Less Cytotoxic Protoflavones as Antiviral Agents: Protoapigenone 1′-O-isopropyl ether Shows Improved Selectivity Against the Epstein–Barr Virus Lytic Cycle

 ,
,  , , , ,
, , , ,  ,
, 
Abstract
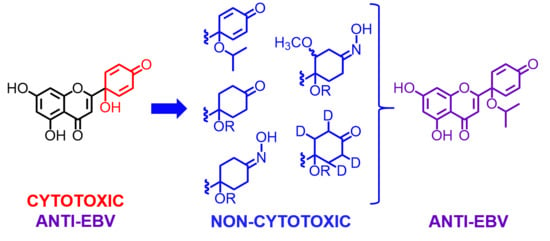
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
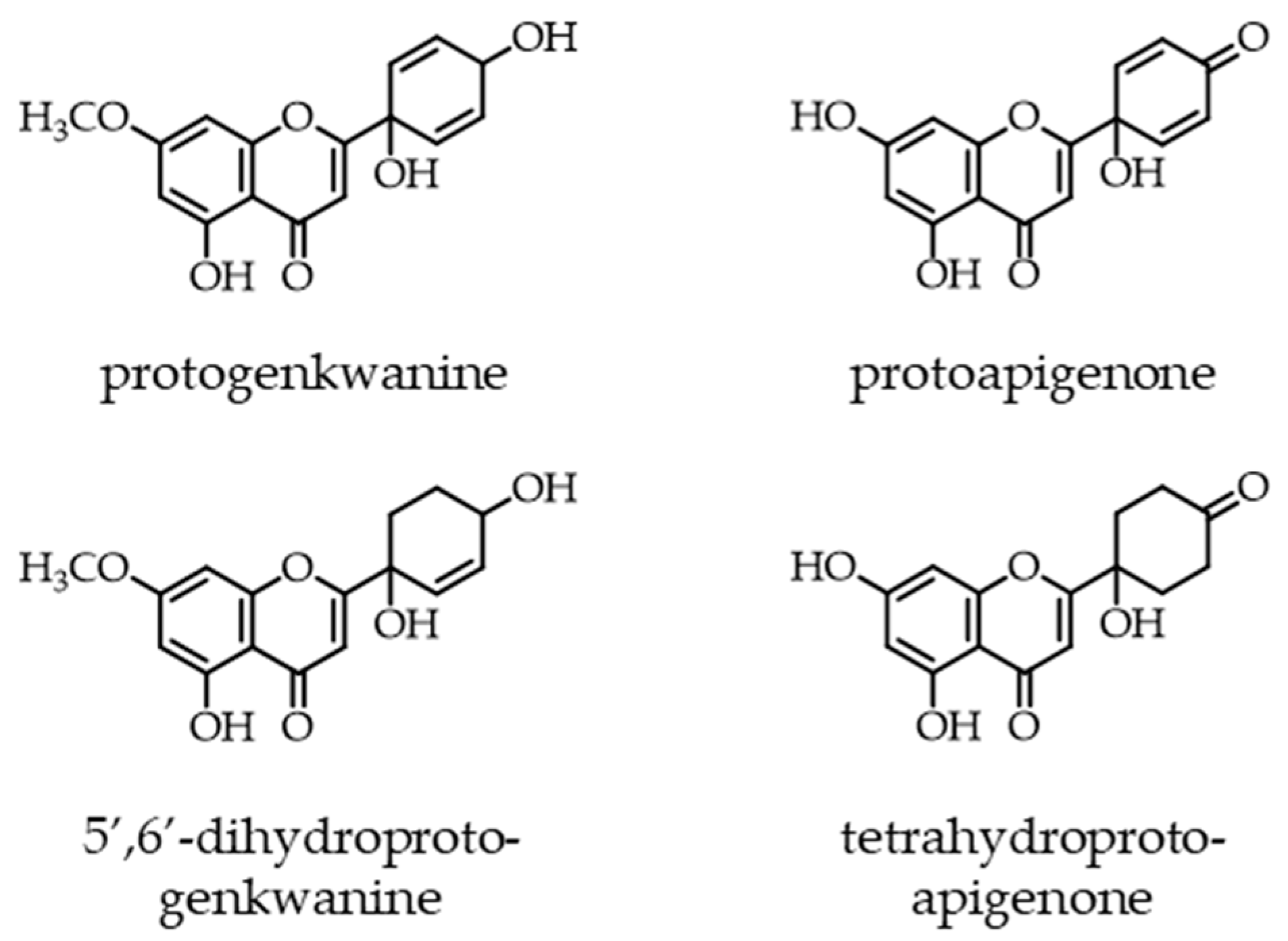
2.1. Synthesis of B-Ring Modified Protoapigenone Analogs
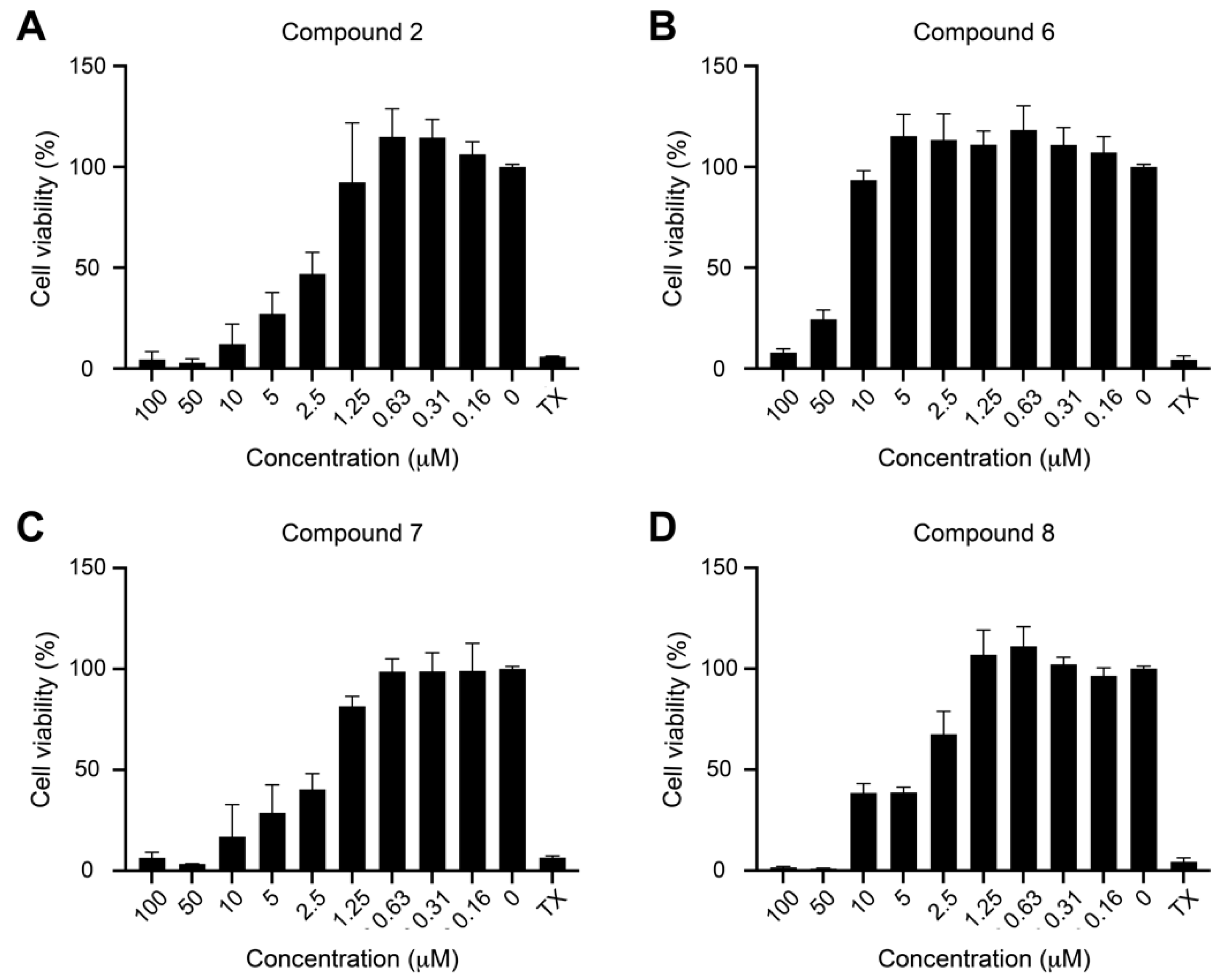
2.2. Biological Activities
2.2.1. Antiretroviral Activity
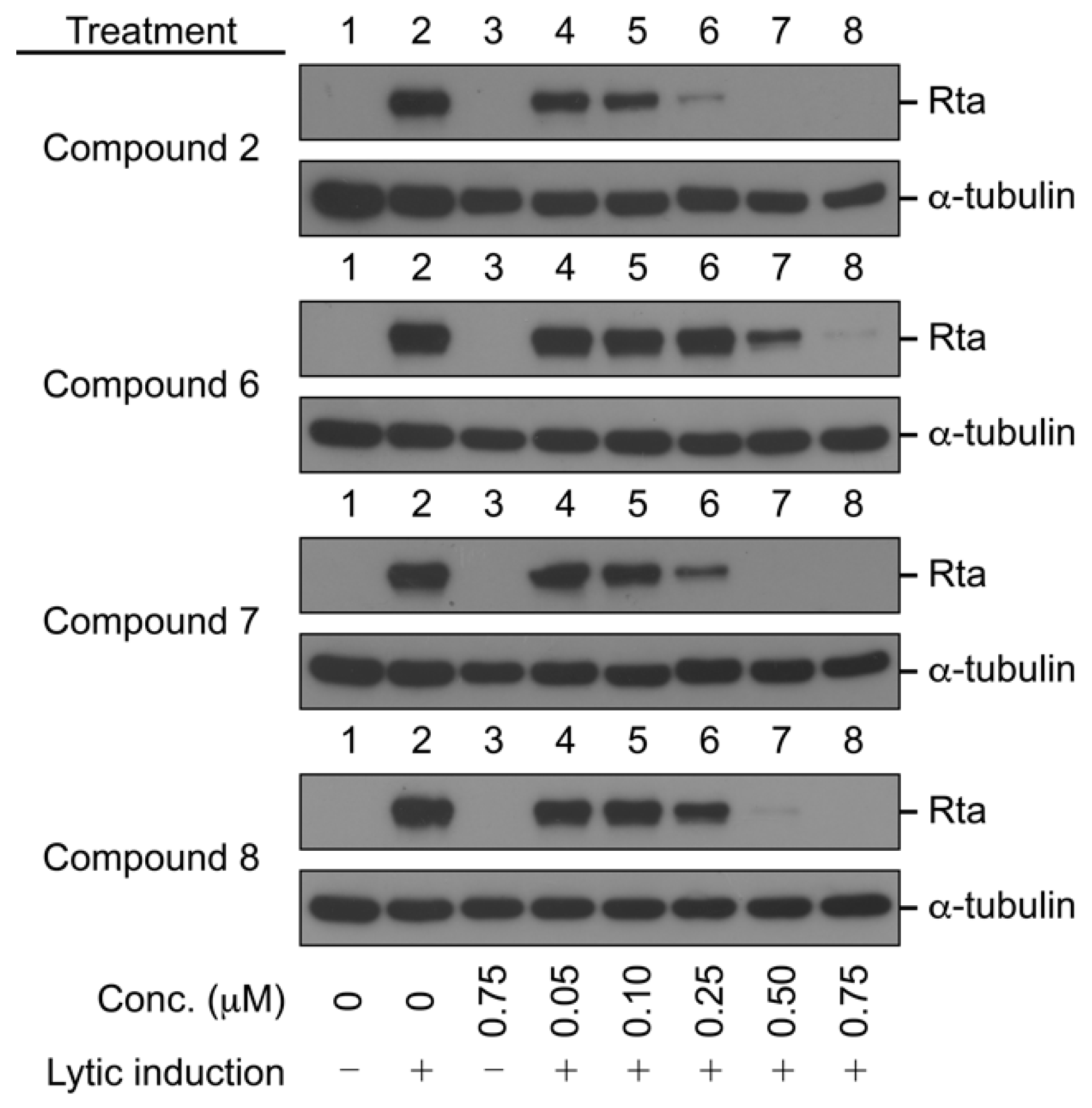
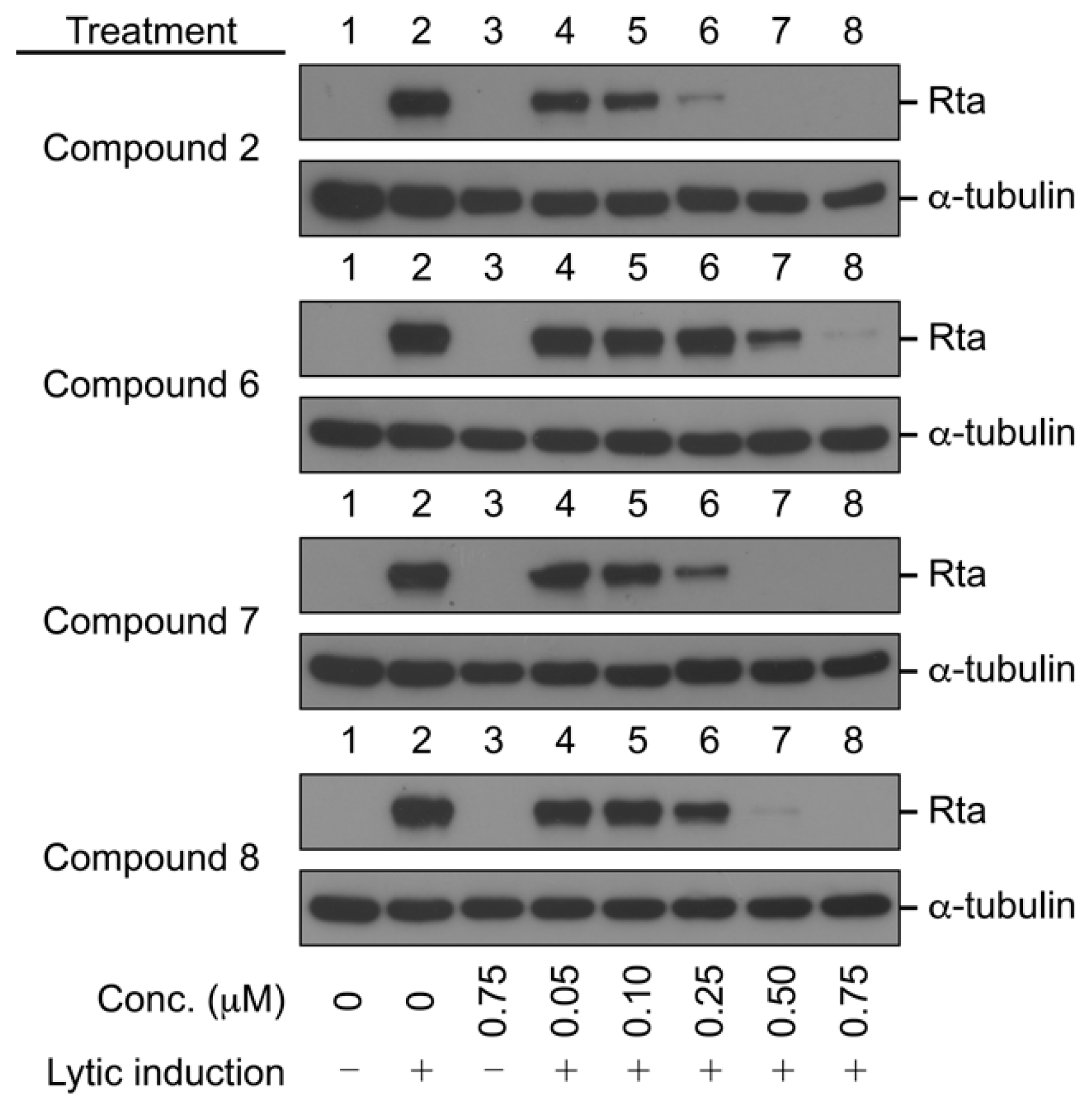
2.2.2. Antiviral Activity against EBV
3. Materials and Methods
3.1. Synthesis and Chromatographic Purification
3.1.1. Preparation of Protoapigenone Derivatives 2–8
3.1.2. Preparation of Tetrahydroprotoapigenone Derivatives 9–14
3.1.3. Preparation of Tetradeuteroprotoapigenone Derivatives 15–20
3.1.4. Preparation of Protoflavone 4′-oxime Derivatives 21–26
3.1.5. Preparation of Tetrahydroprotoflavone 4′-oxime Derivatives 27–28
3.2. Structure Elucidation
3.3. Cell Lines and Viruses
3.4. Anti-HIV Testing
3.5. Immunoblot Analysis of EBV Rta Protein
3.6. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hunyadi, A.; Martins, A.; Danko, B.; Chang, F.R.; Wu, Y.C. Protoflavones: A class of unusual flavonoids as promising novel anticancer agents. Phytochem. Rev. 2014, 13, 69–77. [Google Scholar] [CrossRef]
- Danko, B.; Toth, S.; Martins, A.; Vagvolgyi, M.; Kusz, N.; Molnar, J.; Chang, F.R.; Wu, Y.C.; Szakacs, G.; Hunyadi, A. Synthesis and SAR Study of Anticancer Protoflavone Derivatives: Investigation of Cytotoxicity and Interaction with ABCB1 and ABCG2 Multidrug Efflux Transporters. ChemMedChem 2017, 12, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Stanković, T.; Dankó, B.; Martins, A.; Dragoj, M.; Stojković, S.; Isaković, A.; Wang, H.-C.; Wu, Y.-C.; Hunyadi, A.; Pešić, M. Lower antioxidative capacity of multidrug-resistant cancer cells confers collateral sensitivity to protoflavone derivatives. Cancer Chemother. Pharmacol. 2015, 76, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Lee, A.Y.; Chou, W.C.; Wu, C.C.; Tseng, C.N.; Liu, K.Y.; Lin, W.L.; Chang, F.R.; Chuang, D.W.; Hunyadi, A.; et al. Inhibition of ATR-dependent signaling by protoapigenone and its derivative sensitizes cancer cells to interstrand cross-link-generating agents in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1443–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.S.; Nakagawa-Goto, K.; Chang, F.R.; Yu, D.; Morris-Natschke, S.L.; Wu, C.C.; Chen, S.L.; Wu, Y.C.; Lee, K.H. First total synthesis of protoapigenone and its analogues as potent cytotoxic agents. J. Med. Chem. 2007, 50, 3921–3927. [Google Scholar] [CrossRef] [Green Version]
- Hunyadi, A.; Chuang, D.W.; Danko, B.; Chiang, M.Y.; Lee, C.L.; Wang, H.C.; Wu, C.C.; Chang, F.R.; Wu, Y.C. Direct semi-synthesis of the anticancer lead-drug protoapigenone from apigenin, and synthesis of further new cytotoxic protoflavone derivatives. PLoS ONE 2011, 6, e23922. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.S.; Chang, F.R.; Wu, C.C.; Liaw, C.C.; Wu, Y.C. New cytotoxic flavonoids from Thelypteris torresiana. Planta Med. 2005, 71, 867–870. [Google Scholar] [CrossRef]
- Hunyadi, A.; Martins, A.; Danko, B.; Chuang, D.-W.; Trouillas, P.; Chang, F.-R.; Wu, Y.-C.; Falkay, G. Discovery of the first non-planar flavonoid that can strongly inhibit xanthine oxidase: Protoapigenone 1′-O-propargyl ether. Tetrahedron. Lett. 2013, 54, 6529–6532. [Google Scholar] [CrossRef]
- Tung, C.P.; Chang, F.R.; Wu, Y.C.; Chuang, D.W.; Hunyadi, A.; Liu, S.T. Inhibition of the Epstein-Barr virus lytic cycle by protoapigenone. J. Gen. Virol. 2011, 92, 1760–1768. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Vágvölgyi, M.; Girst, G.; Kuo, C.-Y.; Wang, H.-C.; Fülöp, F.; Hunyadi, A. Synthesis of Nontoxic Protoflavone Derivatives through Selective Continuous-Flow Hydrogenation of the Flavonoid B-Ring. ChemPlusChem 2018, 83, 72–76. [Google Scholar] [CrossRef]
- Ko, Y.-J.; Oh, H.-J.; Ahn, H.-M.; Kang, H.-J.; Kim, J.-H.; Ko, Y.H. Flavonoids as potential inhibitors of retroviral enzymes. J. Korean Soc. Appl. Biol. Chem. 2009, 52, 321–326. [Google Scholar] [CrossRef]
- Kurapati, K.R.V.; Atluri, V.S.; Samikkannu, T.; Garcia, G.; Nair, M.P.N. Natural Products as Anti-HIV Agents and Role in HIV-Associated Neurocognitive Disorders (HAND): A Brief Overview. Front. Microbiol. 2016, 6, 1444. [Google Scholar] [CrossRef] [PubMed]
- Áy, É.; Hunyadi, A.; Mezei, M.; Minárovits, J.; Hohmann, J. Flavonol 7-O-Glucoside Herbacitrin Inhibits HIV-1 Replication through Simultaneous Integrase and Reverse Transcriptase Inhibition. Evid. Based Complement. Altern. Med. 2019, 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, J.; Sleno, L.; Volmer, D.A. Isotopic labeling of metabolites in drug discovery applications. Curr. Drug Metab. 2012, 13, 1213–1225. [Google Scholar] [CrossRef]
- Robins, R.J.; Billault, I.; Duan, J.-R.; Guiet, S.; Pionnier, S.; Zhang, B.-L. Measurement of 2H distribution in natural products by quantitative 2H NMR: An approach to understanding metabolism and enzyme mechanism? Phytochem. Rev. 2003, 2, 87–102. [Google Scholar] [CrossRef]
- Krumbiegel, P. Large deuterium isotope effects and their use: A historical review. Isot. Environ. Health Stud. 2011, 47, 1–17. [Google Scholar] [CrossRef]
- Gant, T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Hsieh, C.-T.; Wu, Y.-C.; Li, J.-H.; Chang, F.-R.; Fülöp, F. Continuous-Flow Synthesis of Deuterium-Labeled Antidiabetic Chalcones: Studies towards the Selective Deuteration of the Alkynone Core. Molecular 2016, 21, 318. [Google Scholar] [CrossRef] [Green Version]
- Otvos, S.B.; Mandity, I.M.; Fulop, F. Highly selective deuteration of pharmaceutically relevant nitrogen-containing heterocycles: A flow chemistry approach. Mol. Divers. 2011, 15, 605–611. [Google Scholar] [CrossRef]
- Hsieh, C.-T.; Ötvös, S.B.; Wu, Y.-C.; Mándity, I.M.; Chang, F.-R.; Fülöp, F. Highly Selective Continuous-Flow Synthesis of Potentially Bioactive Deuterated Chalcone Derivatives. ChemPlusChem 2015, 80, 859–864. [Google Scholar] [CrossRef]
- Kozłowska, J.; Grela, E.; Baczyńska, D.; Grabowiecka, A.; Anioł, M. Novel O-alkyl Derivatives of Naringenin and Their Oximes with Antimicrobial and Anticancer Activity. Molecules 2019, 24, 679. [Google Scholar] [CrossRef] [Green Version]
- Türkkan, B.; Özyürek, M.; Bener, M.; Güçlü, K.; Apak, R. Synthesis, characterization and antioxidant capacity of naringenin-oxime. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 85, 235–240. [Google Scholar] [CrossRef]
- Latif, A.D.; Gonda, T.; Vágvölgyi, M.; Kúsz, N.; Kulmány, Á.; Ocsovszki, I.; Zomborszki, Z.P.; Zupkó, I.; Hunyadi, A. Synthesis and In Vitro Antitumor Activity of Naringenin Oxime and Oxime Ether Derivatives. Int. J. Mol. Sci. 2019, 20, 2184. [Google Scholar] [CrossRef] [Green Version]
- Arendt, V.; Amand, M.; Iserentant, G.; Lemaire, M.; Masquelier, C.; Ndayisaba, G.F.; Verhofstede, C.; Karita, E.; Allen, S.; Chevigné, A.; et al. Predominance of the heterozygous CCR5 delta-24 deletion in African individuals resistant to HIV infection might be related to a defect in CCR5 addressing at the cell surface. J. Int. AIDS Soc. 2019, 22, e25384. [Google Scholar] [CrossRef]
- Chang, L.K.; Lee, Y.H.; Cheng, T.S.; Hong, Y.R.; Lu, P.J.; Wang, J.J.; Wang, W.H.; Kuo, C.W.; Li, S.S.; Liu, S.T. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J. Biol. Chem. 2004, 279, 38803–38812. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.C.; Yoshikai, Y.; Hsu, S.W.; Saitoh, H.; Chang, L.K. Role of RNF4 in the ubiquitination of Rta of Epstein-Barr virus. J. Biol. Chem. 2013, 288, 12866–12879. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vágvölgyi, M.; Girst, G.; Kúsz, N.; Ötvös, S.B.; Fülöp, F.; Hohmann, J.; Servais, J.-Y.; Seguin-Devaux, C.; Chang, F.-R.; Chen, M.S.; et al. Less Cytotoxic Protoflavones as Antiviral Agents: Protoapigenone 1′-O-isopropyl ether Shows Improved Selectivity Against the Epstein–Barr Virus Lytic Cycle. Int. J. Mol. Sci. 2019, 20, 6269. https://doi.org/10.3390/ijms20246269
Vágvölgyi M, Girst G, Kúsz N, Ötvös SB, Fülöp F, Hohmann J, Servais J-Y, Seguin-Devaux C, Chang F-R, Chen MS, et al. Less Cytotoxic Protoflavones as Antiviral Agents: Protoapigenone 1′-O-isopropyl ether Shows Improved Selectivity Against the Epstein–Barr Virus Lytic Cycle. International Journal of Molecular Sciences. 2019; 20(24):6269. https://doi.org/10.3390/ijms20246269
Chicago/Turabian StyleVágvölgyi, Máté, Gábor Girst, Norbert Kúsz, Sándor B. Ötvös, Ferenc Fülöp, Judit Hohmann, Jean-Yves Servais, Carole Seguin-Devaux, Fang-Rong Chang, Michael S. Chen, and et al. 2019. "Less Cytotoxic Protoflavones as Antiviral Agents: Protoapigenone 1′-O-isopropyl ether Shows Improved Selectivity Against the Epstein–Barr Virus Lytic Cycle" International Journal of Molecular Sciences 20, no. 24: 6269. https://doi.org/10.3390/ijms20246269
APA StyleVágvölgyi, M., Girst, G., Kúsz, N., Ötvös, S. B., Fülöp, F., Hohmann, J., Servais, J.-Y., Seguin-Devaux, C., Chang, F.-R., Chen, M. S., Chang, L.-K., & Hunyadi, A. (2019). Less Cytotoxic Protoflavones as Antiviral Agents: Protoapigenone 1′-O-isopropyl ether Shows Improved Selectivity Against the Epstein–Barr Virus Lytic Cycle. International Journal of Molecular Sciences, 20(24), 6269. https://doi.org/10.3390/ijms20246269