Genome-Wide Analysis of Whole Human Glycoside Hydrolases by Data-Driven Analysis in Silico

 , and
, and 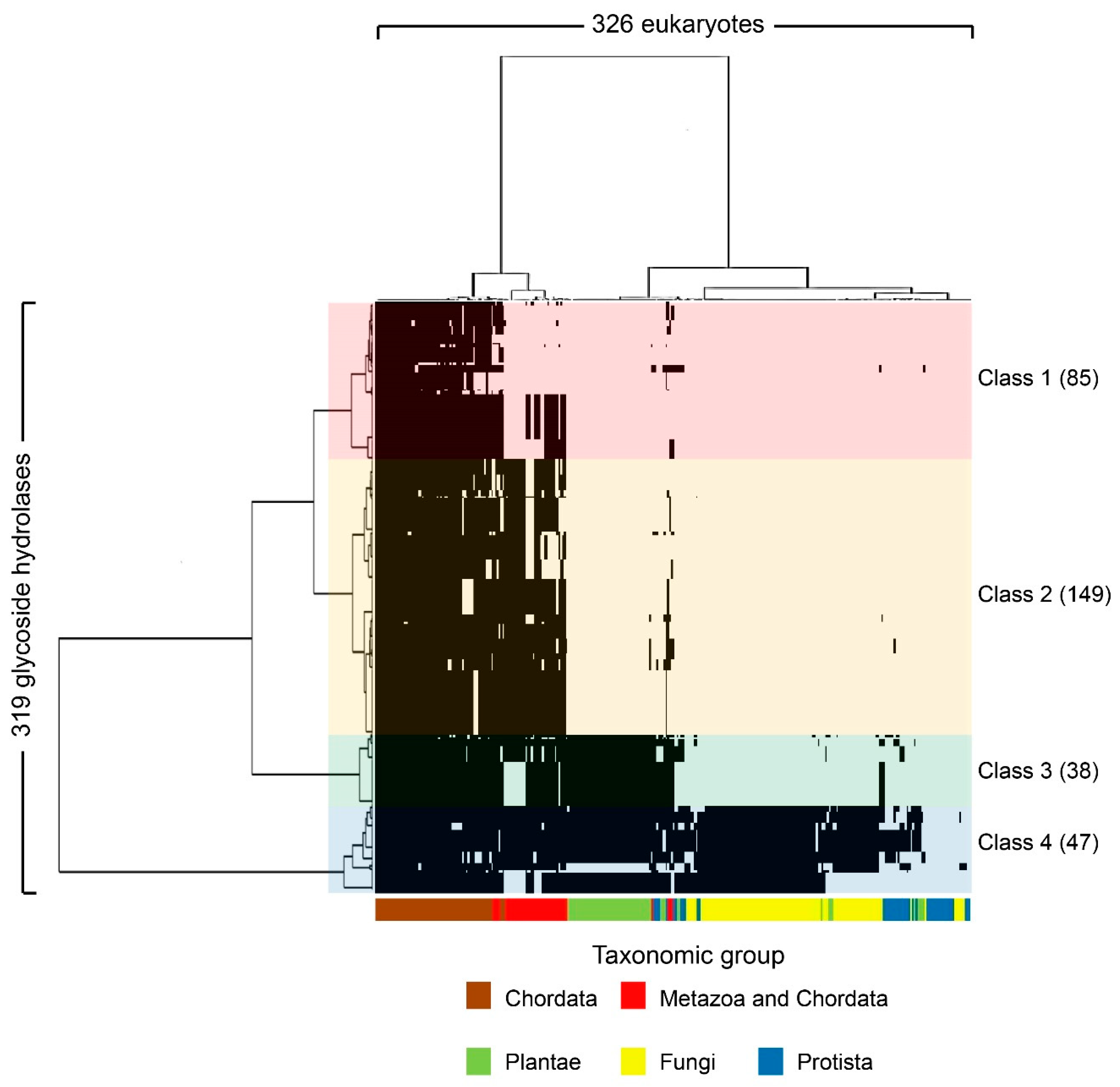{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Human Glycoside Hydrolase Dataset
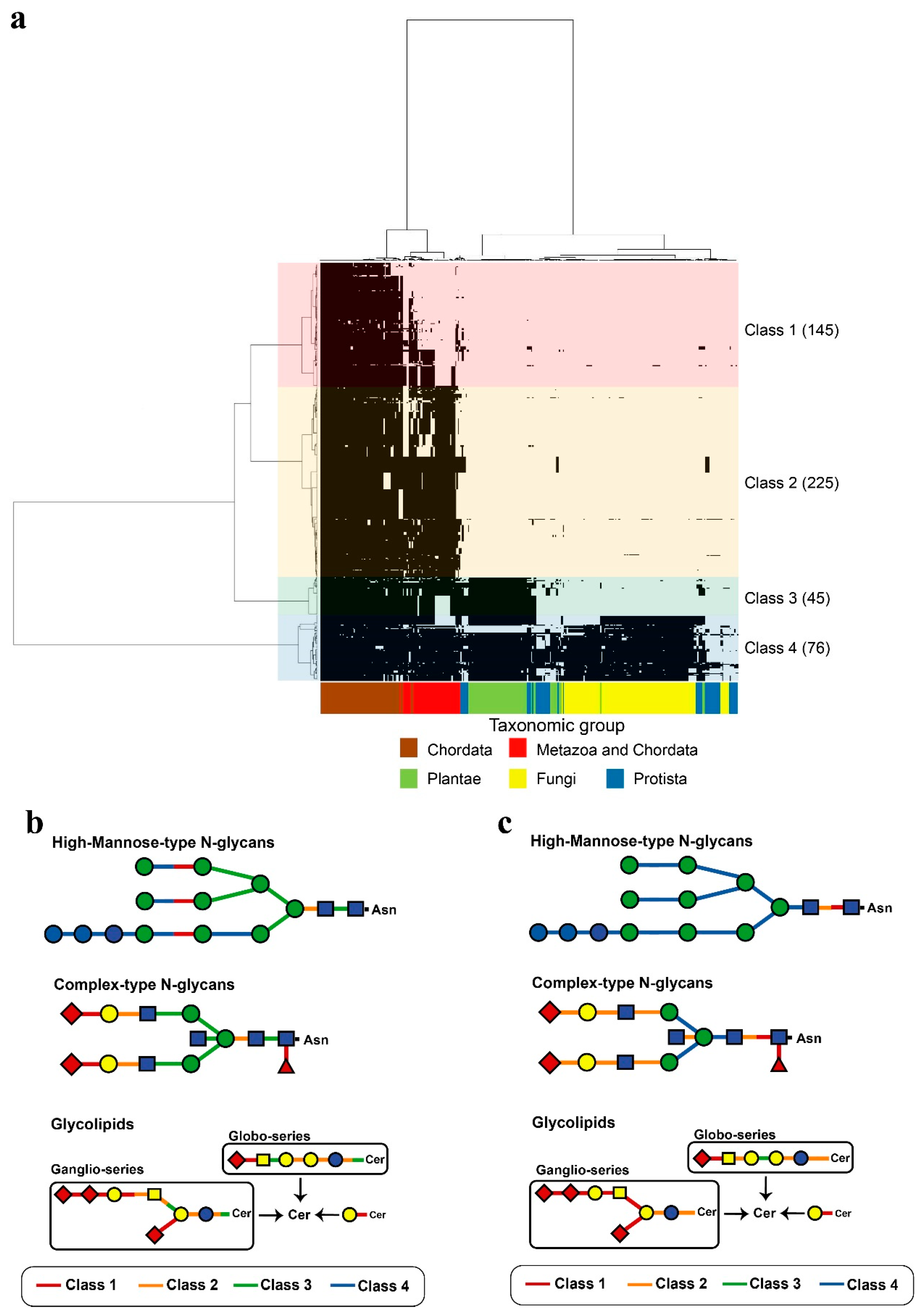
2.2. Human Glycoside Hydrolases Belong to Four Evolutionary Classes
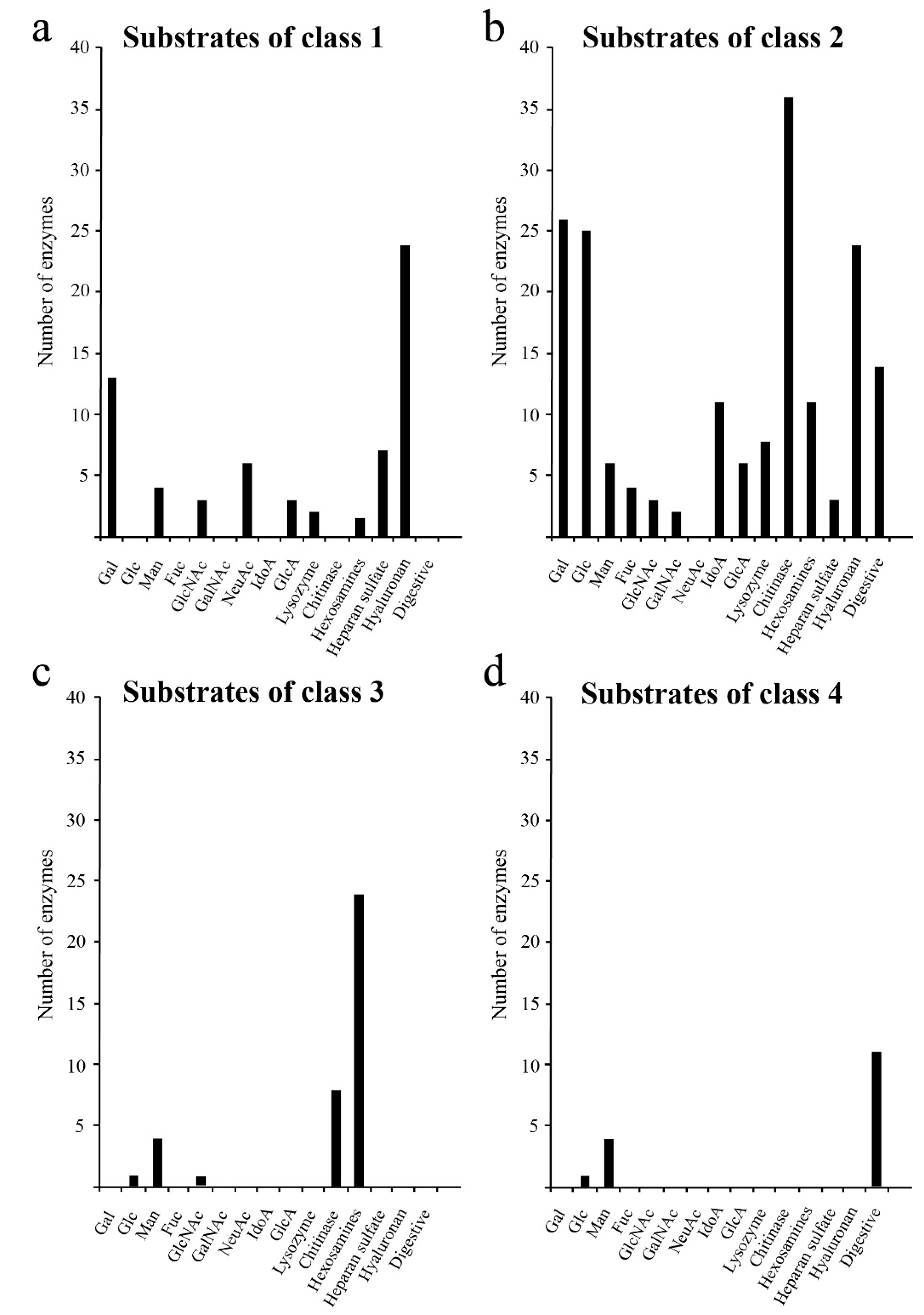
2.3. Functions of Human Glycoside Hydrolases Differ Among Classes
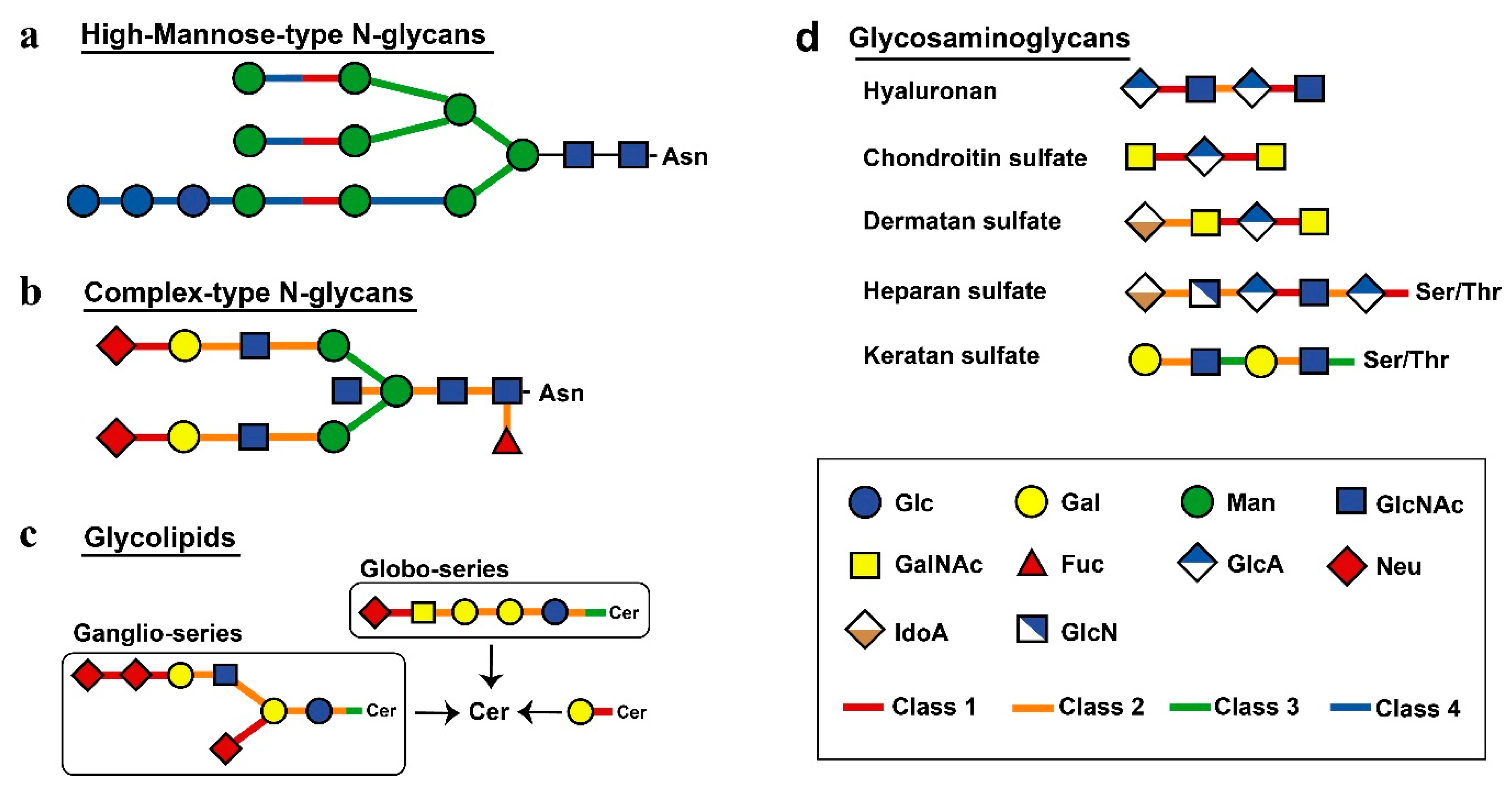
2.4. Comparison of Decomposition Substrates Among Classes of Glycoside Hydrolases
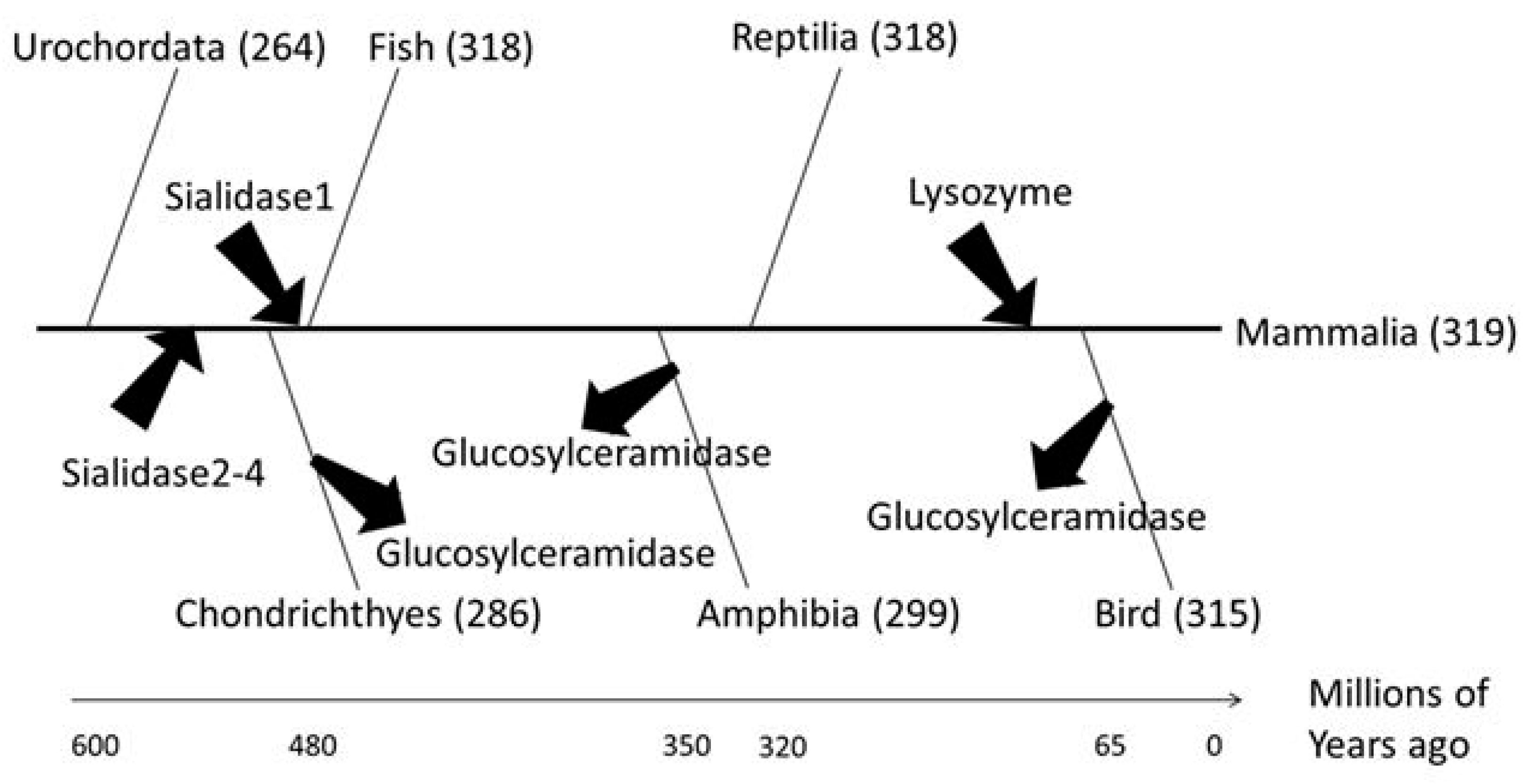
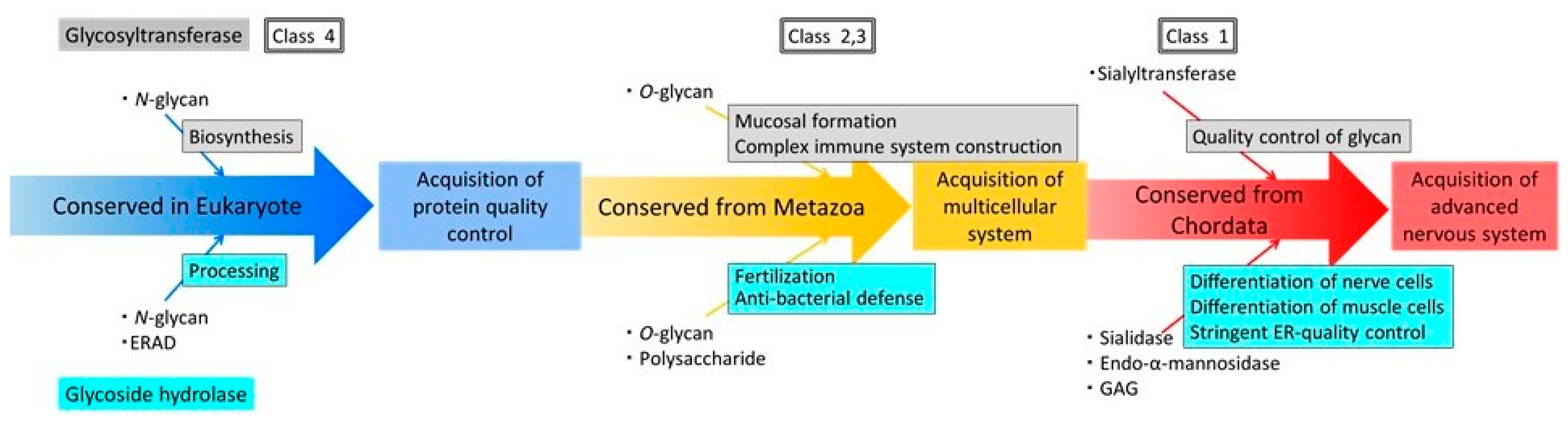
2.5. Identification of Glycoside Hydrolases Important for the Evolution to Mammals
2.6. Evolution of Glycosyltransferases and Glycoside Hydrolases
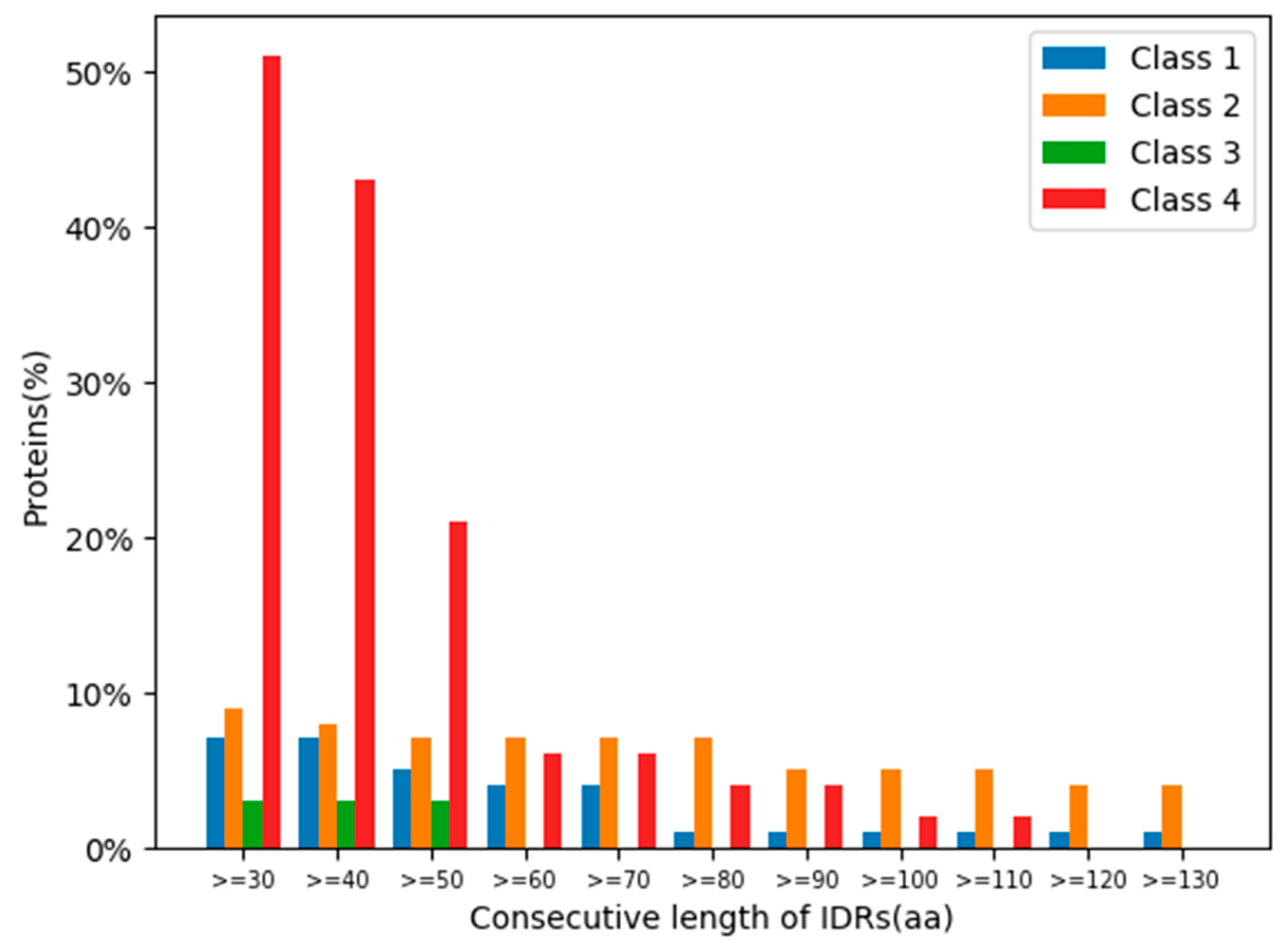
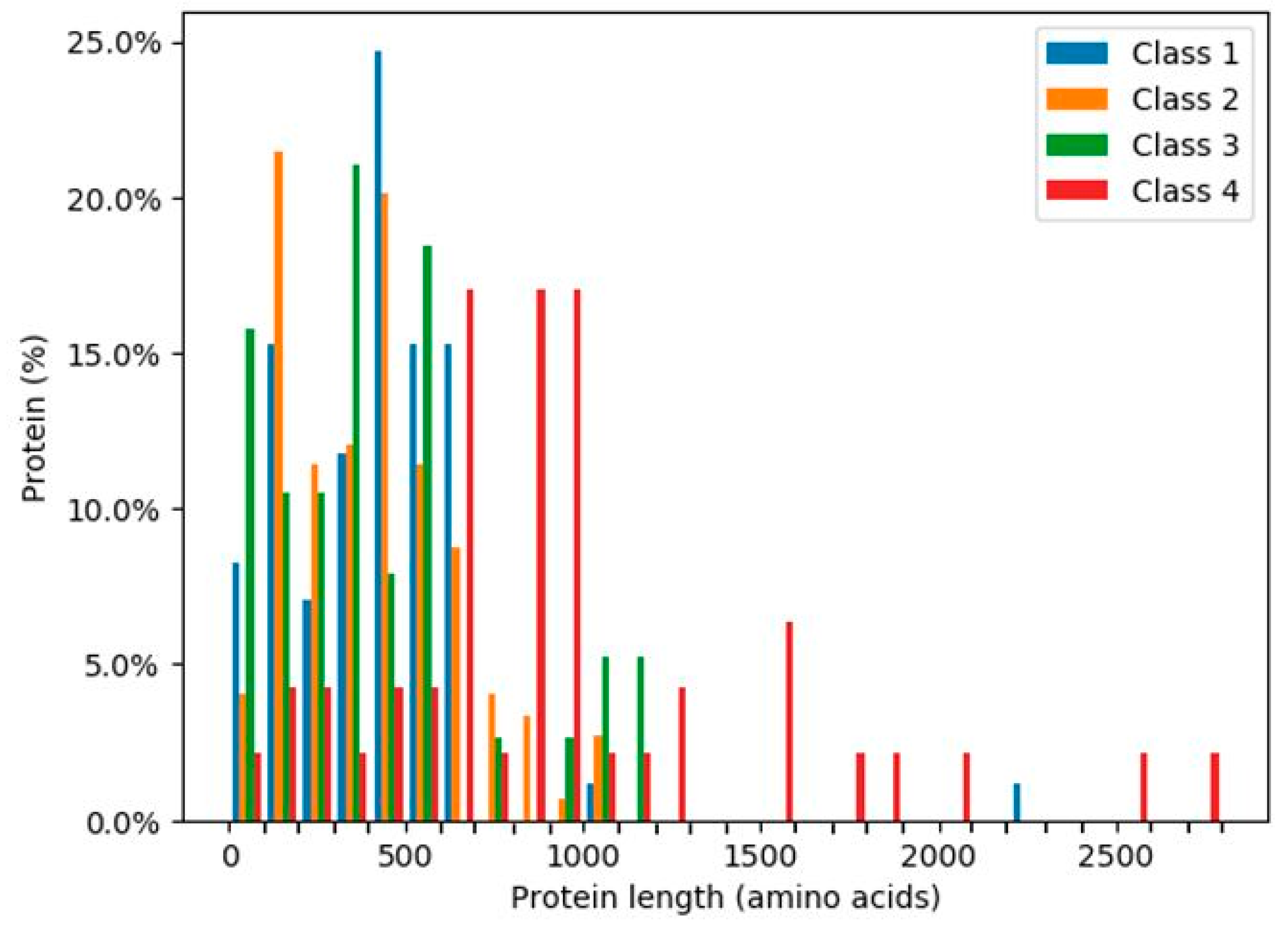
2.7. Intrinsically Disordered Regions of Glycoside Hydrolases
3. Discussion
4. Materials and Methods
4.1. Human Glycoside Hydrolase Dataset
4.2. Phylogenetic Profiling and Cluster Analyses
4.3. Molecular Phylogenetic Analysis
4.4. Protein IDR Analysis
4.5. Source Code
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| UniProt | The Universal Protein Resource |
| CAZy | Carbohydrate-Active Enzymes |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| O-linked | Oxygen-linked-type |
| ECM | Extracellular matrix |
| N-linked | Nitrogen-linked-type |
| GO | Gene Ontology |
| GHs | Glycosyl hydrolases |
| ERAD | Endoplasmic reticulum-associated degradation |
| MGAT3 | β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase |
| GlcNAc | N-acetylglucosamine |
| N-glycan | N-linked glycan |
| O-glycan | O-linked glycan |
| IDRs | Intrinsically disordered regions |
| EDEMs | ER degradation-enhancing mannosidase-like proteins |
| BovB | Bovine-B |
| IDPs | Intrinsically disordered proteins |
References
- Schwarz, F.; Aebi, M. Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 2011, 21, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Freeze, H.H.; Gagneux, P. Evolution of glycan diversity. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2009; pp. 281–292. [Google Scholar]
- Tomono, T.; Kojima, H.; Fukuchi, S.; Tohsato, Y.; Ito, M. Investigation of glycan evolution based on a comprehensive analysis of glycosyltransferases using phylogenetic profiling. Biophys. Physicobiol. 2015, 12, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Day, A.J.; Prestwich, G.D. Hyaluronan-binding proteins: Tying up the giant. J. Biol. Chem. 2002, 277, 4585–4588. [Google Scholar] [CrossRef] [PubMed]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Ann. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Song, L.; Qin, X. Glycan changes: Cancer metastasis and anti-cancer vaccines. J. Biosci. 2010, 35, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Mony, V.K.; Benjamin, S.; O’Rourke, E.J. A lysosome-centered view of nutrient homeostasis. Autophagy 2016, 12, 619–631. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Yang, Y.; Kelly, T.; MacLeod, V.; Dai, Y.; Theus, A. Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: Growth regulation and the prospect of new cancer therapies. J. Cell. Biochem. 2005, 96, 897–905. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 684–696. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Daughdrill, G.W. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol. 2002, 27, 609–621. [Google Scholar] [CrossRef]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 2006, 5, 879–887. [Google Scholar] [CrossRef]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, M.; Ito, M. Evolutionary approach of intrinsically disordered CIP/KIP proteins. Sci. Rep. 2019, 9, 1575. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, M.; Yasui, G.; Seki, K.; Katayama, S.; Kaneko-Kawano, T.; Inazu, T.; Kubota, Y.; Ito, M. In silico study of rett syndrome treatment-related genes, mecp2, cdkl5, and foxg1, by evolutionary classification and disordered region assessment. Int. J. Mol. Sci. 2019, 20, 5593. [Google Scholar] [CrossRef]
- Goldman, N.; Thorne, J.L.; Jones, D.T. Assessing the impact of secondary structure and solvent accessibility on protein evolution. Genetics 1998, 149, 445–458. [Google Scholar] [PubMed]
- Bellay, J.; Michaut, M.; Kim, T.; Han, S.; Colak, R.; Myers, C.L.; Kim, P.M. An omics perspective of protein disorder. Mol. BioSyst. 2012, 8, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alam-Faruque, Y.; Antunes, R.; Barrell, D.; Bely, B.; Bingley, M.; Binns, D.; et al. The universal protein resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013, 42, D490–D495. [Google Scholar] [CrossRef]
- Placzek, S.; Schomburg, I.; Chang, A.; Jeske, L.; Ulbrich, M.; Tillack, J.; Schomburg, D. BRENDA in 2017: New perspectives and new tools in BRENDA. Nucleic Acids Res. 2016, 45, D380–D388. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2008, 37, D211–D215. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef]
- Thompson, A.J.; Williams, R.J.; Hakki, Z.; Alonzi, D.S.; Wennekes, T.; Gloster, T.M.; Songsrirote, K.; Thomas-Oates, J.E.; Wrodnigg, T.M.; Spreitz, J.; et al. Structural and mechanistic insight into N-glycan processing by endo--mannosidase. Proc. Natl. Acad. Sci. USA 2012, 109, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Marin, M.B.; Ghenea, S.; Spiridon, L.N.; Chiritoiu, G.N.; Petrescu, A.J.; Petrescu, S.M. Tyrosinase degradation is prevented when EDEM1 lacks the intrinsically disordered region. PLoS ONE 2012, 7, e42998. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Tohsato, Y.; Sugisawa, H.; Kohara, S.; Fukuchi, S.; Nishikawa, I.; Nishikawa, K. Intrinsically disordered proteins in human mitochondria. Genes Cells 2012, 17, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Harada, Y.; Hosomi, A.; Masahara-Negishi, Y.; Seino, J.; Fujihira, H.; Funakoshi, Y.; Suzuki, T.; Dohmae, N.; Suzuki, T. Endo-N-acetylglucosaminidase forms N-GlcNAc protein aggregates during ER-associated degradation in Ngly1-defective cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1398–1403. [Google Scholar] [CrossRef]
- Kornfeld, R.; Kornfeld, S. Assembly of asparagine-linked oligosaccharides. Ann. Rev. Biochem. 1985, 54, 631–664. [Google Scholar] [CrossRef]
- Mukherjee, S.; Vaishnava, S.; Hooper, L.V. Multi-layered regulation of intestinal antimicrobial defense. Cell. Mol. Life Sci. 2008, 65, 3019–3027. [Google Scholar] [CrossRef]
- Paoletti, M.G.; Norberto, L.; Damini, R.; Musumeci, S. Human gastric juice contains chitinase that can degrade chitin. Ann. Nutr. Metab. 2007, 51, 244–251. [Google Scholar] [CrossRef]
- Modelski, M.J.; Menlah, G.; Wang, Y.; Dash, S.; Wu, K.; Galileo, D.S.; Martin-DeLeon, P.A. Hyaluronidase 2: A novel germ cell hyaluronidase with epididymal expression and functional roles in mammalian sperm. Biol. Reprod. 2014, 91, 109. [Google Scholar] [CrossRef]
- Funderburgh, J.L. Keratan sulfate biosynthesis. IUBMB Life 2002, 54, 187–194. [Google Scholar] [CrossRef]
- Carvalho, S.; Catarino, T.A.; Dias, A.M.; Kato, M.; Almeida, A.; Hessling, B.; Figueiredo, J.; Gärtner, F.; Sanches, J.M.; Ruppert, T.; et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene 2016, 35, 1619. [Google Scholar] [CrossRef]
- Olivari, S.; Molinari, M. Glycoprotein folding and the role of EDEM1, EDEM2 and EDEM3 in degradation of folding-defective glycoproteins. FEBS let. 2007, 581, 3658–3664. [Google Scholar] [CrossRef] [PubMed]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Baalsrud, H.T.; Tørresen, O.K.; Solbakken, M.H.; Salzburger, W.; Hanel, R.; Jakobsen, K.S.; Jentoft, S. De novo gene evolution of antifreeze glycoproteins in codfishes revealed by whole genome sequence data. Mol. Biol. Evol. 2017, 35, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread horizontal transfer of retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Eyun, S.I.; Wang, H.; Pauchet, Y.; Benson, A.K.; Valencia-Jiménez, A.; Moriyama, E.N.; Siegfried, B.D. Molecular evolution of glycoside hydrolase genes in the western corn rootworm (Diabrotica virgifera virgifera). PLoS ONE 2014, 9, e9405. [Google Scholar] [CrossRef] [PubMed]
- German, J.B.; Freeman, S.L.; Lebrilla, C.B.; Mills, D.A. Human milk oligosaccharides: Evolution, structures and bioselectivity as substrates for intestinal bacteria. In Personalized nutrition for the diverse needs of infants and children. Karger Publ. 2008, 62, 205–222. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef]
- Ward Jr, J.H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Rowe, T. Chordate phylogeny and development. In Assembling the Tree of Life; Cracraft, J., Donoghue, M.J., Eds.; Oxford University Press: Oxford, UK, 2009; pp. 384–409. [Google Scholar]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D2P2: Database of disordered protein predictions. Nucleic Acids Res. 2012, 41, D508–D516. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdo˝s, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic’, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, T.; Fahmi, M.; Tanaka, J.; Seki, K.; Kubota, Y.; Ito, M. Genome-Wide Analysis of Whole Human Glycoside Hydrolases by Data-Driven Analysis in Silico. Int. J. Mol. Sci. 2019, 20, 6290. https://doi.org/10.3390/ijms20246290
Nakamura T, Fahmi M, Tanaka J, Seki K, Kubota Y, Ito M. Genome-Wide Analysis of Whole Human Glycoside Hydrolases by Data-Driven Analysis in Silico. International Journal of Molecular Sciences. 2019; 20(24):6290. https://doi.org/10.3390/ijms20246290
Chicago/Turabian StyleNakamura, Takahiro, Muhamad Fahmi, Jun Tanaka, Kaito Seki, Yukihiro Kubota, and Masahiro Ito. 2019. "Genome-Wide Analysis of Whole Human Glycoside Hydrolases by Data-Driven Analysis in Silico" International Journal of Molecular Sciences 20, no. 24: 6290. https://doi.org/10.3390/ijms20246290
APA StyleNakamura, T., Fahmi, M., Tanaka, J., Seki, K., Kubota, Y., & Ito, M. (2019). Genome-Wide Analysis of Whole Human Glycoside Hydrolases by Data-Driven Analysis in Silico. International Journal of Molecular Sciences, 20(24), 6290. https://doi.org/10.3390/ijms20246290