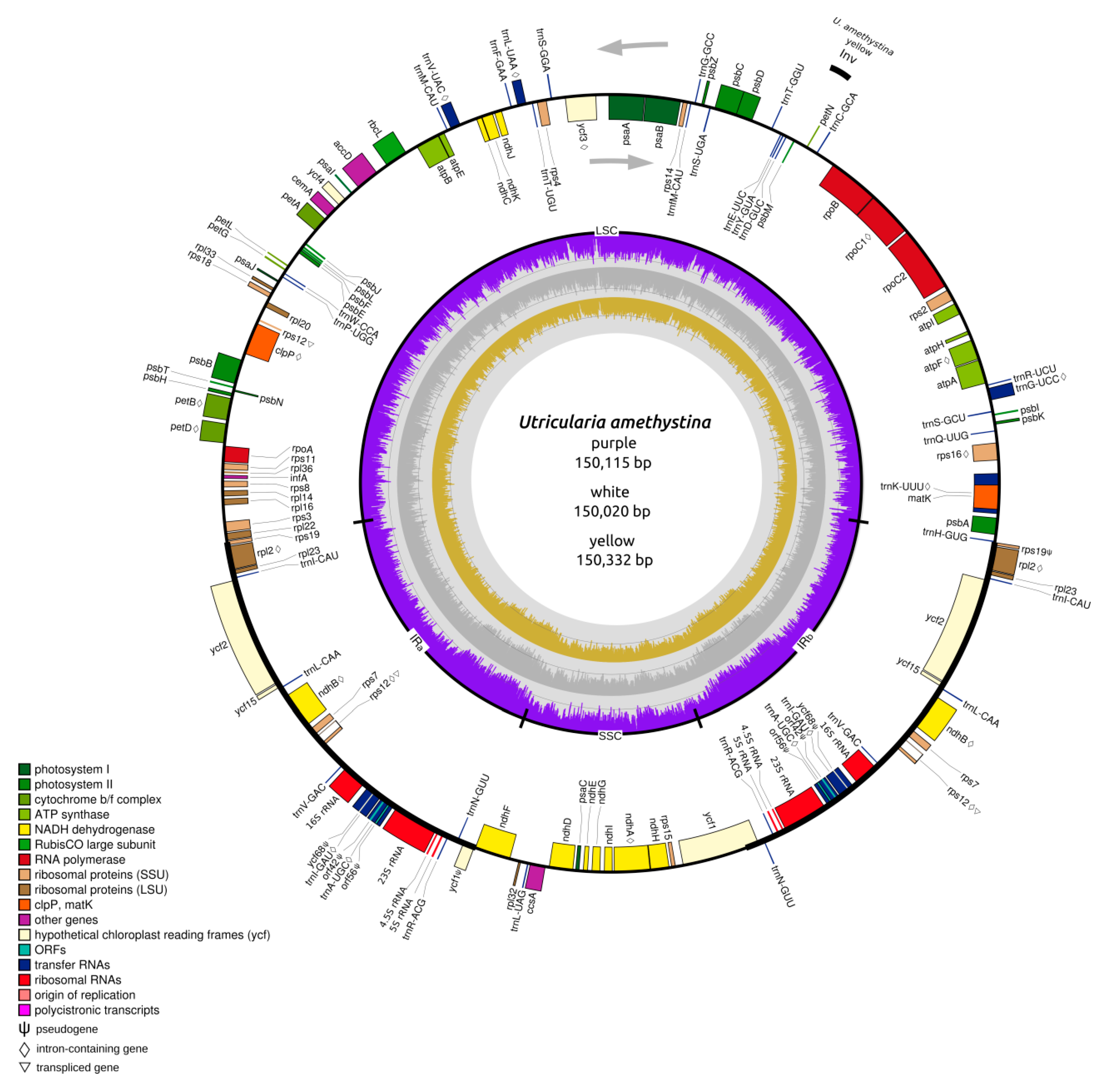
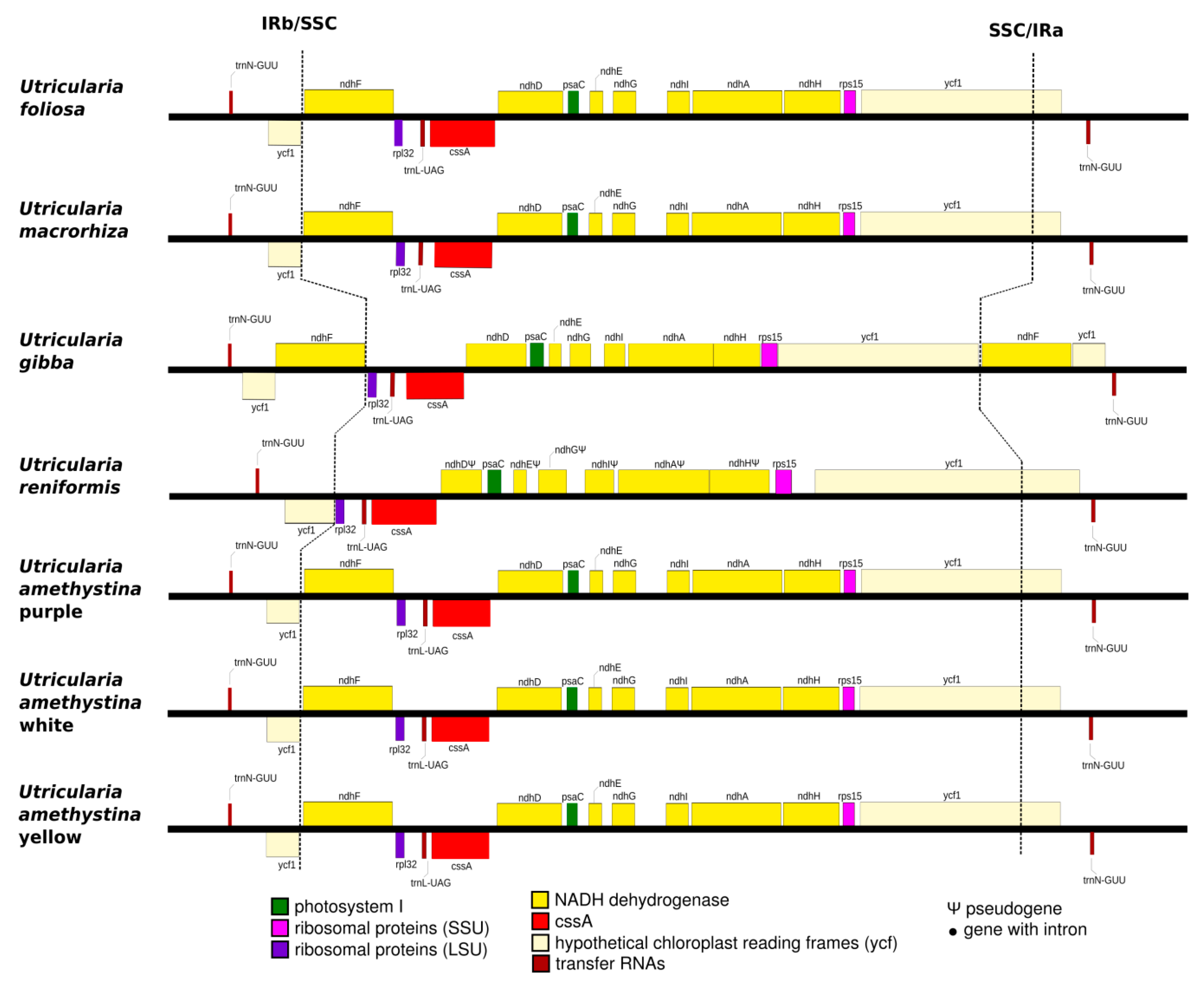
2.1. Structure of Chloroplasts in Utricularia amethystina
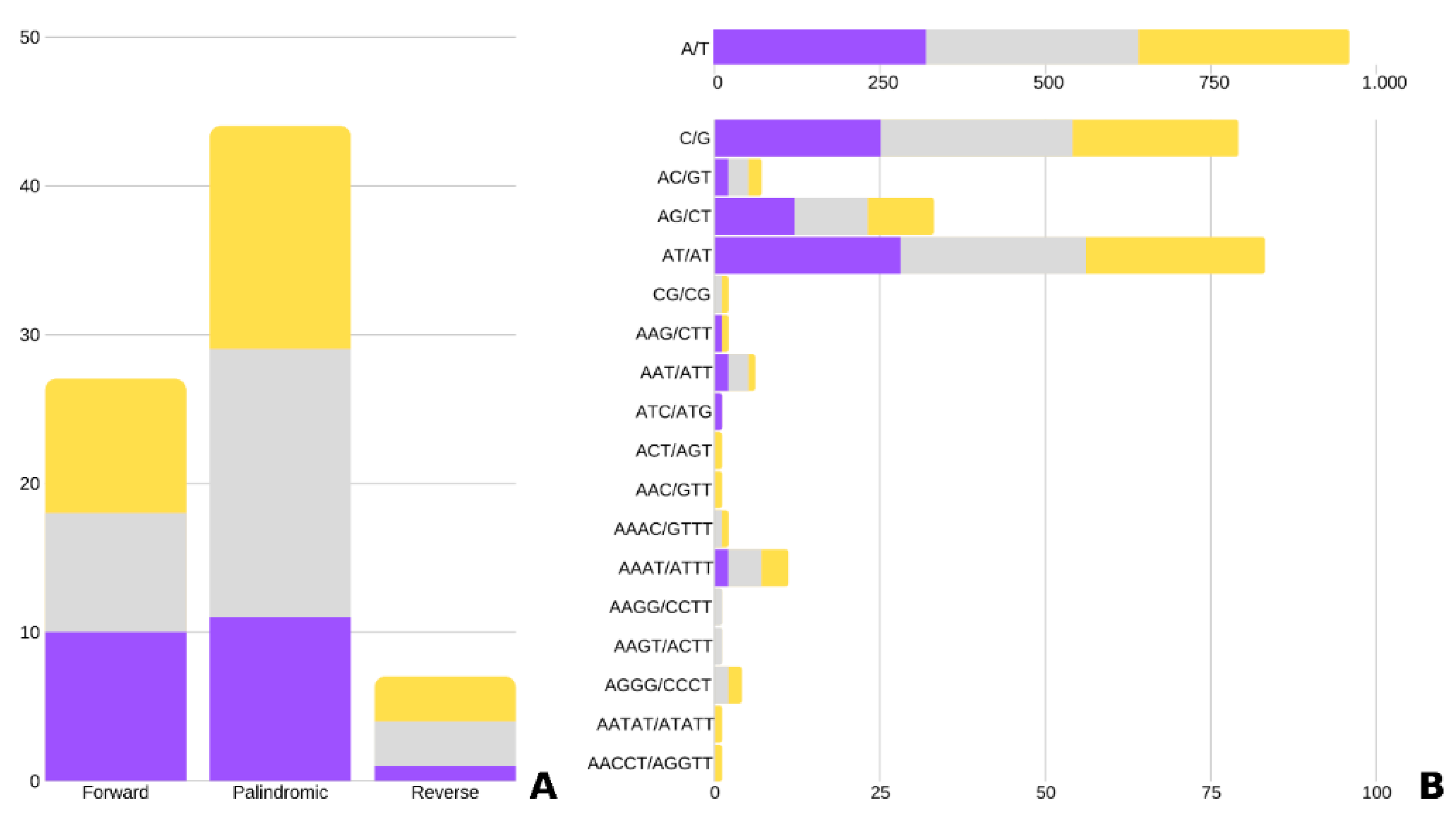
2.2. Repeats and Chloroplast Microsatellites (cpSSR)
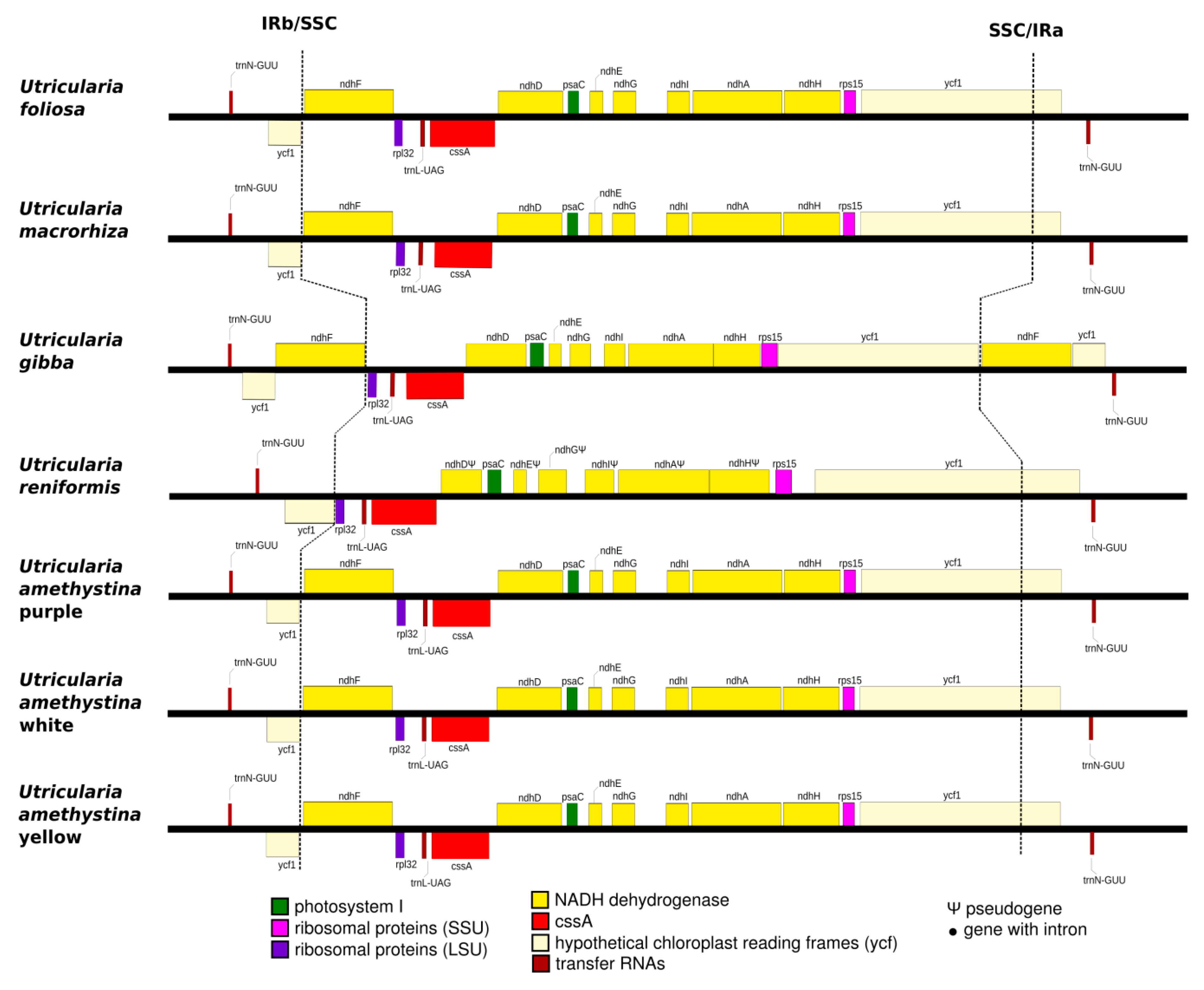
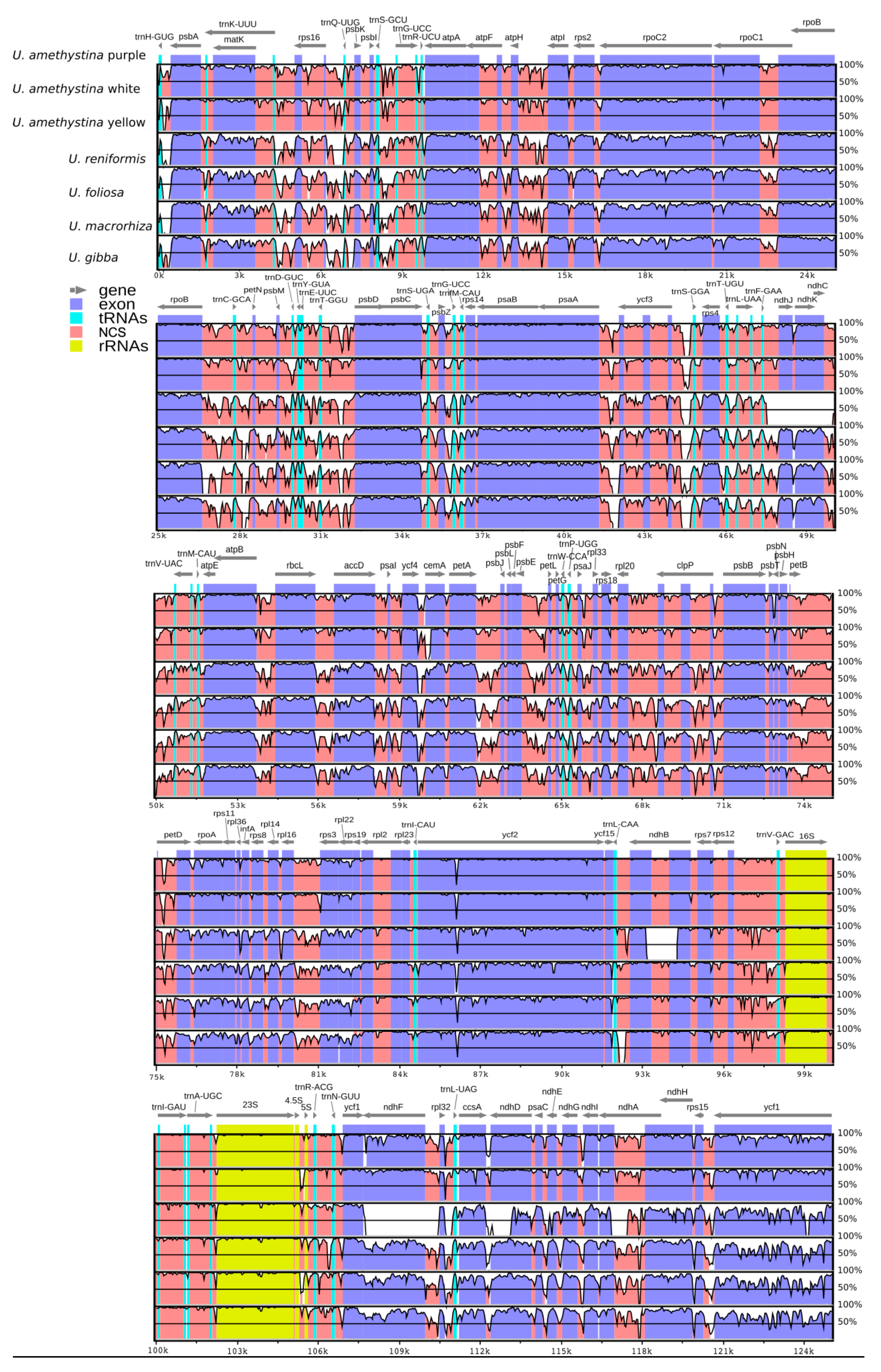
2.3. Interspecific Comparison
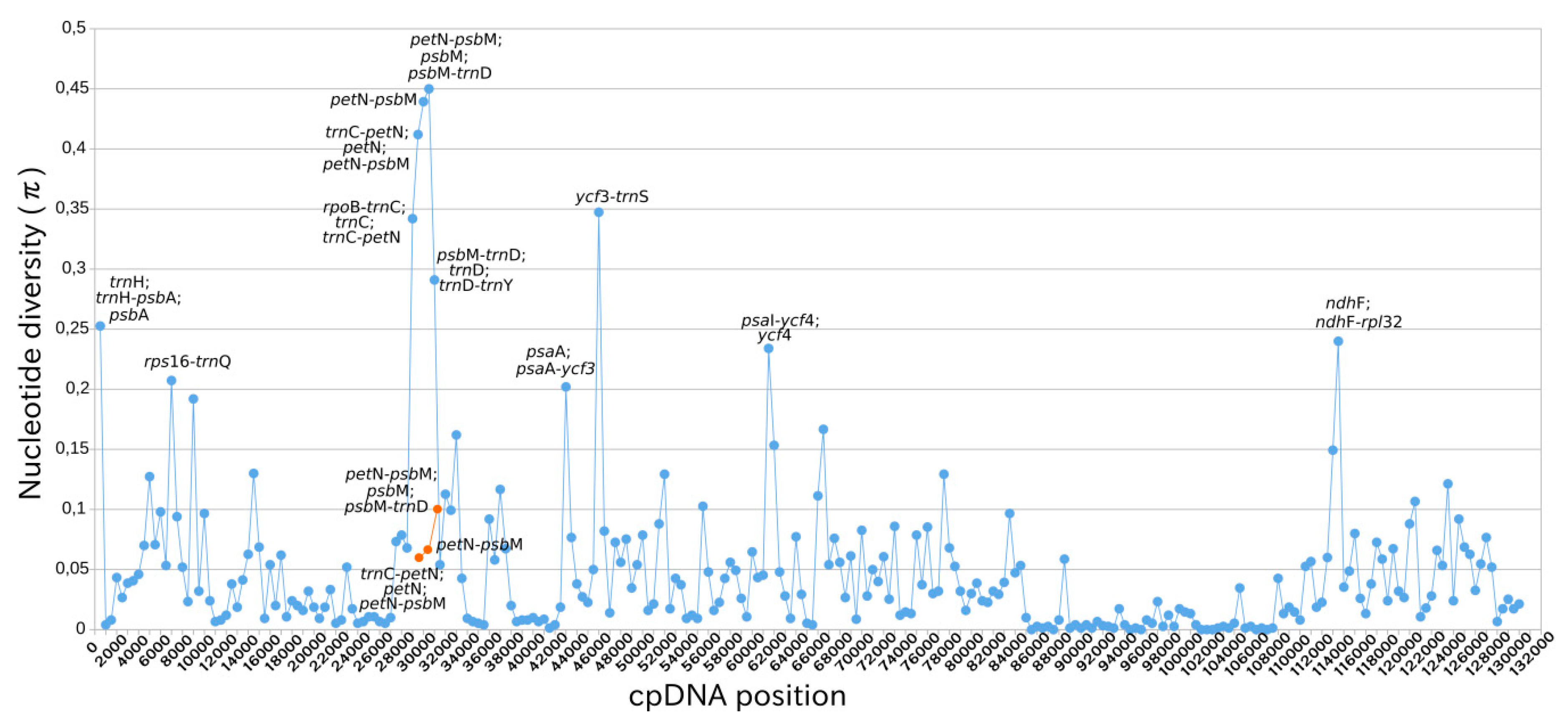
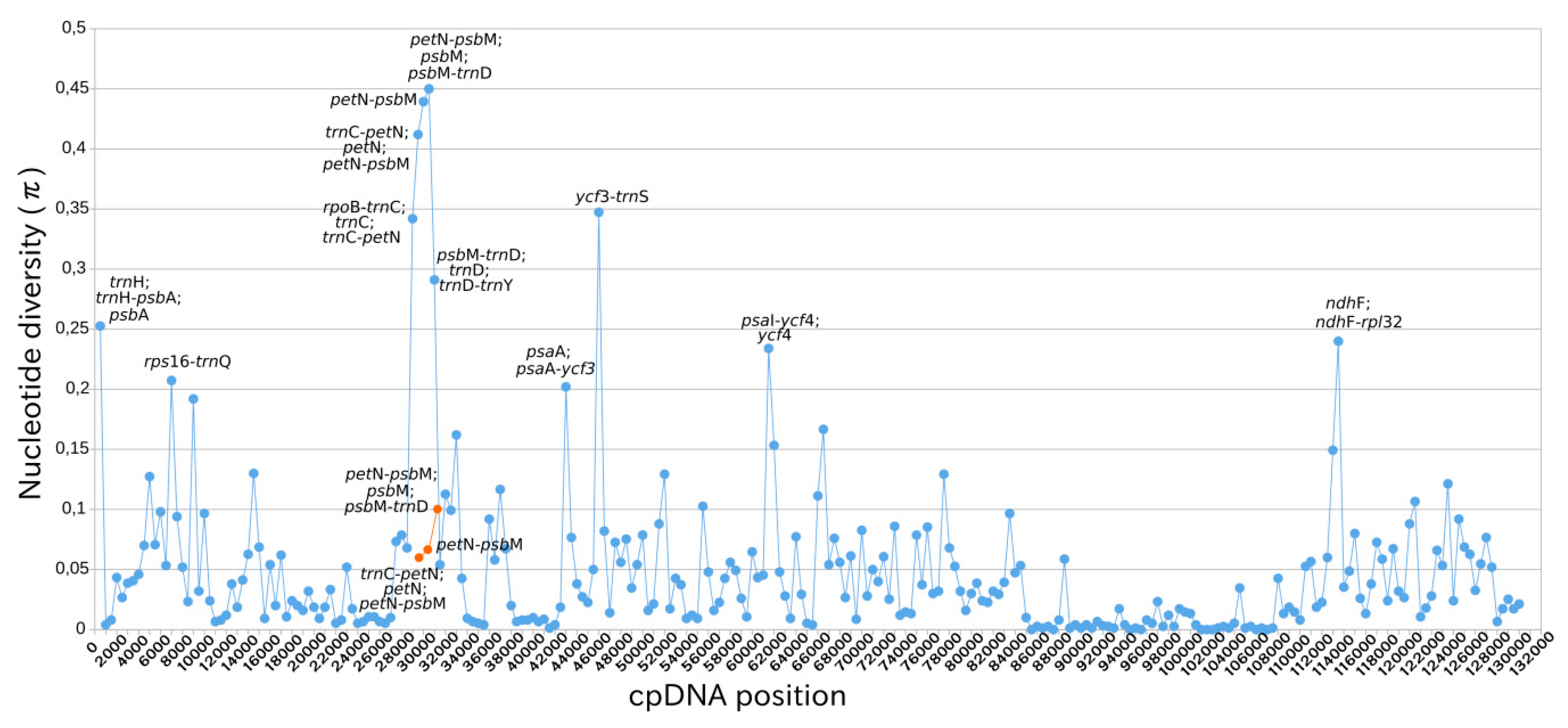
2.4. Intraspecific Comparison: Species Polymorphism
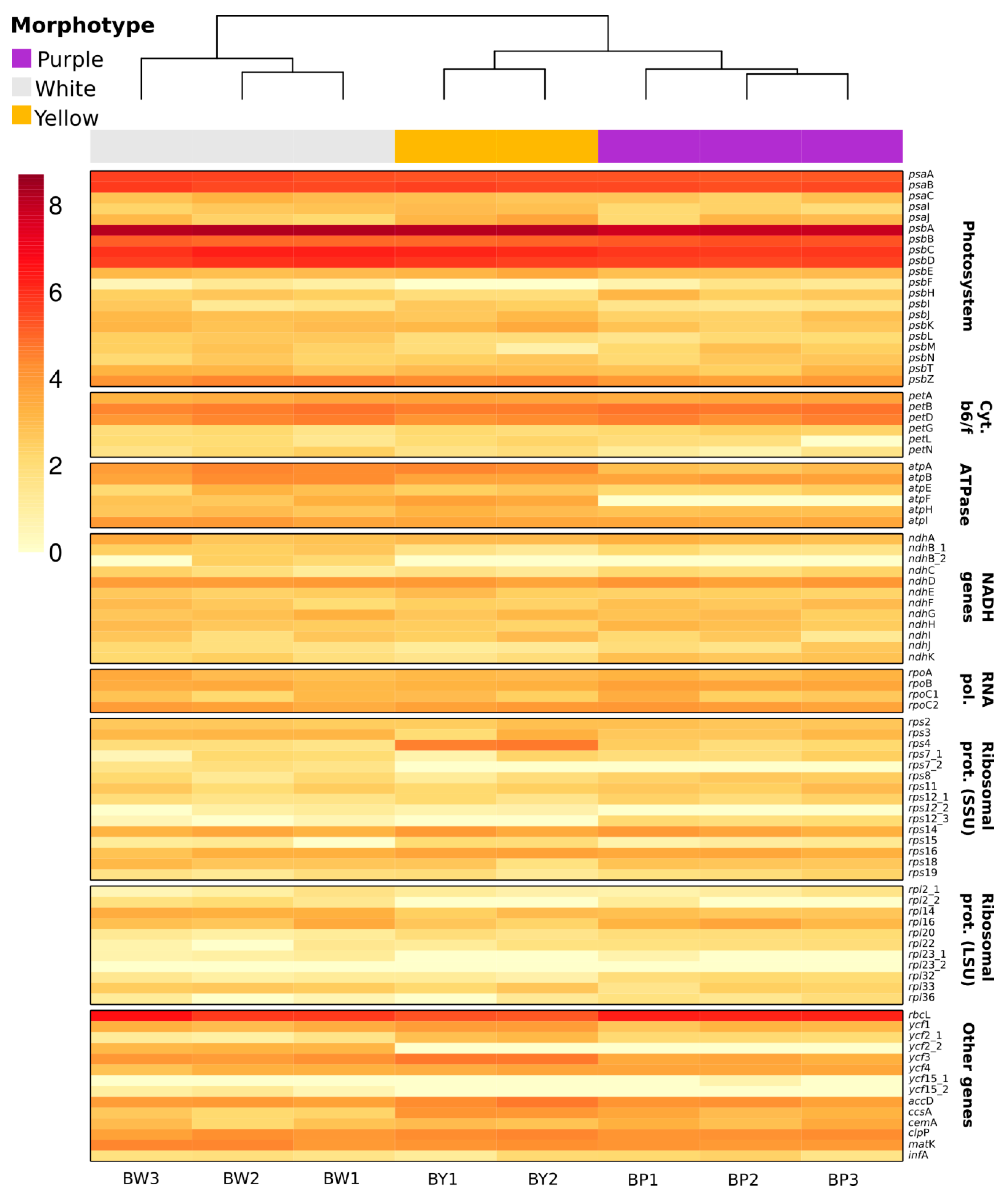
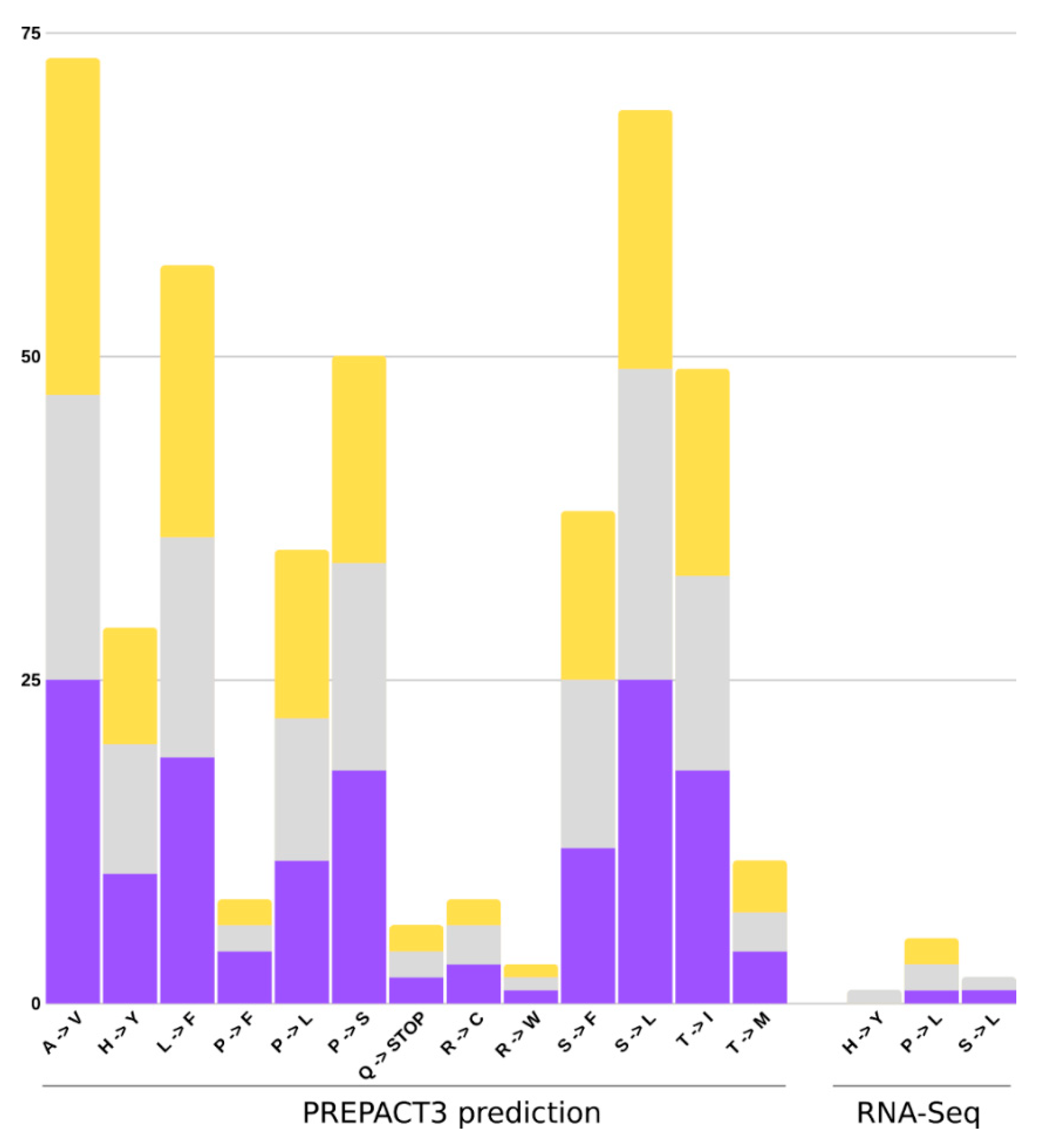
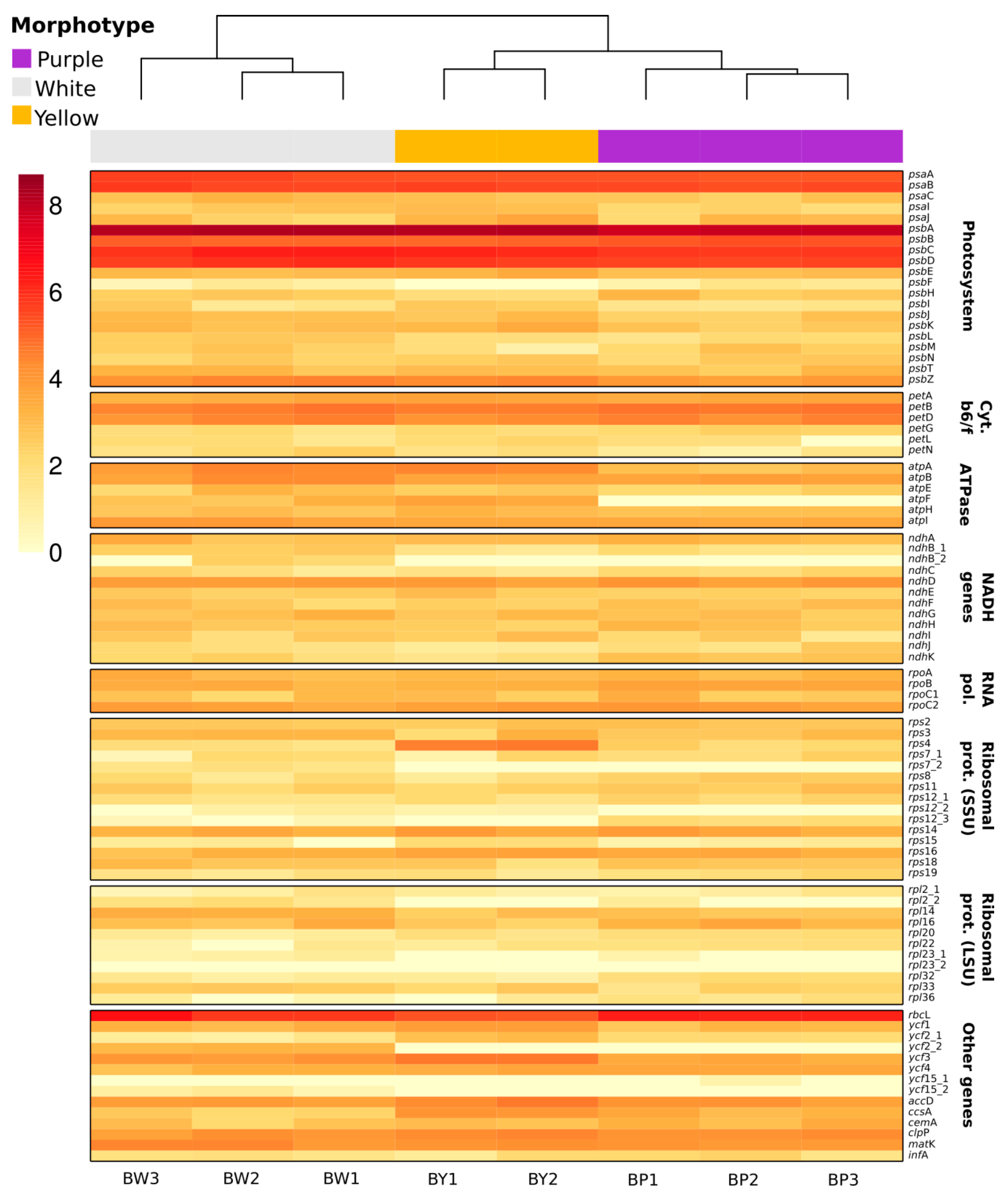
2.5. Chloroplast Expression
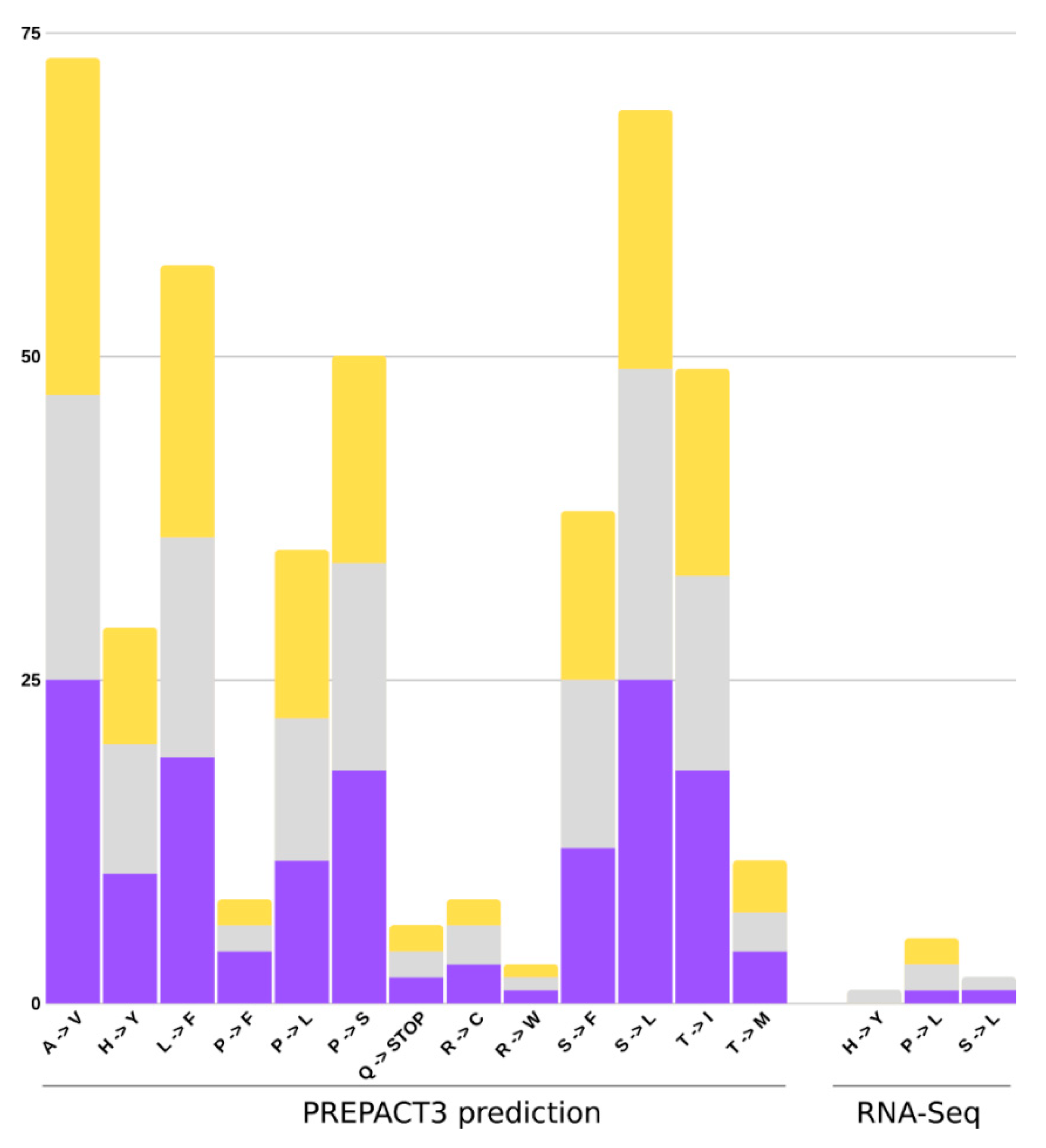
2.6. RNA Edit
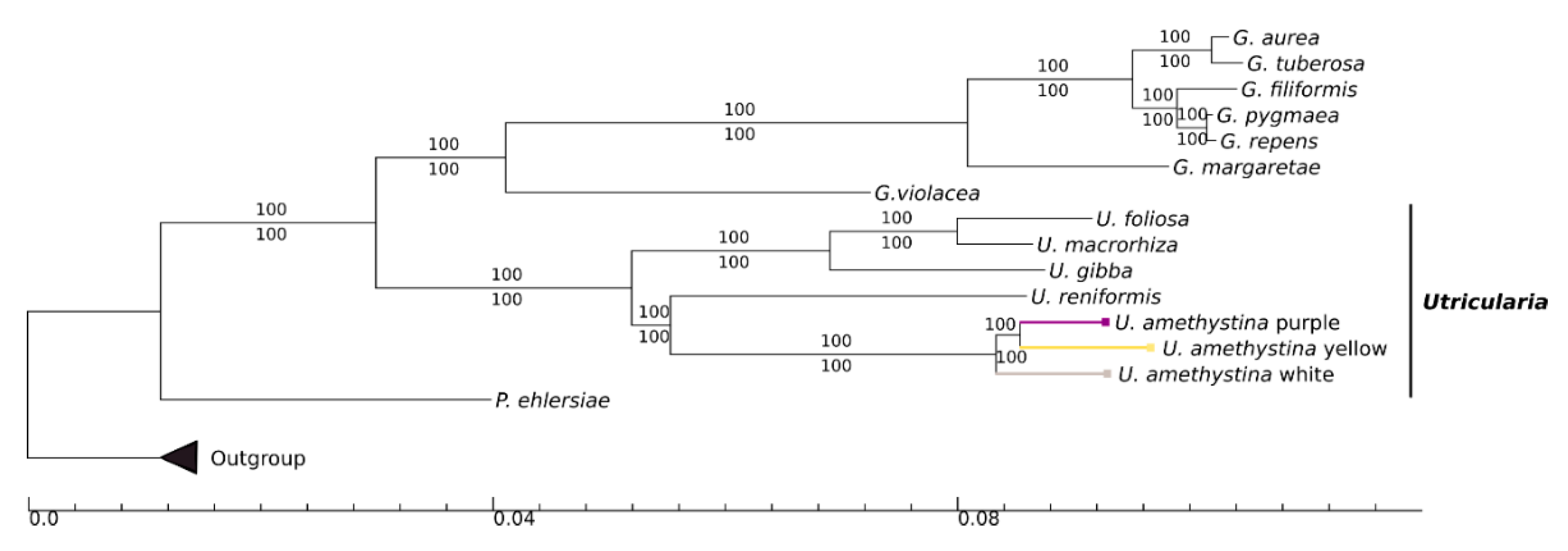
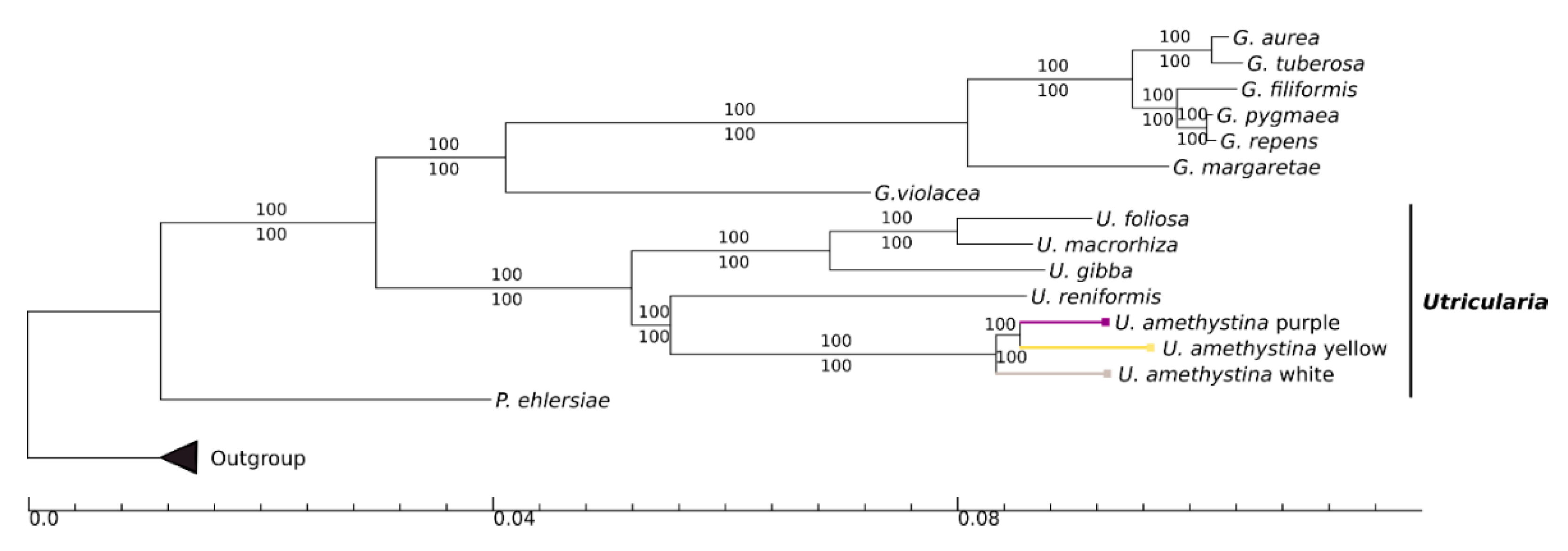
2.7. Phylogeny
3. Discussion
4. Materials and Methods
4.1. Sampling and DNA Extraction
4.2. Organellar Genome Sequencing, Assembly and Annotation
4.3. Repeats and SSR Analyses
4.4. Phylogenetic Reconstruction
4.5. Intraspecific Polymorphism Analyses
4.6. Interspecific Comparison
4.7. RNA Extraction, Sequencing and RNA Editing Site Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Statements
References
- Müller, K.F.; Borsch, T.; Legendre, L.; Porembski, S.; Barthlott, W. Recent progress in understanding the evolution of carnivorous Lentibulariaceae (Lamiales). Plant Biol. (Stuttg) 2006, 8, 748–757. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Gibson, R.; Adamec, L.; Domínguez, Y.; Miranda, V.F.O. Molecular phylogeny of bladderworts: A wide approach of Utricularia (Lentibulariaceae) species relationships based on six plastidial and nuclear DNA sequences. Mol. Phylogenet. Evol. 2018, 118, 244–264. [Google Scholar] [CrossRef] [Green Version]
- Rutishauser, R.; Isler, B. Developmental genetics and morphological evolution of flowering plants, especially bladderworts (Utricularia): Fuzzy Arberian Morphology complements Classical Morphology. Ann. Bot. 2001, 88, 1173–1202. [Google Scholar] [CrossRef] [Green Version]
- Reut, M.S.; Płachno, B.J. Unusual developmental morphology and anatomy of vegetative organs in Utricularia dichotoma – leaf, shoot and root dynamics. Protoplasma 2019. [Google Scholar] [CrossRef] [Green Version]
- Albert, V.A.; Jobson, R.W.; Michael, T.P.; Taylor, D.J. The carnivorous bladderwort (Utricularia, Lentibulariaceae): A system inflates. J. Exp. Bot. 2010, 61, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Veleba, A.; Bureš, P.; Adamec, L.; Šmarda, P.; Lipnerová, I.; Horová, L. Genome size and genomic GC content evolution in the miniature genome-sized family Lentibulariaceae. New Phytol. 2014, 203, 22–28. [Google Scholar] [CrossRef]
- Taylor, P. The Genus Utricularia—A Taxonomic Monograph; The Royal Botanic Gardens, Kew: London, UK, 1989. [Google Scholar]
- Seine, R.; Porembski, S.; Barthlott, W. A neglected habitat of carnivorous plants: Inselbergs. Feddes Repert. 1996, 106, 555–562. [Google Scholar] [CrossRef]
- BFG Brazilian Flora 2020: Innovation and collaboration to meet Target 1 of the Global Strategy for Plant Conservation (GSPC). Rodriguésia 2018, 69, 1513–1527. [CrossRef] [Green Version]
- Vellozo, J.M.C. 19. Utricularia. In Florae Fluminensis, Seu, Descriptionum Plantarum Praefectura Fluminensi Sponte Mascentium Liber Primus ad Systema Sexuale Concinnatus; Typographia National: Rio de Janeiro, Brazil, 1825; p. 18. [Google Scholar]
- Saint-Hilaire, A.F.C.P.; Girard, F. Comptes Rendus Hebdomadaires des Séances de L’académie des Sciences; French Academy of Sciences: Paris, France, 1838; Volume 7, ISBN 0001-4036. [Google Scholar]
- Baleeiro, P.C.; Jobson, R.W.; Sano, P.T. Morphometric approach to address taxonomic problems: The case of Utricularia sect. Foliosa (Lentibulariaceae). J. Syst. Evol. 2016, 54, 175–186. [Google Scholar] [CrossRef]
- Baleeiro, P.C.; Sano, P.T.; Jobson, R.W. Molecular Phylogeny of the Utricularia amethystina Complex (Utricularia sect. Foliosa) Assessed Using Plastid and Nuclear Sequence Data. Syst. Bot. 2019, 44, 398–404. [Google Scholar]
- Zhang, Y.; Iaffaldano, B.J.; Zhuang, X.; Cardina, J.; Cornish, K. Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biol. 2017, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.H.; Suh, H.J.; Park, J.; Kim, Y.; Kim, S. The complete chloroplast genome sequence of a morphotype of Goodyera schlechtendaliana (Orchidaceae) with the column appendages. Mitochondrial DNA Part B Resour. 2019, 4, 626–627. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria. Advances in Photosynthesis and Respiration (Including Bioenergy and Related Processes); Bock, R., Knoop, V., Eds.; Springer: Dordrecht, The Netherlands, 2012; ISBN 978-94-007-2920-9. [Google Scholar]
- Westwood, J.H.; Yoder, J.I.; Timko, M.P.; DePamphilis, C.W. The evolution of parasitism in plants. Trends Plant Sci. 2010, 15, 227–235. [Google Scholar] [CrossRef]
- Delannoy, E.; Fujii, S.; Colas Des Francs-Small, C.; Brundrett, M.; Small, I. Rampant Gene loss in the underground orchid Rhizanthella Gardneri highlights evolutionary constraints on plastid genomes. Mol. Biol. Evol. 2011, 28, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Braukmann, T.; Stefanović, S. Plastid genome evolution in mycoheterotrophic Ericaceae. Plant Mol. Biol. 2012, 79, 5–20. [Google Scholar] [CrossRef]
- Gruzdev, E.V.; Kadnikov, V.V.; Beletsky, A.V.; Kochieva, E.Z.; Mardanov, A.V.; Skryabin, K.G.; Ravin, N.V. Plastid Genomes of Carnivorous Plants Drosera rotundifolia and Nepenthes × ventrata Reveal Evolutionary Patterns Resembling Those Observed in Parasitic Plants. Int. J. Mol. Sci. 2019, 20, 4107. [Google Scholar] [CrossRef] [Green Version]
- Nevill, P.G.; Howell, K.A.; Cross, A.T.; Williams, A.V.; Zhong, X.; Tonti-Filippini, J.; Boykin, L.M.; Dixon, K.W.; Small, I. Plastome-wide rearrangements and gene losses in carnivorous droseraceae. Genome Biol. Evol. 2019, 11, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Martín, M.; Sabater, B. Plastid ndh genes in plant evolution. Plant Physiol. Biochem. 2010, 48, 636–645. [Google Scholar] [CrossRef]
- Ruhlman, T.A.; Chang, W.-J.; Chen, J.J.W.; Huang, Y.-T.; Chan, M.-T.; Zhang, J.; Liao, D.-C.; Blazier, J.C.; Jin, X.; Shih, M.-C.; et al. NDH expression marks major transitions in plant evolution and reveals coordinate intracellular gene loss. BMC Plant Biol. 2015, 15, 100. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schäferhoff, B.; Depamphilis, C.W.; Müller, K.F. Disproportional plastome-wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Mol. Biol. Evol. 2014, 31, 529–545. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Michael, T.P.; Meer, E.J.; Pinheiro, D.G.; Varani, A.M.; Miranda, V.F.O. Comparative genomic analysis of Genlisea (corkscrew plants—Lentibulariaceae) chloroplast genomes reveals an increasing loss of the ndh genes. PLoS ONE 2018, 13, e0190321. [Google Scholar] [CrossRef] [Green Version]
- Ibarra-Laclette, E.; Lyons, E.; Hernández-Guzmán, G.; Pérez-Torres, C.A.; Carretero-Paulet, L.; Chang, T.H.; Lan, T.; Welch, A.J.; Juárez, M.J.A.; Simpson, J.; et al. Architecture and evolution of a minute plant genome. Nature 2013, 498, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Diaz, Y.C.A.; Penha, H.A.; Pinheiro, D.G.; Fernandes, C.C.; Miranda, V.F.O.; Michael, T.P.; Varani, A.M. The chloroplast genome of Utricularia reniformis sheds light on the evolution of the ndh gene complex of terrestrial carnivorous plants from the Lentibulariaceae family. PLoS ONE 2016, 11, e0165176. [Google Scholar] [CrossRef]
- Silva, S.R.; Pinheiro, D.G.; Meer, E.J.; Michael, T.P.; Varani, A.M.; Miranda, V.F.O. The complete chloroplast genome sequence of the leafy bladderwort, Utricularia foliosa L. (Lentibulariaceae). Conserv. Genet. Resour. 2017, 9, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.R.; Alvarenga, D.O.; Aranguren, Y.; Penha, H.A.; Fernandes, C.C.; Pinheiro, D.G.; Oliveira, M.T.; Michael, T.P.; Miranda, V.F.O.; Varani, A.M. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary landmarks. PLoS ONE 2017, 12, e0180484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jin, J.; Moore, M.J.; Yi, T.; Li, D. Plastome characteristics of Cannabaceae. Plant Divers. 2018, 40, 127–137. [Google Scholar] [CrossRef]
- Gargano, D.; Scotti, N.; Vezzi, A.; Bilardi, A.; Valle, G.; Grillo, S.; Cozzolino, S.; Cardi, T. Genome-wide analysis of plastome sequence variation and development of plastidial CAPS markers in common potato and related Solanum species. Genet. Resour. Crop Evol. 2012, 59, 419–430. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, Y.; Wang, Z.W.; Su, Y.J.; Wang, T. Evolution of the rpoB-psbZ region in fern plastid genomes: Notable structural rearrangements and highly variable intergenic spacers. BMC Plant Biol. 2011, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Yi, X.; Yang, Y.X.; Su, Y.J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.; Roper, J.; Duffy, A. The evolution of chloroplast genome structure in ferns. Genome 2010, 53, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Shikanai, T. The NAD(P)H Dehydrogenase Complex in Photosynthetic Organisms: Subunit Composition and Physiological Function. Funct. Plant Sci. Biotechnol. 2007, 1, 129–137. [Google Scholar]
- Sazanov, L.A.; Burrows, P.A.; Nixon, P.J. The plastid ndh genes code for an NADH-specific dehydrogenase: Isolation of a complex I analogue from pea thylakoid membranes. Proc. Natl. Acad. Sci. USA 2002, 95, 1319–1324. [Google Scholar] [CrossRef] [Green Version]
- Pearson, C.E.; Zorbas, H.; Price, G.B.; Zannis-Hadjopoulos, M. Inverted repeats, stem-loops, and cruciforms: Significance for initiation of DNA replication. J. Cell. Biochem. 1996, 63, 1–22. [Google Scholar] [CrossRef]
- Gupta, P.K.; Balyan, H.S.; Sharma, P.C.; Ramesh, B. Microsatellites in plants: A new class of molecular markers. Curr. Sci. 1996, 70, 45–54. [Google Scholar]
- Gao, C.; Ren, X.; Mason, A.S.; Li, J.; Wang, W.; Xiao, M.; Fu, D. Revisiting an important component of plant genomes: Microsatellites. Funct. Plant Biol. 2013, 40, 645–661. [Google Scholar] [CrossRef]
- Lawson, M.J.; Zhang, L. Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biol. 2006, 7, R14. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef]
- Astuti, G.; Petroni, G.; Adamec, L.; Miranda, V.F.O.; Peruzzi, L. DNA barcoding approach fails to discriminate Central European bladderworts (Utricularia, Lentibulariaceae), but provides insights concerning their evolution. Plant Biosyst. 2019. [Google Scholar] [CrossRef]
- Kress, W.J.; Wurdack, K.J.; Zimmer, E.A.; Weigt, L.A.; Janzen, D.H. Use of DNA barcodes to identify flowering plants. Proc. Natl. Acad. Sci. USA 2005, 102, 8369–8374. [Google Scholar] [CrossRef] [Green Version]
- Rorbach, J.; Bobrowicz, A.; Pearce, S.; Minczuk, M. Polyadenylation in bacteria and organelles. In Methods in Molecular Biology; Rorbach, J., Bobrowicz, A., Eds.; Humana Press: Totowa, NJ, USA, 2014; Volume 1125, pp. 211–227. [Google Scholar]
- Saschenbrecker, S.; Bracher, A.; Rao, K.V.; Rao, B.V.; Hartl, F.U.; Hayer-Hartl, M. Structure and Function of RbcX, an Assembly Chaperone for Hexadecameric Rubisco. Cell 2007, 129, 1189–1200. [Google Scholar] [CrossRef] [Green Version]
- Andrews, T.J.; Lorimer, G.H. Rubisco: Structure, Mechanisms, and Prospects for Improvement. In Photosynthesis; Hatch, M.D., Boardman, N.K., Eds.; Academic Press: Cambridge, MA, USA, 1987; pp. 131–218. ISBN 9780126754100. [Google Scholar]
- Nelson, N.; Yocum, C.F. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006, 57, 521–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesnicar, M.; Bendall, D.S. The photochemical activities and electron carriers of developing barley leaves. Biochem. J. 1973, 136, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.R.; Mason, H.S.; Mullet, J.E. Light-regulated translation of chloroplast proteins. I. Transcripts of psaA-psaB, psbA, and rbcL are associated with polysomes in dark-grown and illuminated barley seedlings. J. Cell Biol. 1988, 106, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Bock, R. Chloroplast translation: Structural and functional organization, operational control, and regulation. Plant Cell 2018, 30, 745–770. [Google Scholar] [CrossRef] [Green Version]
- Wolf, P.G.; Rowe, C.A.; Hasebe, M. High levels of RNA editing in a vascular plant chloroplast genome: Analysis of transcripts from the fern Adiantum capillus-veneris. Gene 2004, 339, 89–97. [Google Scholar] [CrossRef]
- Betts, M.J.; Russell, R.B. Amino-Acid Properties and Consequences of Substitutions. In Bioinformatics for Geneticists: A Bioinformatics Primer for the Analysis of Genetic Data, 2nd ed.; Barnes, M.R., Gray, I.C., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; ISBN 9780470026199. [Google Scholar]
- Wang, M.; Liu, H.; Ge, L.; Xing, G.; Wang, M.; Weining, S.; Nie, X. Identification and analysis of RNA editing sites in the chloroplast transcripts of Aegilops tauschii L. Genes (Basel) 2017, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Sugita, M.; Sugiura, M. Regulation of gene expression in chloroplasts of higher plants. Plant Mol. Biol. 1996, 32, 315–326. [Google Scholar] [CrossRef]
- Müller, K.; Borsch, T. Phylogenetics of Utricularia (Lentibulariaceae) and molecular evolution of the trnK intron in a lineage with high substitutional rates. Plant Syst. Evol. 2005, 250, 39–67. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Scheleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Nylander, J.A.A. MrModeltest v2.3. Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Lenz, H.; Hein, A.; Knoop, V. Plant organelle RNA editing and its specificity factors: Enhancements of analyses and new database features in PREPACT 3.0. BMC Bioinform. 2018, 19, 255. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U. amethystina Morphotypes | Purple | White | Yellow |
|---|---|---|---|
| Genbank accession number | MN223721 | MN223722 | MN223720 |
| Genome size (bp) | 150,115 | 150,332 | 150,020 |
| Large single copy (LSC) length (bp) | 82,388 (54.9%) | 82,561 (54.9%) | 82,256 (54.8%) |
| Small single copy (SSC) length (bp) | 16,969 (11.3%) | 17,070 (11.4%) | 16,870 (11.3%) |
| Inverted repeats (IRa+IRb) length (bp) | 25,399 (34.7%) | 25,350 (33.8%) | 25,447 (34.0%) |
| Noncoding regions (bp) | 40,199 | 41,901 | 41,259 |
| Intronic regions(bp) | 18,052 | 19,260 | 20,280 |
| %GC | 37.5 | 37.5 | 37.7 |
| Coverage | 84× | 119× | 123× |
| Morphotype | Gene | Genome (cpDNA) | Codon | Codon Position | Amino Acid | Editing Level | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (Each Biorep) | |||||||||||
| Purple | Position | Strand | from | to | from | to | P1 | P2 | P3 | ||
| rps14 * | 36,640 | - | UCA | UUA | 2 | S | L | 96 | 100 | 100 | |
| petB | 74,797 | + | CCA | CUA | 2 | P | L | 100 | 100 | 97 | |
| White | W1 | W2 | W3 | ||||||||
| ndhB | 138,239 | + | CCA | CUA | 2 | H | Y | 92 | 82 | 0 | |
| ndhD | 113,305 | - | CUA | UUA | 1 | Synonym | 100 | 0 | 100 | ||
| rbcL | 55,651 | + | GCC | GCU | 3 | Synonym | 100 | 100 | 0 | ||
| 55,777 | + | AUC | AUU | 3 | Synonym | 100 | 100 | 0 | |||
| rps14 * | 36,572 | - | UCA | UUA | 2 | S | L | 38 | 14 | 75 | |
| 36,497 | - | CCA | CUA | 2 | P | L | 100 | 0 | 72 | ||
| 36,553 | - | AAC | AAU | 3 | Synonym | 100 | 100 | 0 | |||
| psbB | 72,140 | + | GGC | GGU | 3 | Synonym | 100 | 100 | 0 | ||
| 71,372 | + | UAC | UAU | 3 | Synonym | 100 | 0 | 100 | |||
| psaA | 39,370 | - | AUC | AUU | 3 | Synonym | 100 | 100 | 100 | ||
| petB | 74,816 | + | CCA | CUA | 2 | P | L | 100 | 0 | 97 | |
| Yellow | Y1 | Y2 | - | ||||||||
| rps14 | 36,439 | - | CCA | CUA | 2 | P | L | 69 | 71 | - | |
| ccsA | 111,950 | + | CUA | UUA | 1 | Synonym | 18 | 16 | - | ||
| petB | 74,948 | + | CCA | CUA | 2 | P | L | 100 | 100 | - | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.R.; Pinheiro, D.G.; Penha, H.A.; Płachno, B.J.; Michael, T.P.; Meer, E.J.; Miranda, V.F.O.; Varani, A.M. Intraspecific Variation within the Utricularia amethystina Species Morphotypes Based on Chloroplast Genomes. Int. J. Mol. Sci. 2019, 20, 6130. https://doi.org/10.3390/ijms20246130
Silva SR, Pinheiro DG, Penha HA, Płachno BJ, Michael TP, Meer EJ, Miranda VFO, Varani AM. Intraspecific Variation within the Utricularia amethystina Species Morphotypes Based on Chloroplast Genomes. International Journal of Molecular Sciences. 2019; 20(24):6130. https://doi.org/10.3390/ijms20246130
Chicago/Turabian StyleSilva, Saura R., Daniel G. Pinheiro, Helen A. Penha, Bartosz J. Płachno, Todd P. Michael, Elliott J. Meer, Vitor F. O. Miranda, and Alessandro M. Varani. 2019. "Intraspecific Variation within the Utricularia amethystina Species Morphotypes Based on Chloroplast Genomes" International Journal of Molecular Sciences 20, no. 24: 6130. https://doi.org/10.3390/ijms20246130
APA StyleSilva, S. R., Pinheiro, D. G., Penha, H. A., Płachno, B. J., Michael, T. P., Meer, E. J., Miranda, V. F. O., & Varani, A. M. (2019). Intraspecific Variation within the Utricularia amethystina Species Morphotypes Based on Chloroplast Genomes. International Journal of Molecular Sciences, 20(24), 6130. https://doi.org/10.3390/ijms20246130