The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease



Abstract
1. Alzheimer’s Disease and the Hypothesis of Its Onset
2. Oxidative Stress Theory and AD
3. Why Vitamin E as Treatment for AD?
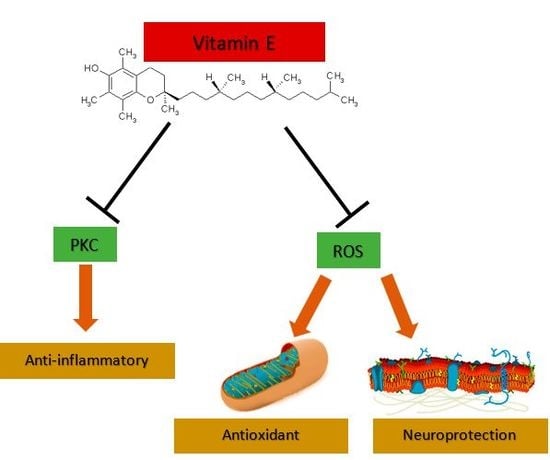
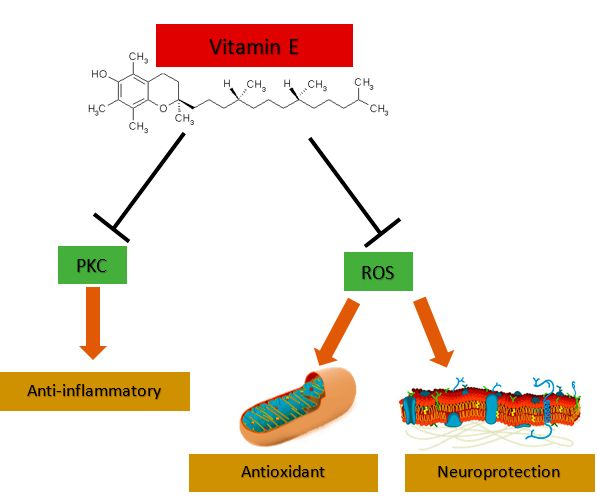
3.1. Vitamin E Is an Antioxidant and Neuroprotector
3.2. Vitamin E as an Anti-Inflammatory and Cell Signaling
3.3. Levels of Vitamin E in AD Are Low
3.4. Vitamin E and Prevention of Cognitive Decline

{kind=link}
{kind=link}
{kind=link}
| Authors and Publication Year | Isoform | Method | Number of Patients and Diagnosis | Results |
|---|---|---|---|---|
| Schippling et al, 2000 [98] | α-tocopherol | HPLC | 29 patients | The difference of α-tocopherol levels among AD patients and controls were no significant |
| von Arnim et al., 2012 [99] | α-tocopherol | HPLC | 74 MCI patients | No association was found for vitamin E levels and dementia |
| Charlton el at., 2004 [100] | α-tocopherol | HPLC | 15 AD patients | No differences in the vitamin E levels between AD patients and controls |
| Ryglewicz et al., 2002 [101] | α-tocopherol | HPLC | 26 AD patients | Levels of vitamin E was significantly lower in patients with vascular dementia in comparison to patients with AD and controls |
4. Is Vitamin E Effective as Treatment for AD? An Approach to Main Trials
5. Why Does Vitamin E Fail to Treat AD?
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.J.; Visser, P.J. Prevalence of Cerebral Amyloid Pathology in Persons Without Dementia A Meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Multhaup, G.; Simms, G.; Pottgiesser, J.; Martins, R.N.; Beyreuther, K. Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985, 4, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.D.; Golde, T.E.; Younkin, S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science 1993, 259, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Müller-Schiffmann, A.; Herring, A.; Abdel-Hafiz, L.; Chepkova, A.N.; Schäble, S.; Wedel, D.; Horn, A.H.; Sticht, H.; de Souza Silva, M.A.; Gottmann, K.; et al. Amyloid-β dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain 2016, 139, 509–525. [Google Scholar] [CrossRef]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef]
- Maccioni, R.B.; Muñoz, J.P.; Barbeito, L. The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 2001, 32, 367–381. [Google Scholar] [CrossRef]
- Ghoshal, N.; García-Sierra, F.; Wuu, J.; Leurgans, S.; Bennett, D.A.; Berry, R.W.; Binder, L.I. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 2002, 177, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Bierer, L.M.; Haroutunian, V.; Gabriel, S.; Knott, P.J.; Carlin, L.S.; Purohit, D.P.; Perl, D.P.; Schmeidler, J.; Kanof, P.; Davis, K.L. Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J. Neurochem. 1995, 64, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Lavados, M.; Guillón, M.; Mujica, C.; Bosch, R.; Farías, G.; Fuentes, P. Anomalously phosphorylated tau and Abeta fragments in the CSF correlates with cognitive impairment in MCI subjects. Neurobiol. Aging 2006, 27, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Kuret, J.; Congdon, E.E. Disaggregation of tau as a therapeutic approach to tauopathies. Curr. Alzheimer Res. 2010, 7, 40. [Google Scholar] [CrossRef]
- Fillit, H.; Ding, W.H.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320. [Google Scholar] [CrossRef]
- Strauss, S.; Bauer, J.; Ganter, U.; Jonas, U.; Berger, M.; Volk, B. Detection of interleukin-6 and alpha 2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer’s disease patients. Lab. Invest. 1992, 66, 223–230. [Google Scholar] [PubMed]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2005, 2. [Google Scholar] [CrossRef]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017, 13, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Fasolato, C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, J.C. The vascular hypothesis of Alzheimer’s disease: Bench to bedside and beyond. Neurodegener. Dis. 2010, 7, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Biochemistry of Oxidative Stress. Angew. Chem. Int. Ed. 1986, 25, 1058–1071. [Google Scholar] [CrossRef]
- Perry, G.; Nunomura, A.; Siedlak, S.L.; Harris, P.L.R.; Zhu, X.; Castellani, R.J.; Aliev, G.; Smith, M.A. Oxidant and antioxidant responses in Alzheimer disease. Recent Res. Dev. Biophys. Biochem. 2001, 1, 35–41. [Google Scholar]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Castellani, R.J.; Harris, P.L.R.; Sayre, L.M.; Fujii, J.; Taniguchi, N.; Vitek, M.P.; Founds, H.; Atwood, C.S.; Perry, G.; Smith, M.A. Advanced glycation in neurofibrillary pathology of Alzheimer disease: Ne-(carboxymethyl) lysine and hexitol-lysine. Free Radic. Biol. Med. 2001, 31, 175–180. [Google Scholar] [CrossRef]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.R.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef]
- Smith, M.A.; Sayre, L.M.; Anderson, V.E.; Harris, P.L.; Beal, M.F.; Kowall, N.; Perry, G. Cytochemical demonstration of oxidative damage in Alzheimer disease by immunochemical enhancement of the carbonyl reaction with 2,4-dinitrophenylhydrazine. J. Histochem. Cytochem. 1998, 46, 731–735. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [CrossRef]
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar] [CrossRef]
- Reiter, E.; Jiang, Q.; Christen, S. Anti-inflammatory properties of α- and γ-tocopherol. Mol. Asp. Med. 2007, 28, 668–691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Quinn, P. Vitamin E and its Function in Membranes. Prog. Lipid Res. 1999, 38, 309–336. [Google Scholar] [CrossRef]
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, A.; Han, D.; Packer, L. Vitamin E recycling in Human Erythrocyte Membranes. J. Biol. Chem. 1993, 268, 10906–10913. [Google Scholar]
- Maguire, J.J.; Wilson, D.S.; Packer, L. Mitochondrial Electron Transport-Linked Tocopheroxyl Radical Reduction. J. Biol. Chem. 1989, 264, 21462–21465. [Google Scholar] [PubMed]
- Thiele, J.J.; Schroeter, C.; Hsieh, S.N.; Podda, M.; Packer, L. Antixoidant Network of the Stratum Corneum. Curr. Probl. Dermatol. 2001, 29, 26–42. [Google Scholar] [PubMed]
- Brigelius-Flohe, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med. 2009, 46, 543–554. [Google Scholar] [CrossRef]
- Xu, L.; Davis, T.A.; Porter, N.A. Rate constants for peroxidation of polyunsaturated fatty acids and sterols in solution and in liposomes. J. Am. Chem. Soc. 2009, 131, 13037–13044. [Google Scholar] [CrossRef] [PubMed]
- Ham, A.J.; Liebler, D.C. Antioxidant reactions of vitamin E in the perfused rat liver: Product distribution and effect of dietary vitamin E supplementation. Arch. Biochem. Biophys. 1997, 339, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Copp, R.P.; Wisniewski, T.; Hentati, F.; Larnaout, A.; Ben Hamida, M.; Kayden, H.J. Localization of alpha-tocopherol transfer protein in the brains of patients with ataxia with vitamin e deficiency and other oxidative stress related neurodegenerative disorders. Brain Res. 1999, 822, 80–87. [Google Scholar] [CrossRef]
- Ulatowski, L.M.; Manor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E.; Matthews, S.; Jones, S.; Ellis, C.J.; Booth, I.W.; Muller, D.P. Spinocerebellar degeneration associated with a selective defect of vitamin E absorption. N. Engl. J. Med. 1985, 313, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Cavalier, L.; Ouahchi, K.; Kayden, H.J.; Di Donato, S.; Reutenauer, L.; Mandel, J.L.; Koenig, M. Ataxia with isolated vitamin E deficiency: Heterogeneity of mutations and phenotypic variability in a large number of families. Am. J. Hum. Genet. 1998, 62, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Manor, D. Vitamin E trafficking in neurologic health and disease. Annu. Rev. Nutr. 2013, 33, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Dreussi, C.; Noy, N.; Barnholtz-Sloan, J.; Klein, E.; Manor, D. Expression of the α-tocopherol transfer protein gene is regulated by oxidative stress and common single-nucleotide polymorphisms. Free Radic. Biol. Med. 2012, 53, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015, 151979. [Google Scholar] [CrossRef]
- Gamblin, T.C.; King, M.E.; Kuret, J.; Berry, R.W.; Binder, L.I. Oxidative regulation of fatty acid-induced tau polymerization. Biochemistry 2000, 39, 14203–14210. [Google Scholar] [CrossRef]
- Pérez, M.; Cuadros, R.; Smith, M.A.; Perry, G.; Avila, J. Phosphorylated, but not native, tau protein assembles following reaction with the lipid peroxidation product, 4-hydroxy-2-nonenal. FEBS Lett. 2000, 486, 270–274. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimers Dis. 2013, 33, S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Viña, J. Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Biol. 2014, 2, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, I.; Garwood, C.; Hanger, D.P.; Anderton, B.H.; Noble, W. Kinase activities increase during the development of tauopathy in htau mice. J. Neurochem. 2007, 103, 2256–2267. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Man-Fan Wan, J. Vitamin E Supplementation Improves Cell-Mediated Immunity and Oxidative Stress of Asian Men and Women. J. Nutr. 2000, 130, 2932–2937. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente, M.; Hernanz, A.; Guayerbas, N.; Victor, V.M.; Arnalich, F. Vitamin E ingestion improves several immune functions in elderly men and women. Free Radic. Res. 2008, 42, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Montano Velazquez, B.B.; Jauregui-Renaud, K.; Banuelos Arias Adel, C.; Ayala, J.C.; Martinez, M.D.; Campillo Navarrete, R.; Rosalia, I.S.; Salazar Mdel, R.; Serrano, H.A.; Mondragon, A.O.; et al. Vitamin E effects on nasal symptoms and serum specific IgE levels in patients with perennial allergic rhinitis. Ann. Allergy Asthma Immunol. 2006, 96, 45–50. [Google Scholar] [CrossRef]
- Wolvers, D.A.; van Herpen-Broekmans, W.M.; Logman, M.H.; van der Wielen, R.P.; Albers, R. Effect of a mixture of micronutrients, but not of bovine colostrum concentrate, on immune function parameters in healthy volunteers: A randomized placebo-controlled study. Nutr. J. 2006, 5, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hemila, H.; Kaprio, J. Modification of the effect of vitamin E supplementation on the mortality of male smokers by age and dietary vitamin C. Am. J. Epidemiol. 2009, 169, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Hemila, H.; Kaprio, J. Vitamin E may affect the life expectancy of men, depending on dietary vitamin C intake and smoking. Age Ageing 2011, 40, 215–220. [Google Scholar] [CrossRef]
- Jiang, Q.; Elson-Schwab, I.; Courtemanche, C.; Ames, B.N. Gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc. Natl. Acad. Sci. USA 2000, 97, 11494–11499. [Google Scholar] [CrossRef]
- Jiang, Q.; Yin, X.; Lill, M.A.; Danielson, M.L.; Freiser, H.; Huang, J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc. Natl. Acad. Sci. USA 2008, 105, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yin, X.; Jiang, Q. Natural forms of vitamin E and 13′-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J. Immunol. 2011, 186, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Tasinato, A.; Boscoboinik, D.; Bartoli, G.M.; Maroni, P.; Azzi, A. d-α-Tocopherol Inhibition of Vascular Smooth Muscle Cell Proliferation Occurs at Physiological Correlates with Protein Kinase C Inhibition and Is Independent of Its Antioxidant properties. Proc. Natl. Acad. Sci. USA 1995, 92, 12190–12194. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Tasinato, A.; Clement, S.; Ozer, N.K.; Boscoboinik, D.; Azzi, A. α-Tocopherol Specifically Inactivates Cellular Protein Kinase C Alpha by Changing Its Phosphorylation State. Biochem. J. 1998, 334, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Tasinato, A.; Boscoboinik, D. The Effect of Alpha Tocopherol on the Synthesis, Phosphorylation and Activity of Protein Kinase C in Smooth Muscle Cells After Phorbol 12-Myristate 13-Acetate Down-Regulation. Eur. J. Biochem. 1997, 246, 745–749. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Azzi, A. α-Tocopherol (Vitamin E) regulates vascular smooth muscle cell proliferation and protein kinase C activity. Arch. Biochem. Biophys. 1991, 286, 264–269. [Google Scholar] [CrossRef]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of cell proliferation by α-Tocopherol. Role of protein kinase C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar]
- Miscia, S.; Ciccocioppo, F.; Lanuti, P.; Velluto, L.; Bascelli, A.; Pierdomenico, L.; Genovesi, D.; Di Siena, A.; Santavenere, E.; Gambi, F.; et al. Aβ1–42 stimulated T cells express P-PKC-δ and P-PKC-ζ in Alzheimer disease. Neurobiol. Aging 2009, 30, 394–406. [Google Scholar] [CrossRef]
- Skovronsky, D.M.; Moore, D.B.; Milla, M.E.; Doms, R.W.; Lee, V.M. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J. Biol. Chem. 2000, 275, 2568–2575. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Thiery, J.; Seidel, D. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 1999, 102, 82–87. [Google Scholar] [CrossRef]
- Teupser, D.; Stein, O.; Burkhardt, R.; Nebendahl, K.; Stein, Y.; Thiery, J. Scavenger receptor activity is increased in macrophages from rabbit with low atherosclerotic response: Studies in normocholesterolemic high and low atherosclerotic response rabbits. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Koga, T.; Martin, K.R.; Meydani, M. Effect of Vitamin E on human aortic endothelial cell production of chemokines and adhesion to monocytes. Atherosclerosis 1999, 147, 297–307. [Google Scholar] [CrossRef]
- Jeandel, C.; Nicolas, M.B.; Dubois, F.; Nabet-Belleville, F.; Penin, F.; Cuny, G. Lipid peroxidation and free radical scavengers in Alzheimer’s disease. Gerontology 1989, 35, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lopes da Silva, S.; Vellas, B.; Elemans, S.; Luchsinger, J.; Kamphuis, P.; Yaffe, K.; Sijben, J.; Groenendijk, M.; Stijnen, T. Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimers Dement. 2014, 10, 485–502. [Google Scholar] [CrossRef] [PubMed]
- de Wilde, M.C.; Vellas, B.; Girault, E.; Yavuz, A.C.; Sijben, J.W. Lower brain and blood nutrient status in Alzheimer’s disease: Results from meta-analyses. Alzheimers Dement. 2017, 3, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Chen, X.; Liu, Y.; Shu, Y.; Chen, T.; Xu, L.; Li, M.; Guan, X. Do low-serum vitamin E levels increase the risk of Alzheimer disease in older people? Evidence from a meta-analysis of case-control studies. Int. J. Geriatr. Psychiatry 2018, 33, e257–e263. [Google Scholar] [CrossRef]
- Zaman, Z.; Roche, S.; Fielden, P.; Frost, P.G.; Niriella, D.C.; Cayley, A.C. Plasma concentrations of vitamins A and E and carotenoids in Alzheimer’s disease. Age Ageing 1992, 21, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Jiménez, F.J.; de Bustos, F.; Molina, J.A.; Benito-León, J.; Tallón-Barranco, A.; Gasalla, T.; Ortí-Pareja, M.; Guillamón, F.; Rubio, J.C.; Arenas, J.; et al. Cerebrospinal fluid levels of alpha-tocopherol (vitamin E) in Alzheimer’s disease. J. Neural Transm. 1997, 104, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J.; Bayer, A.J.; Johnston, J.; Warner, C.; Maxwell, S.R. Altered plasma antioxidant status in subjects with Alzheimer’s disease and vascular dementia. Int. J. Geriatr. Psychiatry 1998, 13, 840–845. [Google Scholar] [CrossRef]
- Foy, C.J.; Passmore, A.P.; Vahidassr, M.D.; Young, I.S.; Lawson, J.T. Plasma chain-breaking antioxidants in Alzheimer’s disease, vascular dementia and Parkinson’s disease. Q. J. Med. 1999, 92, 39–45. [Google Scholar] [CrossRef]
- Bourdel-Marchasson, I.; Delmas-Beauvieux, M.C.; Peuchant, E.; Richard-Harston, S.; Decamps, A.; Reignier, B.; Emeriau, J.P.; Rainfray, M. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing 2001, 30, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mecocci, P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J. Alzheimers Dis. 2002, 4, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.; Cherubini, A.; Ingegni, T.; Mattioli, P.; Catani, M.; Rinaldi, P.; Cecchetti, R.; Stahl, W.; Senin, U.; et al. Lymphocyte oxidative DNA damage and plasma antioxidants in Alzheimer disease. Arch. Neurol. 2002, 59, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Giavarotti, L.; Simon, K.A.; Azzalis, L.A.; Fonseca, F.L.A.; Lima, A.F.; Freitas, M.C.V.; Brunialti, M.K.C.; Salomao, R.; Moscardi, A.A.V.S.; Montano, M.B.M.M.; et al. Mild systemic oxidative stress in the subclinical stage of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 609019. [Google Scholar] [CrossRef]
- Mullan, K.; Williams, M.A.; Cardwell, C.R.; McGuinness, B.; Passmore, P.; Silvestri, G.; Woodside, J.V.; McKay, G.J. Serum concentrations of vitamin E and carotenoids are altered in Alzheimer’s disease: A case-control study. Alzheimers. Dement. Trans. Res. Clin. Interv. 2017, 3, 432–439. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Y.; Jin, S.; Hu, Y.; Wang, T.; Tian, R.; Han, Z.; Xu, D.; Jiang, Q. Circulating vitamin E levels and Alzheimer’s disease: A Mendelian randomization study. Neurobiol. Aging 2018, 72, e1–e189. [Google Scholar] [CrossRef]
- Morris, M.C.; Beckett, L.A.; Scherr, P.A.; Hebert, L.E.; Bennett, D.A.; Field, T.S.; Evans, D.A. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 121–126. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Wilson, R.S.; Scherr, P.A. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 2002, 287, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Devore, E.E.; Grodstein, F.; van Rooij, F.J.; Hofman, A.; Stampfer, M.J.; Witteman, J.C.; Breteler, M.M. Dietary antioxidants and long-term risk of dementia. Arch. Neurol. 2010, 67, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Basambombo, L.L.; Carmichael, P.H.; Côté, S.; Laurin, D. Use of vitamin E and C supplements for the prevention of cognitive decline. Ann. Pharmacother. 2017, 51, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.L.; Anderson, M.L.; Crane, P.K.; Breitner, J.C.; McCormick, W.; Bowen, J.D.; Teri, L.; Larson, E. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. J. Am. Geriatr. Soc. 2008, 56, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Masaki, K.H.; Losonczy, K.G.; Izmirlian, G.; Foley, D.J.; Ross, G.W.; Petrovitch, H.; Havlik, R.; White, L.R. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology 2000, 54, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch. Neurol. 2003, 60, 203–208. [Google Scholar] [CrossRef]
- Schippling, S.; Kontush, A.; Arlt, S.; Buhmann, C.; Sturenburg, H.J.; Mann, U.; Muller-Thomsen, T.; Beisiegel, U. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic. Biol. Med. 2000, 28, 351–360. [Google Scholar] [CrossRef]
- von Arnim, C.A.; Herbolsheimer, F.; Nikolaus, T.; Peter, R.; Biesalski, H.K.; Ludolph, A.C.; Riepe, M.; Nagel, G. Dietary antioxidants and dementia in a population-based case-control study among older people in South Germany. J. Alzheimers Dis. 2012, 31, 717–724. [Google Scholar] [CrossRef]
- Charlton, K.E.; Rabinowitz, T.L.; Geffen, L.N.; Dhansay, M.A. Lowered plasma vitamin C, but not vitamin E, concentration in dementia patients. J. Nutr. Health Aging 2004, 8, 99–107. [Google Scholar]
- Ryglewicz, D.; Rodo, M.; Kunicki, P.K.; Bednarska-Makaruk, M.; Graban, A.; Lojkowska, W.; Wehr, H. Plasma antioxidant activity and vascular dementia. J. Neurol. Sci. 2002, 203–204, 195–197. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A.; et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badía, M.C.; Mora, N.J.; Pallardó, F.V.; Alonso, M.D.; Viña, J. Vitamin E paradox in Alzheimer’s disease: It does not prevent loss of cognition and may even be detrimental. J. Alzheimers Dis. 2009, 17, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J.; et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Ibrahima, N.F.; Yanagisawa, D.; Durani, L.W.; Hamezah, H.S.; Damanhuri, H.A.; Ngah, W.Z.W.; Tsuji, M.; Kiuchi, Y.; Ono, K.; Tooyama, I. Tocotrienol-Rich Fraction Modulates Amyloid Pathology and Improves Cognitive Function in AβPP/PS1 Mice. J. Alzheimers Dis. 2017, 55, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Huang, X.; Zhen, J.; Van Halm-Lutterodt, N.; Wang, J.; Zhou, C.; Yuan, L. Dietary Vitamin E Status Dictates Oxidative Stress Outcomes by Modulating Effects of Fish Oil Supplementation in Alzheimer Disease Model APPswe/PS1dE9 Mice. Mol. Neurobiol. 2018, 55, 9204–9219. [Google Scholar] [CrossRef]
- Halliwell, B. Free radicals and antioxidants: Updating a personal view. Nutr. Rev. 2012, 70, 257–265. [Google Scholar] [CrossRef]
- Halliwell, B. The antioxidant paradox: Less paradoxical now? Br. J. Clin. Pharmacol. 2013, 75, 637–644. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 2014, 1840, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Hardy, M.; Michalski, R.; Sikora, A.; Zielonka, M.; Cheng, G.; Ouari, O.; Podsiadły, R.; Kalyanaraman, B. Recent developments in the probes and assays for measurement of the activity of NADPH oxidases. Cell Biochem. Biophys. 2017, 75, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Ribou, A.C. Synthetic Sensors for Reactive Oxygen Species Detection and Quantification: A Critical Review of Current Methods. Antioxid. Redox Signal. 2016, 25, 520–533. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Richelle, M.; Enslen, M.; Hager, C.; Groux, M.; Tavazzi, I.; Godin, J.P.; Berger, A.; Métairon, S.; Quaile, S.; Piguet-Welsch, C.; et al. Both free and esterified plant sterols reduce cholesterol absorption and the bioavailability of beta-carotene and alpha-tocopherol in normocholesterolemic humans. Am. J. Clin. Nutr. 2004, 80, 171–177. [Google Scholar] [CrossRef]
- Bjørneboe, A.; Bjørneboe, G.E.; Drevon, C.A. Absorption, transport and distribution of vitamin E. J. Nutr. 1990, 120, 233–242. [Google Scholar] [CrossRef]
- Bieri, J.G.; Wu, A.L.; Tolliver, T.J. Reduced intestinal absorption of vitamin E by low dietary levels of retinoic acid in rats. J. Nutr. 1981, 111, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.L.; Vuksan, V.; Jenkins, D.J. Fiber in the treatment of hyperlipidemia. In CRC Handbook of Dietary Fiber in Human Nutrition, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2001; pp. 358–380. [Google Scholar]
- Kolleck, I.; Schlame, M.; Fechner, H.; Looman, A.C.; Wissel, H.; Rüstow, B. HDL is the major source of vitamin E for type II pneumocytes. Free Radic. Biol. Med. 1999, 27, 882–890. [Google Scholar] [CrossRef]
- Goti, D.; Hammer, A.; Galla, H.J.; Malle, E.; Sattler, W. Uptake of lipoprotein-associated alpha-tocopherol by primary porcine brain capillary endothelial cells. J. Neurochem. 2000, 74, 1374–1383. [Google Scholar] [CrossRef]
- Campbell, D.; Bunker, V.W.; Thomas, A.J.; Clayton, B.E. Selenium and vitamin E status of healthy and institutionalized elderly subjects: Analysis of plasma, erythrocytes and platelets. Br. J. Nutr. 1989, 62, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.W.; Paterson, E.; Atkinson, J.K.; Ramakrishnan, R.; Cross, C.E.; Traber, M.G. Studies in humans using deuterium-labeled alpha- and gamma-tocopherols demonstrate faster plasma gamma-tocopherol disappearance and greater gamma-metabolite production. Free Radic. Biol. Med. 2005, 38, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Al-Azemi, M.K.; Omu, A.E.; Fatinikun, T.; Mannazhath, N.; Abraham, S. Factors contributing to gender differences in serum retinol and alpha-tocopherol in infertile couples. Reprod. Biomed. Online 2009, 19, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Khand, F.; Khand, T.U. Effect of smoking on serum xanthine oxidase, malondialdehyde, ascorbic acid and α-tocopherol levels in healthy male subjects. Pak. J. Med. Sci. 2015, 31, 146–149. [Google Scholar] [CrossRef]
- Galan, P.; Viteri, F.E.; Bertrais, S.; Czernichow, S.; Faure, H.; Arnaud, J.; Ruffieux, D.; Chenal, S.; Arnault, N.; Favier, A.; et al. Serum concentrations of beta-carotene, vitamins C and E, zinc and selenium are influenced by sex, age, diet, smoking status, alcohol consumption and corpulence in a general French adult population. Eur. J. Clin. Nutr. 2005, 59, 1181–1190. [Google Scholar] [CrossRef] [PubMed]
- Gunanti, I.R.; Marks, G.C.; Al-Mamun, A.; Long, K.Z. Low serum concentrations of carotenoids and vitamin E are associated with high adiposity in Mexican-American children. J. Nutr. 2014, 144, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Wallström, P.; Wirfält, E.; Lahmann, P.H.; Gullberg, B.; Janzon, L.; Berglund, G. Serum concentrations of beta-carotene and alpha-tocopherol are associated with diet, smoking, and general and central adiposity. Am. J. Clin. Nutr. 2001, 73, 777–785. [Google Scholar] [CrossRef]
- Ohrvall, M.; Tengblad, S.; Vessby, B. Lower tocopherol serum levels in subjects with abdominal adiposity. J. Intern. Med. 1993, 234, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, S.M.; van’t Veer, P.; Kok, F.; Kardinaal, A.F.; Aro, A. Predictors of adipose tissue tocopherol and toenail selenium levels in nine countries: The EURAMIC study. European Multicentre Case-Control Study on Antioxidants, Myocardial Infarction, and Cancer of the Breast. Eur. J. Clin. Nutr. 1996, 50, 599–606. [Google Scholar]
- Döring, F.; Rimbach, G.; Lodge, J.K. In silico search for single nucleotide polymorphisms in genes important in vitamin E homeostasis. IUBMB Life 2004, 56, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Desmarchelier, C.; Nowicki, M.; Bott, R.; Tourniaire, F. Can genetic variability in α-tocopherol bioavailability explain the heterogeneous response to α-tocopherol supplements? Antioxid. Redox Signal. 2015, 22, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Moussa, M.; Reboul, E.; Lyan, B.; Defoort, C.; Vincent-Baudry, S.; Maillot, M.; Gastaldi, M.; Darmon, M.; Portugal, H.; et al. Human plasma levels of vitamin E and carotenoids are associated with genetic polymorphisms in genes involved in lipid metabolism. J. Nutr. 2007, 137, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Major, J.M.; Yu, K.; Wheeler, W.; Zhang, H.; Cornelis, M.C.; Wright, M.E.; Yeager, M.; Snyder, K.; Weinstein, S.J.; Mondul, A.; et al. Genome-wide association study identifies common variants associated with circulating vitamin E levels. Hum. Mol. Genet. 2011, 20, 3876–3883. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, B.; Ulatowski, L.M. Vitamin E and Alzheimer’s Disease-Is It Time for Personalized Medicine? Antioxidants (Basel) 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Panza, F.; Frisardi, V.; Seripa, D.; Logroscino, G.; Imbimbo, B.P.; Pilotto, A. Diet and Alzheimer’s disease risk factors or prevention: The current evidence. Expert Rev. Neurother. 2011, 11, 677–708. [Google Scholar] [CrossRef]
- Badia, M.C.; Lloret, A.; Giraldo, E.; Dasí, F.; Olaso, G.; Alonso, M.D.; Viña, J. Lymphocytes from young healthy persons carrying the ApoE4 allele overexpress stress-related proteins involved in the pathophysiology of Alzheimer’s disease. J. Alzheimers Dis. 2012, 33, 77–83. [Google Scholar] [CrossRef]


| Authors and Publication Year | Isoform | Method | Number of Patients and Diagnosis | Results |
|---|---|---|---|---|
| Zaman et al., 1992 [78] | High-performance liquid chromatography (HPLC) | 10 AD patients | Lower levels of plasma vitamin E | |
| Jimenez-Jimenez et al., 1997 [79] | α-tocopherol | HPLC | 44 AD patients | Decreased levels of vitamin E both in serum and in CSF |
| Sinclair et al., 1998 [80] | α-tocopherol | HPLC | 25 AD patients | Lower plasma levels of vitamin E |
| Foy et al., 1999 [81] | α-tocopherol | HPLC | 79 patients with AD | Lower plasma levels |
| Bourdel-Marchasson et al., 2001 [82] | α-tocopherol | HPLC | 20 patients | Lower plasma levels of α-tocopherol |
| Polidori et al., 2002 [83] | α-tocopherol | HPLC | 35 patients | Plasma concentration of vitamin E lower and malondialdehyde higher |
| Rinaldi et al., 2003 [84] | α-tocopherol | HPLC | 25 patients with mild cognitive impairment (MCI) and 63 AD patients | Vitamin E decrease |
| Mecocci et al., 2002 [85] | α-tocopherol | HPLC | 40 patients | Vitamin E decrease in plasma |
| Giavarotti et al., 2013 [86] | α-tocopherol | Precolumn and reverse phase C18 column and UV-VIS detector with deuterium lamp, a mobile phase with 80% acetonitrile, 3% methanol and 15% dioxan | 23 patients with AD | Lower plasmatic levels of α-tocopherol |
| Mullan et al., 2017 [87] | α- and γ-tocopherol | HPLC | 251patients with AD | Lower levels of α-tocopherol but γ-tocopherol higher in serum of AD patients |
| Authors and Publication Year | Number of Patients and Diagnosis | Isoform | Doses | Time | Method | Results |
|---|---|---|---|---|---|---|
| Sano et al., 1997 [102] | 341 AD | α-tocopherol | 2000 IU/d for 2 years | Two years | Alzheimer’s Disease Assessment Scale (ADCS); Mini–Mental State Examination (MMSE); Blessed Dementia Scale; Dependence Scale | Decline progression of AD |
| Petersen et al., 2005 [103] | 769 AD | not specified | 2000 IU/d for 3 years | Three years | A battery of fifteen cognitive tests (MMSE, The Clinical Dementia Rating (CDR), ADCS…) | No benefit |
| Lloret et al., 2009 [104] | 33 AD | α-tocopherol | 800 IU/d for 6 months | Six months | MMSE; Blessed-Dementia Scale; Clock Drawing Test | Cognitive status was maintained in some cases but in others it was detrimental in terms of cognition |
| Dysken et al., 2014 [105] | 613 mild to moderate AD | α-tocopherol | 2000 IU/d | 6 months–4 years | Alzheimer’s Disease Cooperative Study/Activities of Daily Living (ADCS-ADL) Inventory; MMSE | Reduced functional decline |
| Kryscio et al., 2017 [106] | 7540 asymptomatic older men | 400 IU/d | 6 years | Memory Impairment Screen (MIS); Consortium to Establish a Registry for AD (CERAD) battery | No prevention of dementia |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879. https://doi.org/10.3390/ijms20040879
Lloret A, Esteve D, Monllor P, Cervera-Ferri A, Lloret A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(4):879. https://doi.org/10.3390/ijms20040879
Chicago/Turabian StyleLloret, Ana, Daniel Esteve, Paloma Monllor, Ana Cervera-Ferri, and Angeles Lloret. 2019. "The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 4: 879. https://doi.org/10.3390/ijms20040879
APA StyleLloret, A., Esteve, D., Monllor, P., Cervera-Ferri, A., & Lloret, A. (2019). The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. International Journal of Molecular Sciences, 20(4), 879. https://doi.org/10.3390/ijms20040879