Identification of Differentially Expressed Genes Involved in the Molecular Mechanism of Pericarp Elongation and Differences in Sucrose and Starch Accumulation between Vegetable and Grain Pea (Pisum sativum L.)

 ,
,
Abstract
:1. Introduction
2. Results
2.1. The Phenotypic Traits of Vegetable and Grain Pea Pericarp at Five Developmental Stages
2.2. RNA-seq of the Pea Pericarp Transcriptome
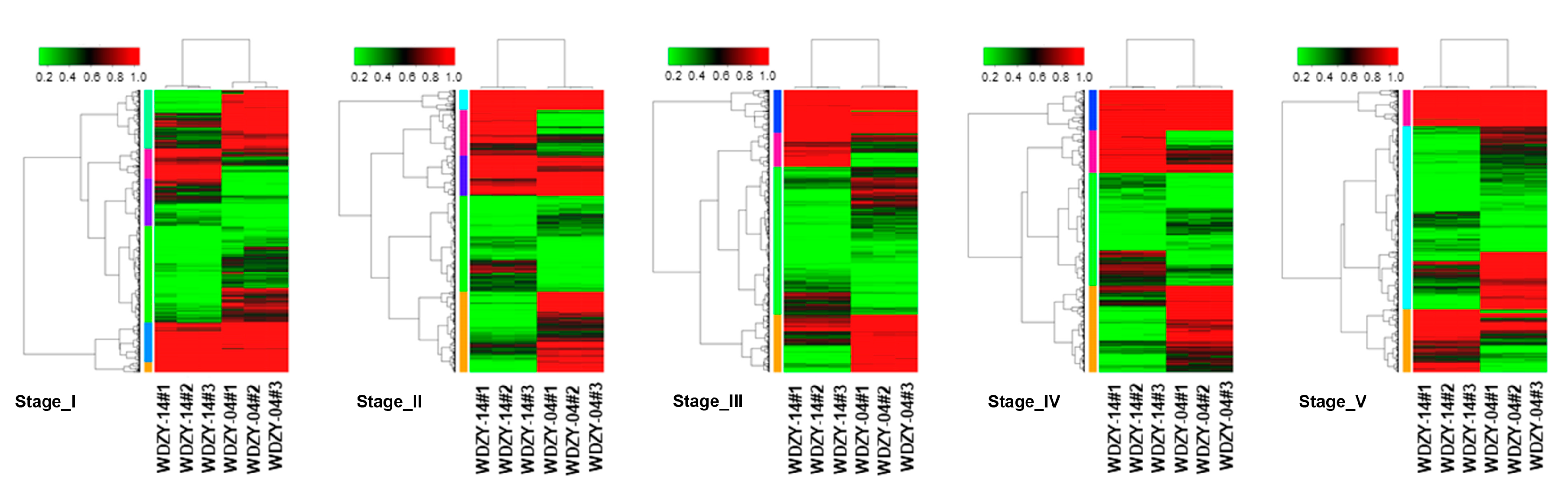
2.3. Analysis of the Expression Level and Differentially Expressed Genes (DEGs)
2.4. GO and KEGG Classification
2.5. DEGs Related to Pod Elongation
2.6. DEGs Related to Pod Sucrose Metabolism
2.7. DEGs Related to Pericarp Starch Synthesis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Measurement of the Phenotypic Traits of the Two Cultivars
4.3. Illumina Sequencing and Mapping
4.4. Expression Level Analysis and Gene Annotation
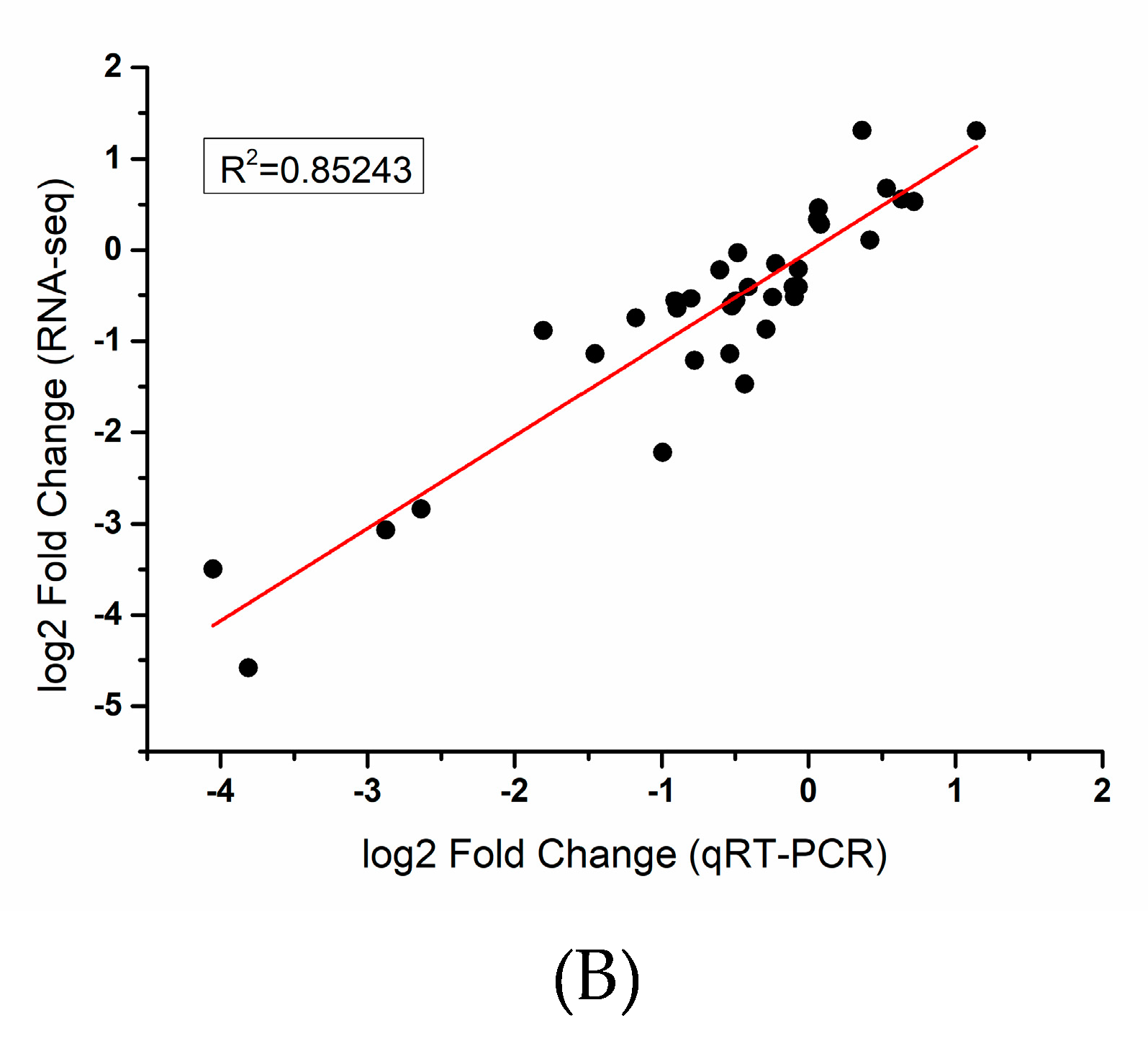
4.5. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bataillon, T. Plant Genetic Resources of Legumes in the Mediterranean. Plant Sci. 2002, 162. [Google Scholar] [CrossRef]
- Vanhala, T.; Normann, K.R.; Lundström, M.; Weller, J.L.; Leino, M.W.; Hagenblad, J. Flowering time adaption in Swedish landrace pea (Pisum sativum L.). BMC Genet. 2016, 17, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonet, M.A.; Valles, J. Ethnobotany of Montseny biosphere reserve (Catalonia, Iberian Peninsula): Plants used in veterinary medicine. J. Ethnopharmacol. 2007, 110, 130–147. [Google Scholar] [CrossRef] [PubMed]
- Zilani, M.N.H.; Sultana, T.; Asabur Rahman, S.M.; Anisuzzman, M.; Islam, M.A.; Shilpi, J.A.; Hossain, M.G. Chemical composition and pharmacological activities of Pisum sativum. BMC Complement. Altern. Med. 2017, 17, 171. [Google Scholar] [CrossRef] [Green Version]
- Rungruangmaitree, R.; Jiraungkoorskul, W. Pea, Pisum sativum, and Its Anticancer Activity. Pharmacogn. Rev. 2017, 11, 39–42. [Google Scholar] [CrossRef] [Green Version]
- FAO (Food and Agriculture Organization of the United Nations). Stat. Database 2017, 12. Available online: http://faostat.fao.org/ (accessed on 28 September 2012).
- Croy, R.R.; Gatehouse, J.A.; Tyler, M.; Boulter, D. The purification and characterization of a third storage protein (convicilin) from the seeds of pea (Pisum sativum L.). Biochem. J. 1980, 191, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Breeding legumes for nutritional value. Proc. Nutr. Soc. 1982, 41, 1–94. [CrossRef] [Green Version]
- Croy, R.R.; Hoque, M.S.; Gatehouse, J.A.; Boulter, D. The major albumin proteins from pea (Pisum sativum L). Purification and some properties. Biochem. J. 1984, 218, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Zinsmeister, J.; Lalanne, D.; Terrasson, E.; Chatelain, E.; Vandecasteele, C.; Vu, B.L.; Dubois-Laurent, C.; Geoffriau, E.; Signor, C.L.; Dalmais, M.; et al. ABI5 Is a Regulator of Seed Maturation and Longevity in Legumes. Plant Cell 2016, 28, 2735–2754. [Google Scholar] [CrossRef] [Green Version]
- Storey, R.; Beevers, L. Enzymology of Glutamine Metabolism Related to Senescence and Seed Development in the Pea (Pisum sativum L.). Plant Physiol. 1978, 61, 494–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Lin, Z.; Guan, L.; Gaughan, G.; Lin, G. Antioxidant enzymes regulate reactive oxygen species during pod elongation in Pisum sativum and Brassica chinensis. PLoS ONE 2014, 9, e87588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.-M.; Xu, S.-C.; Mao, W.-H.; Hu, Q.-Z.; Zhang, G.-W.; Ding, J.; Li, Y.-D. Developing new SSR markers from ESTs of pea (Pisum sativum L.). J. Zhejiang Univ. Sci. B 2010, 11, 702–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teshome, A.; Bryngelsson, T.; Dagne, K.; Geleta, M. Assessment of genetic diversity in Ethiopian field pea (Pisum sativum L.) accessions with newly developed EST-SSR markers. BMC Genet. 2015, 16, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Pembleton, L.W.; Cogan, N.O.I.; Savin, K.W.; Leonforte, T.; Paull, J.; Materne, M.; Forster, J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom. 2012, 13, 104. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Chen, X.-P.; Hu, W.-S.; Zheng, S.-Q. Identification of differentially expressed genes implicated in peel color (red and green) of Dimocarpus confinis. SpringerPlus 2016, 5, 1088. [Google Scholar] [CrossRef] [Green Version]
- Abdul Aziz, N.A.; Wong, L.M.; Bhat, R.; Cheng, L.H. Evaluation of processed green and ripe mango peel and pulp flours (Mangifera indica var. Chokanan) in terms of chemical composition, antioxidant compounds and functional properties. J. Sci. Food Agric. 2012, 92, 557–563. [Google Scholar] [CrossRef]
- Denis, M.C.; Furtos, A.; Dudonne, S.; Montoudis, A.; Garofalo, C.; Desjardins, Y.; Delvin, E.; Levy, E. Apple peel polyphenols and their beneficial actions on oxidative stress and inflammation. PLoS ONE 2013, 8, e53725. [Google Scholar] [CrossRef]
- Taha, K.F.; Hetta, M.H.; Ali, M.E.; Yassin, N.Z.; El Guindi, O.D. The pericarp of Pisum sativum L. (Fabaceae) as a biologically active waste product. Plant. Med. 2011, 12. [Google Scholar] [CrossRef]
- Xue, L.H.; Zhang, J.X.; Yan, X.H.; Li, Q. A study on relationship between growth and seed number and seed weight of soybean pod. J. Xinj. Agric. Univ. 2008, 4, 16–19. [Google Scholar]
- Liu, H.M.; Li, Y.; Bu, G.J.; Lv, W.; Cui, L.; Cang, J.; Wang, X.D. Effects of photosynthate transportation and distrubution in soybean pods on the development of soybean seeds. J. Nucl. Agric. Sci. 2008, 4, 519–523. [Google Scholar]
- Chia, J.M.; Ware, D. Sequencing for the cream of the crop. Nat. Biotechnol. 2011, 29, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Hradilová, I.; Trněný, O.; Válková, M.; Cechová, M.; Janská, A.; Prokešová, L.; Aamir, K.; Krezdorn, N.; Rotter, B.; Winter, P.; et al. A Combined Comparative Transcriptomic, Metabolomic, and Anatomical Analyses of Two Key Domestication Traits: Pod Dehiscence and Seed Dormancy in Pea (Pisum sp.). Front. Plant Sci. 2017, 8, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Zhang, G.; Xu, S.; Mao, W.; Hu, Q.; Gong, Y. Comparative Transcriptomic Analyses of Vegetable and Grain Pea (Pisum sativum L.) Seed Development. Front. Plant Sci. 2015, 6, 1039. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Liszkay, A.; van der Zalm, E.; Schopfer, P. Production of reactive oxygen intermediates (O2˙−, H2O2, and ˙OH) by maize roots and their role in wall loosening and elongation growth. Plant Physiol. 2004, 136, 3114–3123. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.L.; Jin, Y.; Yang, Y.J.; Li, G.J.; Boyer, J.S. Sugar input, metabolism, and signaling mediated by invertase: Roles in development, yield potential, and response to drought and heat. Mol. Plant 2010, 3, 942–955. [Google Scholar] [CrossRef]
- Sturm, A. Invertases. Primary structures, functions, and roles in plant development and sucrose partitioning. Plant Physiol. 1999, 121, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nougué, O.; Corbi, J.; Ball, S.G.; Manicacci, D.; Tenaillon, M.I. Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC Evolut. Boil. 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.Y.; Du, W.G.; Zhang, G.R.; Man, W.Q.; Li, G.Q.; Wang, X.W.; Ge, Q.Y.; Hao, N.B.; Gui, M.Z.; Chen, Y. Observation on photosynthetic structure system of soybean pods. J. Chin. Electron. Microsc. Soc. 2003, 22, 280–285. [Google Scholar]
- Buttery, B.R.; Buzzell, R.I.; Findlay, W.I. Relationships among photosynthetic rate, bean yield and other characters in firld-grown cultivars of soybean. Can. J. Plant Sci. 1981, 61, 191–198. [Google Scholar] [CrossRef]
- Diao, X.N.; Li, W.; Gao, C.Y.; Zhang, Y.P.; Wang, M.; Zhao, J.Z.; Du, W.J.; Yue, A.Q. Dynamic Accumulation Analysis of Nutritional. J. Chin. Cereals Oils Assoc. 2018, 33, 32–37. [Google Scholar]
- Chen, H.; Jiang, B.J.; Zhang, C.; Cai, T.C.; Zeng, J.B.; Deng, Y.; Zhuang, W.J. Construction and primary analysis of peanut pericarp full-length cDNA library. J. Fujian Agric. For. Univ. 2013, 42, 57–62. [Google Scholar]
- Fry, S.C.; Dumville, J.C.; Miller, J.G. Fingerprinting of polysaccharides attacked by hydroxyl radicals in vitro and in the cell walls of ripening pear fruit. Biochem. J. 2001, 357, 729–737. [Google Scholar] [CrossRef]
- Boyer, J.S.; McLaughlin, J.E. Functional reversion to identify controlling genes in multigenic responses: Analysis of floral abortion. J. Exp. Bot. 2007, 58, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Ni, D.A.; Ruan, Y.L. Posttranslational elevation of cell wall invertase activity by silencing its inhibitor in tomato delays leaf senescence and increases seed weight and fruit hexose level. Plant. Cell 2009, 21, 2072–2089. [Google Scholar] [CrossRef] [Green Version]
- Weber, H.; Borisjuk, L.; Wobus, U. Molecular physiology of legume seed development. Ann. Rev. Plant Boil. 2005, 56, 253–279. [Google Scholar] [CrossRef]
- Coleman, H.D.; Yan, J.; Mansfield, S.D. Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 13118–13123. [Google Scholar] [CrossRef] [Green Version]
- Chourey, P.S.; Taliercio, E.W.; Carlson, S.J.; Ruan, Y.L. Genetic evidence that the two isozymes of sucrose synthase present in developing maize endosperm are critical, one for cell wall integrity and the other for starch biosynthesis. Mol. Gen. Genet. MGG 1998, 259, 88–96. [Google Scholar] [PubMed]
- Liu, Y.H.; Cao, J.S.; Li, G.J.; Wu, X.H.; Wang, B.G.; Xu, P.; Hu, T.T.; Lu, Z.F.; Patrick, J.W.; Ruan, Y.L. Genotypic differences in pod wall and seed growth relate to invertase activities and assimilate transport pathways in asparagus bean. Ann. Bot. 2012, 109, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gepts, P.; Beavis, W.D.; Brummer, E.C.; Shoemaker, R.C.; Stalker, H.T.; Weeden, N.F.; Young, N.D. Legumes as a model plant family. Genomics for food and feed report of the Cross-Legume Advances through Genomics Conference. Plant Physiol. 2005, 137, 1228–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakkanong, K.; Yang, J.H.; Zhang, M.F. Starch accumulation and starch related genes expression in novel inter-specific inbred squash line and their parents during fruit development. Sci. Hortic. 2012, 136, 1–8. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptome Sample | Total Clean Reads | Clean Bases (bp) | Q30 (%) | GC (%) | Total Mapped Reads (%) |
|---|---|---|---|---|---|
| WDZY-04_ I#1 | 50,177,200 | 7,327,792,349 | 96.765 | 43.575 | 44,475,396 (88.6%) |
| WDZY-04_ I#2 | 49,061,332 | 7,188,004,754 | 96.825 | 43.245 | 43,387,875 (88.4%) |
| WDZY-04_ I#3 | 50,406,494 | 7,306,478,571 | 96.675 | 43.37 | 44,730,739 (88.7%) |
| WDZY-04_ II#1 | 52,539,172 | 7,662,278,764 | 96.74 | 43.3 | 46,613,759 (88.7%) |
| WDZY-04_ II#2 | 43,217,524 | 6,246,391,018 | 96.585 | 43.475 | 38,215,715 (88.4%) |
| WDZY-04_ II#3 | 64,372,148 | 9,362,931,919 | 96.835 | 43.36 | 57,145,999 (88.8%) |
| WDZY-04_ III#1 | 70,119,664 | 10,202,409,573 | 96.815 | 43.245 | 62,179,404 (88.7%) |
| WDZY-04_ III#2 | 42,849,086 | 6,201,019,405 | 96.655 | 43.22 | 37,836,945 (88.3%) |
| WDZY-04_ III#3 | 50,627,610 | 7,391,282,480 | 97.485 | 43.045 | 45,179,447 (89.2%) |
| WDZY-04_ IV#1 | 46,268,006 | 6,789,317,950 | 96.42 | 43.15 | 40,851,769 (88.3%) |
| WDZY-04_ IV#2 | 55,534,482 | 8,050,533,372 | 96.705 | 43.16 | 49,204,948 (88.6%) |
| WDZY-04_ IV#3 | 44,742,284 | 6,511,953,243 | 97.725 | 43.335 | 39,799,834 (89.0%) |
| WDZY-04_ V#1 | 51,782,254 | 7,463,115,630 | 96.84 | 43.2 | 44,300,719 (85.6%) |
| WDZY-04_ V#2 | 64,122,736 | 9,251,735,817 | 96.825 | 43.435 | 54,682,602 (85.3%) |
| WDZY-04_ V#3 | 51,512,076 | 7,460,292,563 | 96.78 | 43.405 | 43,870,727 (85.2%) |
| WDZY-14_ I#1 | 53,175,366 | 7,763,234,698 | 97.45 | 43.96 | 48,054,402 (90.4%) |
| WDZY-14_ I#2 | 46,601,580 | 6,807,140,735 | 97.435 | 43.93 | 42,061,045 (90.3%) |
| WDZY-14_ I#3 | 48,438,836 | 7,005,642,104 | 97.1 | 43.855 | 43,603,612 (90.0%) |
| WDZY-14_ II#1 | 48,696,214 | 7,105,553,520 | 97.425 | 43.59 | 43,878,335 (90.1%) |
| WDZY-14_ II#2 | 88,115,270 | 12,807,329,980 | 97.37 | 43.63 | 79,369,026 (90.1%) |
| WDZY-14_ II#3 | 47,762,154 | 6,956,660,221 | 97.43 | 43.425 | 43,127,846 (90.3%) |
| WDZY-14_ III#1 | 46,007,202 | 6,562,236,455 | 97.19 | 43.565 | 40,383,101 (87.8%) |
| WDZY-14_ III#2 | 77,181,058 | 11,150,851,050 | 97.51 | 43.715 | 68,087,616 (88.2%) |
| WDZY-14_ III#3 | 73,287,766 | 10,600,678,854 | 97.46 | 43.68 | 64,597,938 (88.1%) |
| WDZY-14_ IV#1 | 46,437,750 | 6,721,682,138 | 96.93 | 43.925 | 41,388,924 (89.1%) |
| WDZY-14_ IV#2 | 52,647,274 | 7,628,273,045 | 97.44 | 43.43 | 47,093,257 (89.5%) |
| WDZY-14_ IV#3 | 56,984,532 | 8,236,432,557 | 96.845 | 43.255 | 50,542,076 (88.7%) |
| WDZY-14_ V#1 | 58,306,756 | 8,466,331,576 | 97.485 | 44.04 | 46,961,785 (80.5%) |
| WDZY-14_ V#2 | 45,359,128 | 6,567,548,081 | 97.49 | 44.155 | 36,311,644 (80.1%) |
| WDZY-14_ V#3 | 65,632,458 | 9,522,635,831 | 97.47 | 44.01 | 52,782,939 (80.4%) |
| Function | Gene_ID (NR database _id) | Growth Stage | WDZY -04#1 (FPKM) | WDZY -04#2 (FPKM) | WDZY -04#3 (FPKM) | WDZY -14#1 (FPKM) | WDZY -14#2 (FPKM) | WDZY -14#3 (FPKM) |
|---|---|---|---|---|---|---|---|---|
| SOD (superoxide dismutase) | XLOC_013249 (CAD42655.1) | Stage III | 0.84 | 0.92 | 1.48 | 0.32 | 0.19 | 0.19 |
| XLOC_030516 (XP_013461079.1) | Stage V | 6.89 | 4.96 | 5.59 | 2.06 | 1.56 | 2.14 | |
| XLOC_038705 (XP_004508271.1) | Stage V | 7.67 | 7.41 | 7.77 | 2.66 | 2.17 | 2.11 | |
| POD (peroxidase) | XLOC_016611 (BAD97435.1) | Stage I | 12.48 | 14.26 | 7.99 | 0.67 | 0.77 | 0.52 |
| XLOC_007148 (BAD97436.1) | Stage I | 156.02 | 161.58 | 124.67 | 51.6 | 55.49 | 54.04 | |
| XLOC_034947 (BAD97436.1) | Stage I | 99.49 | 99.11 | 82.03 | 4.86 | 5.75 | 5.72 | |
| XLOC_037479 (BAD97439.1) | Stage I | 38.62 | 37.61 | 37.81 | 10.94 | 12.67 | 15.38 | |
| XLOC_031402 (CAA09881.1) | Stage I | 259.53 | 233.79 | 187.8 | 60.94 | 63.29 | 70.8 | |
| XLOC_006267 (BAD97438.1) | Stage I | 44.78 | 41.83 | 36.77 | 14.98 | 15.67 | 15.38 | |
| XLOC_006821 (BAD97436.1) | Stage I | 2.94 | 1.66 | 1.73 | 0 | 0.18 | 0 | |
| XLOC_006267 (BAD97438.1) | Stage II | 14.48 | 14.26 | 13.78 | 5.87 | 6.66 | 6.89 | |
| XLOC_016611 (BAD97435.1) | Stage II | 27.55 | 22.79 | 25.32 | 1.18 | 1.55 | 1.83 | |
| XLOC_016611 (BAD97435.1) | Stage III | 26.89 | 28.71 | 30.24 | 2.3 | 2.01 | 1.81 | |
| XLOC_018196 (XP_010104370.1) | Stage III | 0.28 | 0.07 | 0.32 | 0 | 0 | 0 | |
| XLOC_007148 (BAD97436.1) | Stage III | 16.03 | 14.61 | 14.63 | 2.5 | 1.93 | 1.74 | |
| XLOC_034947 (BAD97436.1) | Stage III | 1.18 | 1.42 | 1.45 | 0.38 | 0.09 | 0.15 | |
| XLOC_031402 (CAA09881.1) | Stage III | 40.27 | 39.64 | 36.16 | 6.84 | 6.46 | 5.4 | |
| XLOC_006267 (BAD97438.1) | Stage III | 27.95 | 26.15 | 25.67 | 7.26 | 5.53 | 5.6 | |
| XLOC_016611 (BAD97435.1) | Stage IV | 26.56 | 24.06 | 21.55 | 0.91 | 0.69 | 0.74 | |
| XLOC_034947 (BAD97436.1) | Stage IV | 0.27 | 0.16 | 0.16 | 2.38 | 3.29 | 3.73 | |
| XLOC_037479 (BAD97439.1) | Stage IV | 0.43 | 1 | 0.48 | 1.61 | 1.62 | 1.81 | |
| XLOC_031402 (CAA09881.1) | Stage IV | 0.62 | 0.99 | 0.89 | 2.57 | 2.7 | 4.32 | |
| XLOC_007148 (BAD97436.1) | Stage V | 186.77 | 214.85 | 207.05 | 16.61 | 12.58 | 12.14 | |
| peroxisome | XLOC_010379 (XP_013470503.1) | Stage I | 0.8 | 1.2 | 1.46 | 0.17 | 0.24 | 0.5 |
| XLOC_010379 (XP_013470503.1) | Stage III | 1.48 | 1.55 | 1.92 | 0.33 | 0.47 | 0.51 | |
| XLOC_010379 (XP_013470503.1) | Stage IV | 0.74 | 0.96 | 1.06 | 1.93 | 2.08 | 2.06 | |
| XLOC_010379 (XP_013470503.1) | Stage V | 0.49 | 0.33 | 0.28 | 0.77 | 0.55 | 0.72 | |
| ko(04146) | XLOC_019651 (GAU39672.1) | Stage I | 71.72 | 61.62 | 56.4 | 0 | 0 | 0 |
| XLOC_019651 (GAU39672.1) | Stage II | 36.91 | 42.82 | 46.38 | 0 | 0 | 0 | |
| XLOC_019651 (GAU39672.1) | Stage III | 50.38 | 54.52 | 25.55 | 0 | 0 | 0 | |
| XLOC_019651 (GAU39672.1) | Stage IV | 27.28 | 15.42 | 23.63 | 0 | 0 | 0 | |
| XLOC_019651 (GAU39672.1) | Stage V | 38.71 | 26.16 | 19.8 | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, P.; Li, Z.; Wu, C.; Luo, Y.; Li, J.; Wang, P.; Gao, X.; Gao, J.; Feng, B. Identification of Differentially Expressed Genes Involved in the Molecular Mechanism of Pericarp Elongation and Differences in Sucrose and Starch Accumulation between Vegetable and Grain Pea (Pisum sativum L.). Int. J. Mol. Sci. 2019, 20, 6135. https://doi.org/10.3390/ijms20246135
Yang P, Li Z, Wu C, Luo Y, Li J, Wang P, Gao X, Gao J, Feng B. Identification of Differentially Expressed Genes Involved in the Molecular Mechanism of Pericarp Elongation and Differences in Sucrose and Starch Accumulation between Vegetable and Grain Pea (Pisum sativum L.). International Journal of Molecular Sciences. 2019; 20(24):6135. https://doi.org/10.3390/ijms20246135
Chicago/Turabian StyleYang, Pu, Zhonghao Li, Caoyang Wu, Yan Luo, Jing Li, Pengke Wang, Xiaoli Gao, Jinfeng Gao, and Baili Feng. 2019. "Identification of Differentially Expressed Genes Involved in the Molecular Mechanism of Pericarp Elongation and Differences in Sucrose and Starch Accumulation between Vegetable and Grain Pea (Pisum sativum L.)" International Journal of Molecular Sciences 20, no. 24: 6135. https://doi.org/10.3390/ijms20246135