Semaphorin 3C as a Therapeutic Target in Prostate and Other Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Discovery
3. Structure & Function
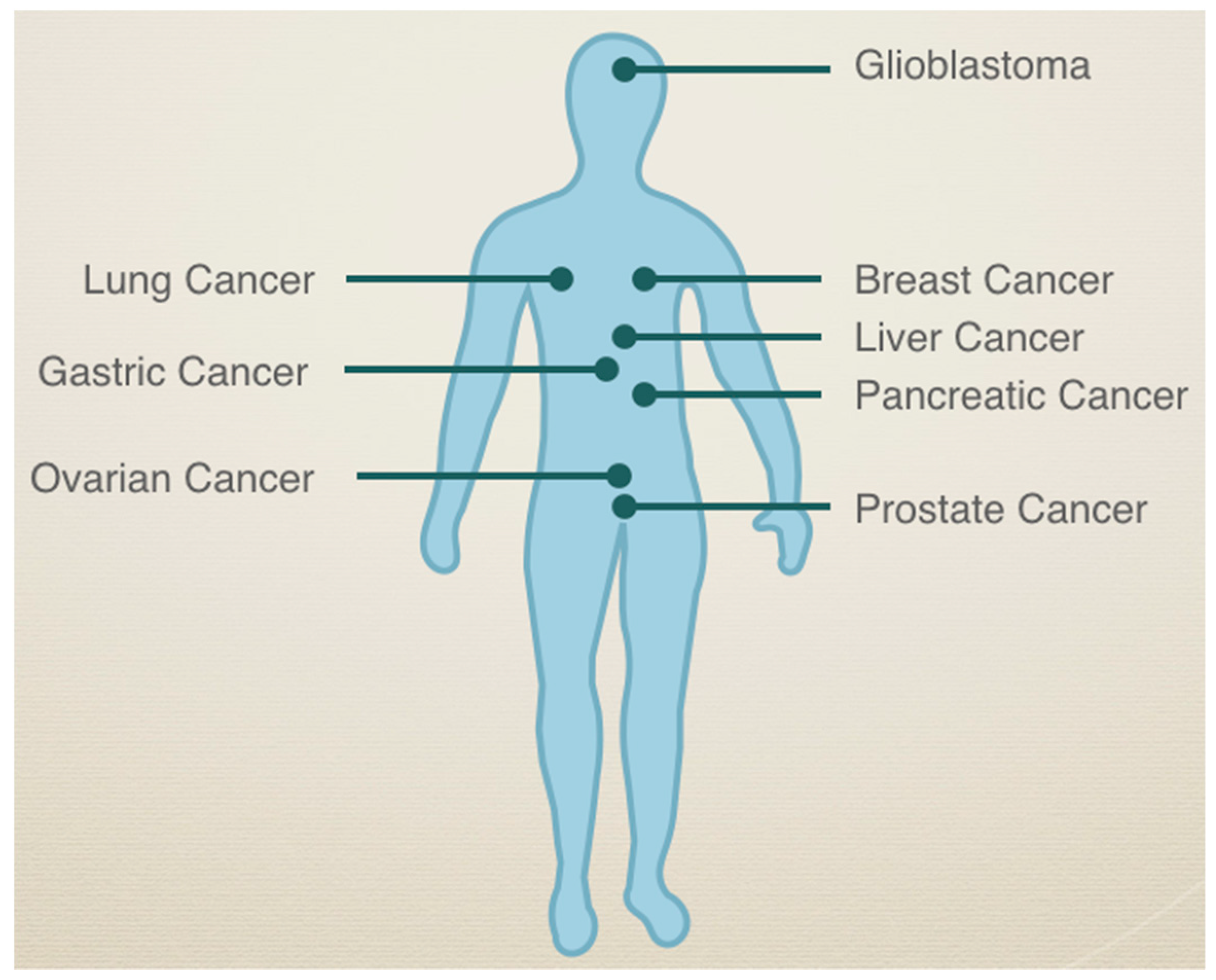
4. SEMA3C by Cancer Type
4.1. Prostate Cancer
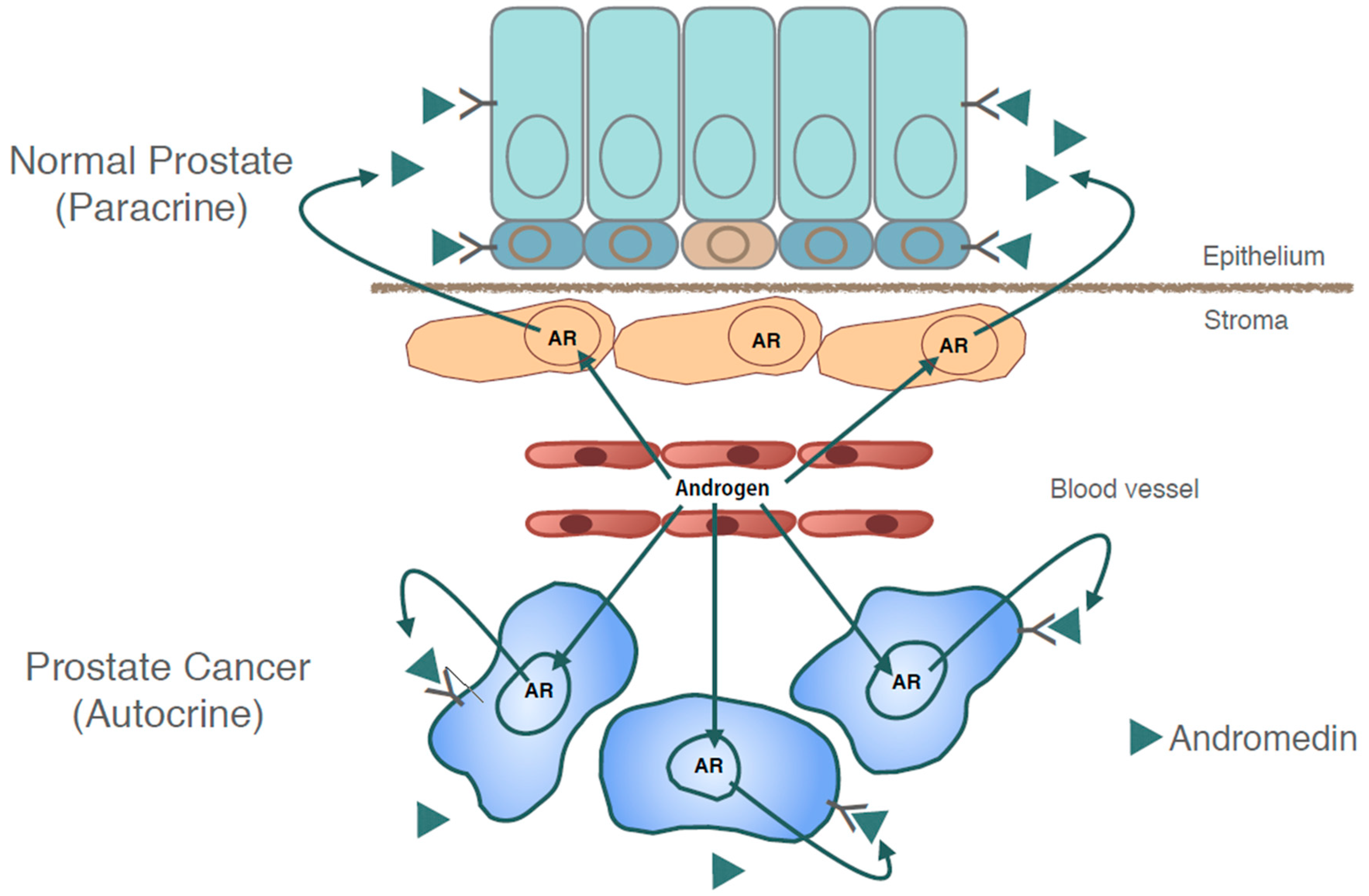
4.1.1. SEMA3C as the First Bona Fide Prostate Cancer Andromedin
4.1.2. AR Reactivation Drives SEMA3C-Induced Growth
4.1.3. SEMA3C and Tumor cell growth via RTK/AR Crosstalk and AR Bypass
4.1.4. SEMA3C and Cancer Stem Cells
4.1.5. SEMA3C and Epithelial-to-Mesenchymal Transition
4.1.6. SEMA3C and RTK Coactivation
4.2. Role of SEMA3C in Other Cancers
4.2.1. Pancreatic Cancer
4.2.2. Brain Cancer
4.2.3. Breast Cancer
4.2.4. Gastric Cancer
4.2.5. Other Cancers
5. Potential Molecular Approaches in the Inhibition of SEMA3C as a Cancer Therapy
5.1. Biologics
5.2. Small Molecule Drugs
5.3. Monoclonal Antibodies
5.4. Antisense Oligonucleotides (ASO)
6. Perspective
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| SEMA3C | semaphorin 3C |
| PLXNB1 | plexin B1 |
| NRP1 | neuropilin 1 |
| PCa | prostate cancer |
| ADT | androgen deprivation therapy |
| AR | androgen receptor |
| FGF | fibroblast growth factor |
| IGF | insulin-like growth factor |
| RTK | receptor tyrosine kinase |
| UBC | University of British Columbia |
| mCRPC | metastatic castration-resistant prostate cancer |
| ARPI | AR pathway inhibitors |
| NEPC | neuroendocrine PCa |
| TKI | tyrosine kinase inhibitors |
| EGFR | epidermal growth factor receptor |
| VEGF | vascular endothelial growth factor |
| CSC | cancer stem cell |
| EMT | epithelial-to-mesenchymal transition |
| TNM | tumor nodes metastasis |
| ER | estrogen receptor |
| PR | progesterone receptor |
| HER2 | human epidermal growth factor receptor 2 |
| mAbs | monoclonal antibodies |
| PCS1 | proteolytic cleavage site 1 |
| ASO | antisense oligonucleotide |
References
- Yazdani, U.; Terman, J.R. The semaphorins. Genome Biol. 2006, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Tamagnone, L. Semaphorins in cancer: Biological mechanisms and therapeutic approaches. Semin. Cell Dev. Biol. 2013, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Capparuccia, L.; Tamagnone, L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment—Two sides of a coin. J. Cell Sci. 2009, 122, 1723–1736. [Google Scholar] [CrossRef]
- Neufeld, G.; Shraga-Heled, N.; Lange, T.; Guttmann-Raviv, N.; Herzog, Y.; Kessler, O. Semaphorins in cancer. Front Biosci.-Landmrk 2005, 10, 751–760. [Google Scholar] [CrossRef]
- Worzfeld, T.; Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 2014, 13, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Yu, J.S. Semaphorin 3C and Its Receptors in Cancer and Cancer Stem-Like Cells. Biomedicines 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Raper, J.A.; Kapfhammer, J.P. The enrichment of a neuronal growth cone collapsing activity from embryonic chick brain. Neuron 1990, 4, 21–29. [Google Scholar] [CrossRef]
- Kolodkin, A.L.; Matthes, D.J.; O’Connor, T.P.; Patel, N.H.; Admon, A.; Bentley, D.; Goodman, C.S. Fasciclin IV: sequence, expression, and function during growth cone guidance in the grasshopper embryo. Neuron 1992, 9, 831–845. [Google Scholar] [CrossRef]
- Luo, Y.L.; Raible, D.; Raper, J.A. Collapsin—A Protein in Brain That Induces the Collapse and Paralysis of Neuronal Growth Cones. Cell 1993, 75, 217–227. [Google Scholar] [CrossRef]
- Puschel, A.W.; Adams, R.H.; Betz, H. Murine Semaphorin-D Collapsin Is a Member of a Diverse Gene Family and Creates Domains Inhibitory for Axonal Extension. Neuron 1995, 14, 941–948. [Google Scholar] [CrossRef]
- Feiner, L.; Webber, A.L.; Brown, C.B.; Lu, M.M.; Jia, L.; Feinstein, P.; Mombaerts, P.; Epstein, J.A.; Raper, J.A. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development 2001, 128, 3061–3070. [Google Scholar] [PubMed]
- Yamada, T.; Endo, R.; Gotoh, M.; Hirohashi, S. Identification of semaphorin E as a non-MDR drug resistance gene of human cancers. Natl. Acad. Sci. USA 1997, 94, 14713–14718. [Google Scholar] [CrossRef]
- Galani, E.; Sgouros, J.; Petropoulou, C.; Janinis, J.; Aravantinos, G.; Dionysiou-Asteriou, D.; Skarlos, D.; Gonos, E. Correlation of MDR-1, nm23-H1 and H sema E gene expression with histopathological findings and clinical outcome in ovarian and breast cancer patients. Anticancer Res. 2002, 22, 2275–2280. [Google Scholar] [PubMed]
- Martin-Satue, M.; Blanco, J. Identification of semaphorin E gene expression in metastatic human lung adenocarcinoma cells by mRNA differential display. J. Surg. Oncol. 1999, 72, 18–23. [Google Scholar] [CrossRef]
- Miyato, H.; Tsuno, N.H.; Kitayama, J. Semaphorin 3C is involved in the progression of gastric cancer. Cancer Sci. 2012, 103, 1961–1966. [Google Scholar] [CrossRef]
- Esselens, C.; Malapeira, J.; Colome, N.; Casal, C.; Rodriguez-Manzaneque, J.C.; Canals, F.; Arribas, J. The cleavage of semaphorin 3C induced by ADAMTS1 promotes cell migration. J. Biol. Chem. 2010, 285, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Meadows, G.G. Increased class 3 semaphorin expression modulates the invasive and adhesive properties of prostate cancer cells. Int. J. Oncol. 2007, 30, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Blanc, V.; Nariculam, J.; Munson, P.; Freeman, A.; Klocker, H.; Masters, J.; Williamson, M. A role for class 3 semaphorins in prostate cancer. Prostate 2011, 71, 649–658. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, Z.; Guo, S.; Li, J.; Liu, S.; You, Y.; Ni, B.; Wang, H.; Bie, P. Increased semaphorin 3c expression promotes tumor growth and metastasis in pancreatic ductal adenocarcinoma by activating the ERK1/2 signaling pathway. Cancer let. 2017, 397, 12–22. [Google Scholar] [CrossRef]
- Rieger, J.; Wick, W.; Weller, M. Human malignant glioma cells express semaphorins and their receptors, neuropilins and plexins. Glia 2003, 42, 379–389. [Google Scholar] [CrossRef]
- Man, J.H.; Shoemake, J.; Zhou, W.C.; Fang, X.G.; Wu, Q.L.; Rizzo, A.; Prayson, R.; Bao, S.D.; Rich, J.N.; Yu, J.S. Sema3C Promotes the Survival and Tumorigenicity of Glioma Stem Cells through Rac1 Activation. Cell Rep. 2014, 9, 1812–1826. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhu, K.L.; Liu, J.H.; Chen, J.; Tang, J.C.; Liang, Y.L.; Jin, E.A.; Liang, X.; Cai, X.J. The evaluative value of Sema3C and MFN2 co-expression detected by immunohistochemistry for prognosis in hepatocellular carcinoma patients after hepatectomy. Oncotargets Ther. 2016, 9, 3213–3221. [Google Scholar]
- Li, K.; Chen, M.K.; Li, L.Y.; Lu, M.H.; Shao, C.K.; Su, Z.L.; He, D.; Pang, J.; Gao, X. The predictive value of semaphorins 3 expression in biopsies for biochemical recurrence of patients with low- and intermediate-risk prostate cancer. Neoplasma 2013, 60, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Goodman, L.D.; Wani, K.M.; Boehling, N.S.; Sharma, D.S.; Mody, R.R.; Gumin, J.; Claus, E.B.; Lang, F.F.; Cloughesy, T.F.; et al. A gene expression signature predicts recurrence-free survival in meningioma. Oncotarget 2018, 9, 16087–16098. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.L.; Sun, Y.M.; Chau, G.Y.; Chau, Y.P.; Lai, T.C.; Wang, J.L.; Horng, J.T.; Hsiao, M.; Tsou, A.P. Identification of SOX4 target genes using phylogenetic footprinting-based prediction from expression microarrays suggests that overexpression of SOX4 potentiates metastasis in hepatocellular carcinoma. Oncogene 2008, 27, 5578–5589. [Google Scholar] [CrossRef]
- Ferreira, G.D.; Capp, E.; Jauckus, J.; Strowitzki, T.; Germeyer, A. Expression of semaphorin class 3 is higher in the proliferative phase on the human endometrium. Arch. Gynecol. Obstet. 2018, 297, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Oinuma, I.; Ishikawa, Y.; Katoh, H.; Negishi, M. The semaphorin 4D receptor plexin-B1 is a GTPase activating protein for R-Ras. Science 2004, 305, 862–865. [Google Scholar] [CrossRef]
- Wang, Y.X.; He, H.W.; Srivastava, N.; Vikarunnessa, S.; Chen, Y.B.; Jiang, J.; Cowan, C.W.; Zhang, X.W. Plexins Are GTPase-Activating Proteins for Rap and Are Activated by Induced Dimerization. Sci. Signal 2012, 5. [Google Scholar] [CrossRef]
- Swiercz, J.M.; Kuner, R.; Behrens, J.; Offermanns, S. Plexin-B1 directly interacts with PDZ-RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 2002, 35, 51–63. [Google Scholar] [CrossRef]
- Aurandt, J.; Vikis, H.G.; Gutkind, J.S.; Ahn, N.; Guan, K.L. The semaphorin receptor plexin-B1 signals through a direct interaction with the Rho-specific nucleotide exchange factor, LARG. Natl. Acad. Sci. USA 2002, 99, 12085–12090. [Google Scholar] [CrossRef] [PubMed]
- Casazza, A.; Finisguerra, V.; Capparuccia, L.; Camperi, A.; Swiercz, J.M.; Rizzolio, S.; Rolny, C.; Christensen, C.; Bertotti, A.; Sarotto, I.; et al. Sema3E-Plexin D1 signaling drives human cancer cell invasiveness and metastatic spreading in mice. J. Clin. Invest. 2010, 120, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Corso, S.; Conrotto, P.; Artigiani, S.; Gilestro, G.; Barberis, D.; Tamagnone, L.; Comoglio, P.M. The Semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 2002, 4, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.R.; Aurandt, J.; Guan, K.L. Semaphorins command cells to move. Nat. Rev. Mol. Cell Biol. 2005, 6, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.H.; Yoshida, Y.; Livet, J.; Reimert, D.V.; Mann, F.; Merte, J.; Henderson, C.E.; Jessell, T.M.; Kolodkin, A.L.; Ginty, D.D. Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 2005, 307, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Smolkin, T.; Nir-Zvi, I.; Duvshani, N.; Mumblat, Y.; Kessler, O.; Neufeld, G. Complexes of plexin-A4 and plexin-D1 convey semaphorin-3C signals to induce cytoskeletal collapse in the absence of neuropilins. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Tamagnone, L.; Comoglio, P.M. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 2000, 10, 377–383. [Google Scholar] [CrossRef]
- Sharma, A.; Verhaagen, J.; Harvey, A.R. Receptor complexes for each of the Class 3 Semaphorins. Front Cell Neurosci. 2012, 6, 28. [Google Scholar] [CrossRef]
- Peacock, J.W.; Takeuchi, A.; Hayashi, N.; Liu, L.; Tam, K.J.; Al Nakouzi, N.; Khazamipour, N.; Tombe, T.; Dejima, T.; Lee, K.C.; et al. SEMA3C drives cancer growth by transactivating multiple receptor tyrosine kinases via Plexin B1. EMBO Mol. Med. 2018, 10, 219–238. [Google Scholar] [CrossRef]
- Delloye-Bourgeois, C.; Bertin, L.; Thoinet, K.; Jarrosson, L.; Kindbeiter, K.; Buffet, T.; Tauszig-Delamasure, S.; Bozon, M.; Marabelle, A.; Combaret, V.; et al. Microenvironment-Driven Shift of Cohesion/Detachment Balance within Tumors Induces a Switch toward Metastasis in Neuroblastoma. Cancer Cell 2017, 32, 427. [Google Scholar] [CrossRef]
- Mire, E.; Hocine, M.; Bazellieres, E.; Jungas, T.; Davy, A.; Chauvet, S.; Mann, F. Developmental Upregulation of Ephrin-B1 Silences Sema3C/Neuropilin-1 Signaling during Post-crossing Navigation of Corpus Callosum Axons. Curr. Biol. 2018, 28, 1768. [Google Scholar] [CrossRef] [PubMed]
- Wiegreffe, C.; Simon, R.; Peschkes, K.; Kling, C.; Strehle, M.; Cheng, J.; Srivatsa, S.; Liu, P.T.; Jenkins, N.A.; Copeland, N.G.; et al. Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 2015, 87, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Sanyas, I.; Bozon, M.; Moret, F.; Castellani, V. Motoneuronal Sema3C is essential for setting stereotyped motor tract positioning in limb-derived chemotropic semaphorins. Development 2012, 139, 3633–3643. [Google Scholar] [CrossRef] [PubMed]
- Ruediger, T.; Zimmer, G.; Barchmann, S.; Castellani, V.; Bagnard, D.; Bolz, J. Integration of Opposing Semaphorin Guidance Cues in Cortical Axons. Cereb. Cortex. 2013, 23, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Tamariz, E.; Diaz-Martinez, N.E.; Diaz, N.F.; Garcia-Pena, C.M.; Velasco, I.; Varela-Echavarria, A. Axon Responses of Embryonic Stem Cell-Derived Dopaminergic Neurons to Semaphorins 3A and 3C. J. Neurosci. Res. 2010, 88, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Montiel, H.L.; Tamariz, E.; Sandoval-Minero, M.T.; Varela-Echavarria, A. Semaphorins 3A, 3C, and 3F in mesencephalic dopaminergic axon pathfinding. J. Comp. Neurol. 2008, 506, 387–397. [Google Scholar] [CrossRef]
- Gonthier, B.; Nasarre, C.; Roth, L.; Perraut, M.; Thomasset, N.; Roussel, G.; Aunis, D.; Bagnard, D. Functional interaction between matrix metalloproteinase-3 and Semaphorin-3C during cortical axonal growth and guidance. Cereb. Cortex. 2007, 17, 1712–1721. [Google Scholar] [CrossRef]
- Moreno-Flores, M.T.; Martin-Aparicio, E.; Martin-Bermejo, M.J.; Agudo, M.; McMahon, S.; Avila, J.; Diaz-Nido, J.; Wandosell, F. Semaphorin 3C preserves survival and induces neuritogenesis of cerebellar granule neurons in culture. J. Neurochem. 2003, 87, 879–890. [Google Scholar] [CrossRef]
- Steup, A.; Lohrum, M.; Hamscho, N.; Savaskan, N.E.; Ninnemann, O.; Nitsch, R.; Fujisawa, H.; Puschel, A.W.; Skutella, T. Sema3C and Netrin-1 differentially affect axon growth in the hippocampal formation. Mol. Cell Neurosci. 2000, 15, 141–155. [Google Scholar] [CrossRef]
- Valdembri, D.; Regano, D.; Maione, F.; Giraudo, E.; Serini, G. Class 3 semaphorins in cardiovascular development. Cell Adhes. Migr. 2016, 10, 641–651. [Google Scholar] [CrossRef]
- Plein, A.; Calmont, A.; Fantin, A.; Denti, L.; Anderson, N.A.; Scambler, P.J.; Ruhrberg, C. Neural crest-derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Invest. 2015, 125, 2661–2676. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Markwald, R.R.; Baldwin, H.S.; SpringerLink Fully Open Access Books; SpringerLINK ebooks—Medicine (2016); SpringerOpen; NCBI Bookshelf; DOAB: Directory of Open Access Books. Etiology and Morphogenesis of Congenital Heart Disease From Gene Function and Cellular Interaction to Morphology; Springer: Tokyo, Japan, 2016; p. 1, online resource. [Google Scholar]
- Kodo, K.; Shibata, S.; Miyagawa-Tomita, S.; Ong, S.G.; Takahashi, H.; Kume, T.; Okano, H.; Matsuoka, R.; Yamagishi, H. Regulation of Sema3c and the Interaction between Cardiac Neural Crest and Second Heart Field during Outflow Tract Development. Sci. Rep.-Uk. 2017, 7, 6711. [Google Scholar] [CrossRef] [PubMed]
- Theveniau-Ruissy, M.; Perez-Pomares, J.M.; Parisot, P.; Baldini, A.; Miquerol, L.; Kelly, R.G. Coronary stem development in wild-type and Tbx1 null mouse hearts. Dev. Dynam. 2016, 245, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Kodo, K.; Nishizawa, T.; Furutani, M.; Arai, S.; Yamamura, E.; Joo, K.; Takahashi, T.; Matsuoka, R.; Yamagishi, H. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Natl. Acad. Sci. USA 2009, 106, 13933–13938. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Yoshida, J.; Sugimoto, T.; Yamamoto, M.; Makino, N.; Takamatsu, H.; Takegahara, N.; Suto, F.; Hori, M.; Fujisawa, H.; et al. Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev. Biol. 2008, 321, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; LeVaillant, C.J.; Plant, G.W.; Harvey, A.R. Changes in expression of Class 3 Semaphorins and their receptors during development of the rat retina and superior colliculus. Bmc Dev. Biol. 2014, 14, 34. [Google Scholar]
- Reidy, K.; Tufro, A. Semaphorins in kidney development and disease: modulators of ureteric bud branching, vascular morphogenesis, and podocyte-endothelial crosstalk. Pediatr. Nephrol. 2011, 26, 1407–1412. [Google Scholar] [CrossRef]
- Liu, C.F.; Angelozzi, M.; Haseeb, A.; Lefebvre, V. SOX9 is dispensable for the initiation of epigenetic remodeling and the activation of marker genes at the onset of chondrogenesis. Development 2018, 145, 1. [Google Scholar] [CrossRef]
- Vadivel, A.; Alphonse, R.S.; Collins, J.J.P.; van Haaften, T.; O’Reilly, M.; Eaton, F.; Thebaud, B. The Axonal Guidance Cue Semaphorin 3C Contributes to Alveolar Growth and Repair. PloS ONE 2013, 8, e67225. [Google Scholar] [CrossRef]
- Walker, S.; Scherer, S.W. Identification of candidate intergenic risk loci in autism spectrum disorder. Bmc Genomics 2013, 14, 499. [Google Scholar] [CrossRef]
- Yamagishi, H.; Kodo, K.; Maeda, J.; Uchida, K.; Tsuchihashi, T.; Shibata, A.; Ishizaki, R.; Yamagishi, C.; Srivastava, D. A History and Interaction of Outflow Progenitor Cells Implicated in “Takao Syndrome”. In Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology; Nakanishi, T., Markwald, R.R., Baldwin, H.S., Keller, B.B., Srivastava, D., Yamagishi, H., Eds.; Springer: Tokyo, Japan, 2016; pp. 201–209. [Google Scholar] [CrossRef]
- Schott, J.M.; Crutch, S.J.; Carrasquillo, M.M.; Uphill, J.; Shakespeare, T.J.; Ryan, N.S.; Yong, K.X.; Lehmann, M.; Ertekin-Taner, N.; Graff-Radford, N.R.; et al. Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement. 2016, 12, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Gunadi; Makhmudi, A.; Agustriani, N.; Rochadi. Effects of SEMA3 polymorphisms in Hirschsprung disease patients. Pediatric Surg. Int. 2016, 32, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. Ca-Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R. Mesenchymal-epithelial interactions: past, present, and future. Differentiation 2008, 76, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R. Tissue interactions between epithelium and mesenchyme of urogenital and integumental origin. Anat. Rec. 1972, 172, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Cuhna, G.R. Epithelio-mesenchymal interactions in developing accessory sexual glands of embryonic mice. Anat. Rec. 1970, 166, 295. [Google Scholar]
- Thomson, A.A.; Cunha, G.R.; Marker, P.C. Prostate development and pathogenesis. Differentiation 2008, 76, 559–564. [Google Scholar] [CrossRef]
- Le, H.; Arnold, J.T.; McFann, K.K.; Blackman, M.R. DHT and testosterone, but not DHEA or E2, differentially modulate IGF-I, IGFBP-2, and IGFBP-3 in human prostatic stromal cells. Am. J. Physiol. Endocrinol. Metabol. 2006, 290, E952–E960. [Google Scholar] [CrossRef]
- Lu, W.; Luo, Y.; Kan, M.; McKeehan, W.L. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J. Biol. Chem. 1999, 274, 12827–12834. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Fukabori, Y.; Nikolaropoulos, S.; Wang, F.; McKeehan, W.L. Heparin-binding keratinocyte growth factor is a candidate stromal-to-epithelial-cell andromedin. Mol. Endocrinol. 1992, 6, 2123–2128. [Google Scholar] [PubMed]
- Gao, J.; Arnold, J.T.; Isaacs, J.T. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001, 61, 5038–5044. [Google Scholar] [PubMed]
- Isaacs, J.T.; Isaacs, W.B. Androgen receptor outwits prostate cancer drugs. Nat. Med. 2004, 10, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.J.; D’Antonio, J.; Gurel, B.; Antony, L.; Demarzo, A.M.; Isaacs, J.T. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate 2010, 70, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.J.; Dalal, K.; Hsing, M.; Cheng, C.W.; Khosravi, S.; Yenki, P.; Tse, C.; Peacock, J.W.; Sharma, A.; Chiang, Y.T.; et al. Androgen receptor transcriptionally regulates semaphorin 3C in a GATA2-dependent manner. Oncotarget 2017, 8, 9617–9633. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Bishop, J.L.; Zoubeidi, A. Castration-Resistant Prostate Cancer. In Precision Molecular Pathology of Prostate Cancer; Robinson, B., Mosquera, M.J., Ro, J.Y., Divatia, M., Eds.; Springer: Cham, Switzerland, 2018; pp. 297–322. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Munnink, T.H.O.; Kruizinga, R.C.; Ananias, H.J.K.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.E.; de Jong, I.J.; et al. CXCR4 Inhibition with AMD3100 Sensitizes Prostate Cancer to Docetaxel Chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Elliott, J.; Salamone, R.J.; Telegeev, G.D.; Stakhovsky, A.E.; Schepotin, I.B.; Yan, F.; Wang, Y.; Bouchez, L.C.; Kularatne, S.A.; et al. CXCR4 Expression in Prostate Cancer Progenitor Cells. PloS ONE 2012, 7, e31226. [Google Scholar] [CrossRef] [PubMed]
- Tam, K.J.; Hui, D.H.F.; Lee, W.W.; Dong, M.; Tombe, T.; Jiao, I.Z.F.; Khosravi, S.; Takeuchi, A.; Peacock, J.W.; Ivanova, L.; et al. Semaphorin 3 C drives epithelial-to-mesenchymal transition, invasiveness, and stem-like characteristics in prostate cells. Sci. Rep.-Uk. 2017, 7, 11501. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Zhou, J.C.; Lin, C.J.; Hernandez, E.; Fazli, L.; Gleave, M.; Hsieh, J.T. Targeting Cancer Stem Cells in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.S.; Kerr, B.A. Prostate Cancer Stem Cell Markers Drive Progression, Therapeutic Resistance, and Bone Metastasis. Stem Cells Int. 2017, 2017, 8629234. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Shen, Y.; Chatterjee, B.; Siegfried, B.H.; Leatherbury, L.; Rosenthal, J.; Lucas, J.F.; Wessels, A.; Spurney, C.F.; Wu, Y.J.; et al. ENU induced mutations causing congenital cardiovascular anomalies. Development 2004, 131, 6211–6223. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Vyse, S.; Huang, P.H. Exploiting receptor tyrosine kinase co-activation for cancer therapy. Drug Discov. Today 2017, 22, 72–84. [Google Scholar] [CrossRef]
- Solit, D.B.; Rosen, N. Targeting HER2 in prostate cancer: where to next? J. Clin. Oncol. 2007, 25, 241–243. [Google Scholar] [CrossRef]
- Gallick, G.E.; Corn, P.G.; Zurita, A.J.; Lin, S.H. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Future Med. Chem. 2012, 4, 107–119. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most Human Carcinomas of the Exocrine Pancreas Contain Mutant C-K-Ras Genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Bednar, F.; Zhang, Y.Q.; Brisset, J.C.; Galban, S.; Galban, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Invest. 2012, 122, 639–653. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef]
- Ji, S.R.; Qin, Y.; Shi, S.; Liu, X.Y.; Hu, H.L.; Zhou, H.; Gao, J.; Zhang, B.; Xu, W.Y.; Liu, J.; et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res. 2015, 25, 561–573. [Google Scholar] [CrossRef]
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatology 2015, 15, 217–225. [Google Scholar] [CrossRef]
- Eser, S.; Schnieke, A.; Schneider, G.; Saur, D. Oncogenic KRAS signalling in pancreatic cancer. Br. J. Cancer 2014, 111, 817–822. [Google Scholar] [CrossRef]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Trejo, C.L.; Silva, J.M.; Gu, S.; Korkola, J.E.; Heiser, L.M.; Charles, R.P.; Rabinovich, B.A.; Hann, B.; Dankort, D.; et al. A Central Role for RAF -> MEK -> ERK Signaling in the Genesis of Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2012, 2, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Ardito, C.M.; Gruner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; DelGiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF Receptor Is Required for KRAS-Induced Pancreatic Tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Navas, C.; Hernandez-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF Receptor Signaling Is Essential for K-Ras Oncogene-Driven Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.C.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6, p11. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data (vol 2, pg 401, 2012). Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Vaitkiene, P.; Skiriute, D.; Steponaitis, G.; Skauminas, K.; Tamasauskas, A.; Kazlauskas, A. High level of Sema3C is associated with glioma malignancy. Diagn. Pathol. 2015, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.D.; Wu, Q.L.; Sathornsumetee, S.; Hao, Y.L.; Li, Z.Z.; Hjelmeland, A.B.; Shi, O.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef]
- Bao, S.D.; Wu, Q.L.; McLendon, R.E.; Hao, Y.L.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.J.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522. [Google Scholar] [CrossRef]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: the way forward. J. Clin. Invest. 2017, 127, 415–426. [Google Scholar] [CrossRef]
- Haar, C.P.; Hebbar, P.; Wallace, G.C.; Das, A.; Vandergrift, W.A.; Smith, J.A.; Giglio, P.; Patel, S.J.; Ray, S.K.; Banik, N.L. Drug Resistance in Glioblastoma: A Mini Review. Neurochem. Res. 2012, 37, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Menard, M.; Weiss, W.A. Neuroblastoma Metastases: Leveraging the Avian Neural Crest. Cancer Cell 2017, 32, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Parise, C.A.; Bauer, K.R.; Brown, M.M.; Caggiano, V. Breast Cancer Subtypes as Defined by the Estrogen Receptor (ER), Progesterone Receptor (PR), and the Human Epidermal Growth Factor Receptor 2 (HER2) among Women with Invasive Breast Cancer in California, 1999-2004. Breast J. 2009, 15, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar]
- Malik, M.F.A.; Satherley, L.K.; Davies, E.L.; Ye, L.; Jiang, W.G. Expression of Semaphorin 3C in Breast Cancer and its Impact on Adhesion and Invasion of Breast Cancer Cells. Anticancer Res. 2016, 36, 1281–1286. [Google Scholar] [PubMed]
- Cole-Healy, Z.; Vergani, P.; Hunter, K.; Brown, N.J.; Reed, M.W.R.; Staton, C.A. The relationship between semaphorin 3C and microvessel density in the progression of breast and oral neoplasia. Exp. Mol. Pathol. 2015, 99, 19–24. [Google Scholar] [CrossRef]
- Zhu, X.F.; Zhang, X.J.; Ye, Z.Q.; Chen, Y.Z.; Lv, L.; Zhang, X.H.; Hu, H.Y. Silencing of semaphorin 3C suppresses cell proliferation and migration in MCF-7 breast cancer cells. Oncol. Lett. 2017, 14, 5913–5917. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Song, H.; Zhu, J.W.; Lu, D.H. Molecular-targeted first-line therapy for advanced gastric cancer. Cochrane Db. Syst. Rev. 2016, 7, CD011461. [Google Scholar] [CrossRef] [PubMed]
- Nienhuser, H.; Schmidt, T. Angiogenesis and Anti-Angiogenic Therapy in Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed]
- Evanno, E.; Godet, J.; Piccirilli, N.; Guilhot, J.; Milin, S.; Gombert, J.M.; Fouchaq, B.; Roche, J. Tri-methylation of H3K79 is decreased in TGF-beta 1-induced epithelial-to-mesenchymal transition in lung cancer. Clin. Epigenetics. 2017, 9, 80. [Google Scholar] [CrossRef]
- Meyer, L.A.; Fritz, J.; Pierdant-Mancera, M.; Bagnard, D. Current drug design to target the Semaphorin/Neuropilin/Plexin complexes. Cell Adh. Migr. 2016, 10, 700–708. [Google Scholar] [CrossRef] [PubMed]
- LaGanke, C.; Samkoff, L.; Edwards, K.; Jung Henson, L.; Repovic, P.; Lynch, S.; Stone, L.; Mattson, D.; Galluzzi, A.; Fisher, T.L.; et al. Safety/tolerability of the anti-semaphorin 4D Antibody VX15/2503 in a randomized phase 1 trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e367. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; LoRusso, P.M.; Messersmith, W.A.; Papadopoulos, K.P.; Gore, L.; Beeram, M.; Ramakrishnan, V.; Kim, A.H.; Beyer, J.C.; Mason Shih, L.; et al. A Phase Ib study evaluating MNRP1685A, a fully human anti-NRP1 monoclonal antibody, in combination with bevacizumab and paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Weekes, C.D.; Beeram, M.; Tolcher, A.W.; Papadopoulos, K.P.; Gore, L.; Hegde, P.; Xin, Y.; Yu, R.; Shih, L.M.; Xiang, H.; et al. A phase I study of the human monoclonal anti-NRP1 antibody MNRP1685A in patients with advanced solid tumors. Investigat. New Drugs 2014, 32, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Luchino, J.; Hocine, M.; Amoureux, M.C.; Gibert, B.; Bernet, A.; Royet, A.; Treilleux, I.; Lecine, P.; Borg, J.P.; Mehlen, P.; et al. Semaphorin 3E Suppresses Tumor Cell Death Triggered by the Plexin D1 Dependence Receptor in Metastatic Breast Cancers. Cancer Cell 2013, 24, 673–685. [Google Scholar] [CrossRef]
- Kaneko, S.; Iwanami, A.; Nakamura, M.; Kishino, A.; Kikuchi, K.; Shibata, S.; Okano, H.J.; Ikegami, T.; Moriya, A.; Konishi, O.; et al. A selective Sema3A inhibitor enhances regenerative responses and functional recovery of the injured spinal cord. Nat. Med. 2006, 12, 1380–1389. [Google Scholar] [CrossRef]
- Kumagai, K.; Hosotani, N.; Kikuchi, K.; Kimura, T.; Saji, I. Xanthofulvin, a novel semaphorin inhibitor produced by a strain of Penicillium. J. Antibiot. 2003, 56, 610–616. [Google Scholar] [CrossRef]
- Lee, C.C.W.; Munuganti, R.S.N.; Peacock, J.W.; Dalal, K.; Jiao, I.Z.F.; Shepherd, A.; Liu, L.; Tam, K.J.; Sedgwick, C.G.; Bhasin, S.; et al. Targeting Semaphorin 3C in Prostate Cancer with Small Molecules. J. Endocrine Society 2018. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Weiss, G.J.; Leonard, J.E.; Rasco, D.W.; Sachdev, J.C.; Fisher, T.L.; Winter, L.A.; Reilly, C.; Parker, R.B.; Mutz, D.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of a Humanized Anti-Semaphorin 4D Antibody, in a First-In-Human Study of Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: from concept to clinical reality. Genome Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Lohrum, M.; Klostermann, A.; Betz, H.; Puschel, A.W. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. Embo. J. 1997, 16, 6077–6086. [Google Scholar] [CrossRef] [PubMed]
- Mumblat, Y.; Kessler, O.; Ilan, N.; Neufeld, G. Full-Length Semaphorin-3C Is an Inhibitor of Tumor Lymphangiogenesis and Metastasis. Cancer Res. 2015, 75, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.J.; Hu, J.H.; Uemura, A.; Tetzlaff, F.; Augustin, H.G.; Fischer, A. Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 2015, 7, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Valiulyte, I.; Preitakaite, V.; Tamasauskas, A.; Kazlauskas, A. Importance of the putative furin recognition site 742RNRR745 for antiangiogenic Sema3C activity in vitro. Braz. J. Med. Biol. Res. 2018, 51, e7786. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hui, D.H.F.; Tam, K.J.; Jiao, I.Z.F.; Ong, C.J. Semaphorin 3C as a Therapeutic Target in Prostate and Other Cancers. Int. J. Mol. Sci. 2019, 20, 774. https://doi.org/10.3390/ijms20030774
Hui DHF, Tam KJ, Jiao IZF, Ong CJ. Semaphorin 3C as a Therapeutic Target in Prostate and Other Cancers. International Journal of Molecular Sciences. 2019; 20(3):774. https://doi.org/10.3390/ijms20030774
Chicago/Turabian StyleHui, Daniel H.F., Kevin J. Tam, Ivy Z.F. Jiao, and Christopher J. Ong. 2019. "Semaphorin 3C as a Therapeutic Target in Prostate and Other Cancers" International Journal of Molecular Sciences 20, no. 3: 774. https://doi.org/10.3390/ijms20030774
APA StyleHui, D. H. F., Tam, K. J., Jiao, I. Z. F., & Ong, C. J. (2019). Semaphorin 3C as a Therapeutic Target in Prostate and Other Cancers. International Journal of Molecular Sciences, 20(3), 774. https://doi.org/10.3390/ijms20030774