Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer

 , , and
, , and
Abstract
:1. Background
2. Results
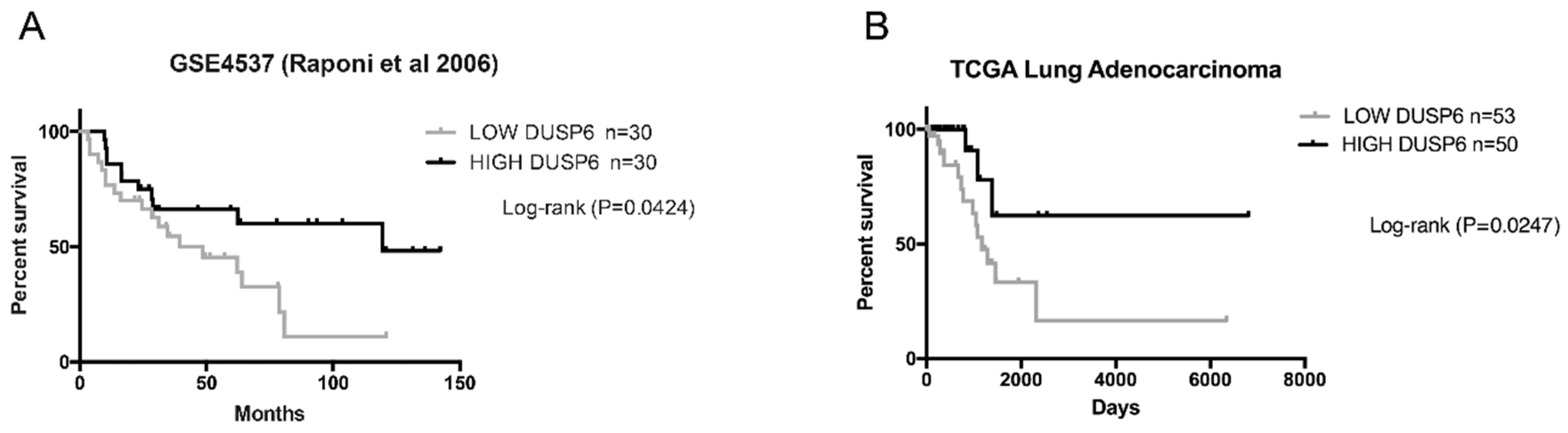
2.1. DUSP6 Acts as a Tumor Suppressor Gene in Lung Adenocarcinoma
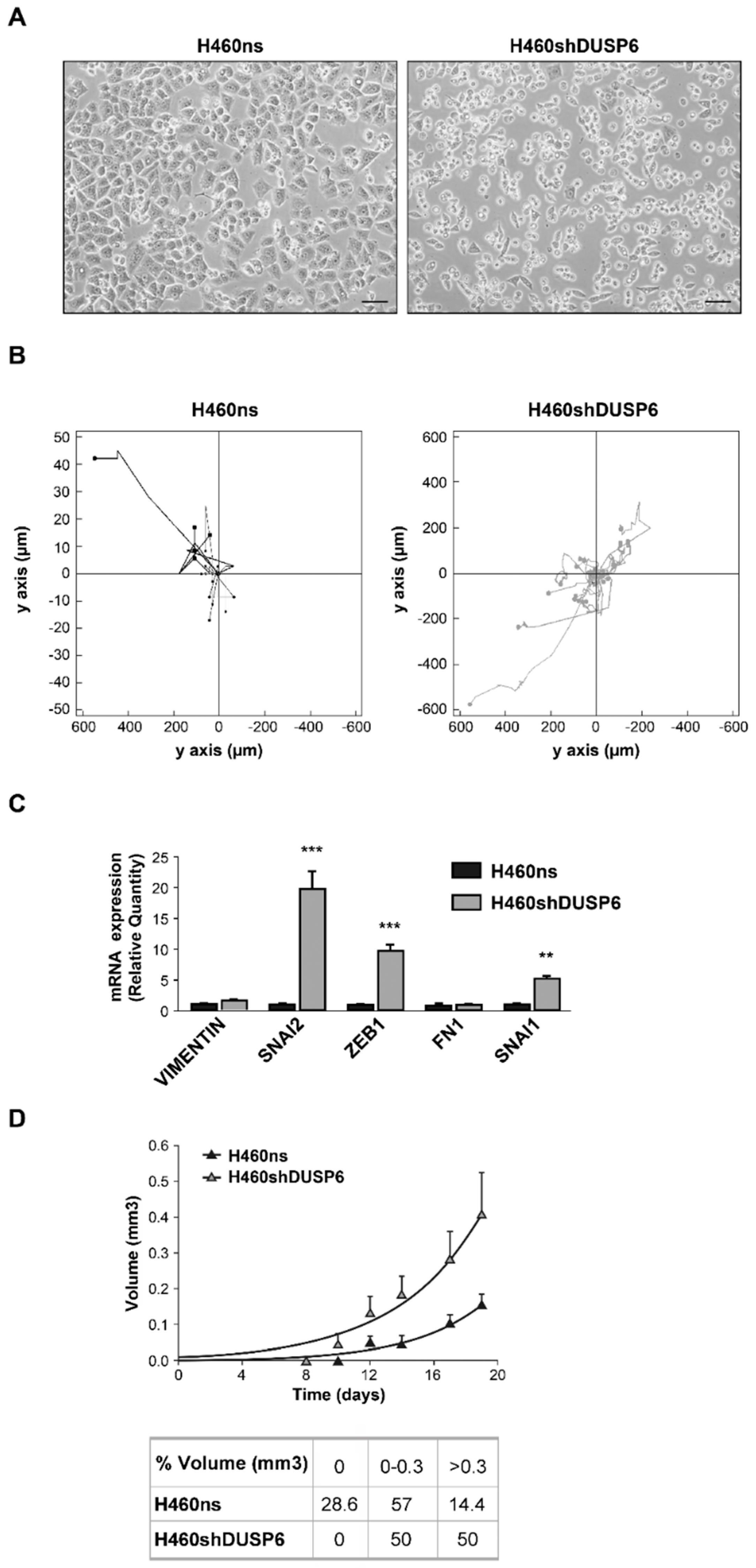
2.2. Inhibition of DUSP6 Expression Induces Changes in Cell Morphology, Anchorage, Motility and Tumorigenesis in Non-Small Cell Lung Cancer Cells
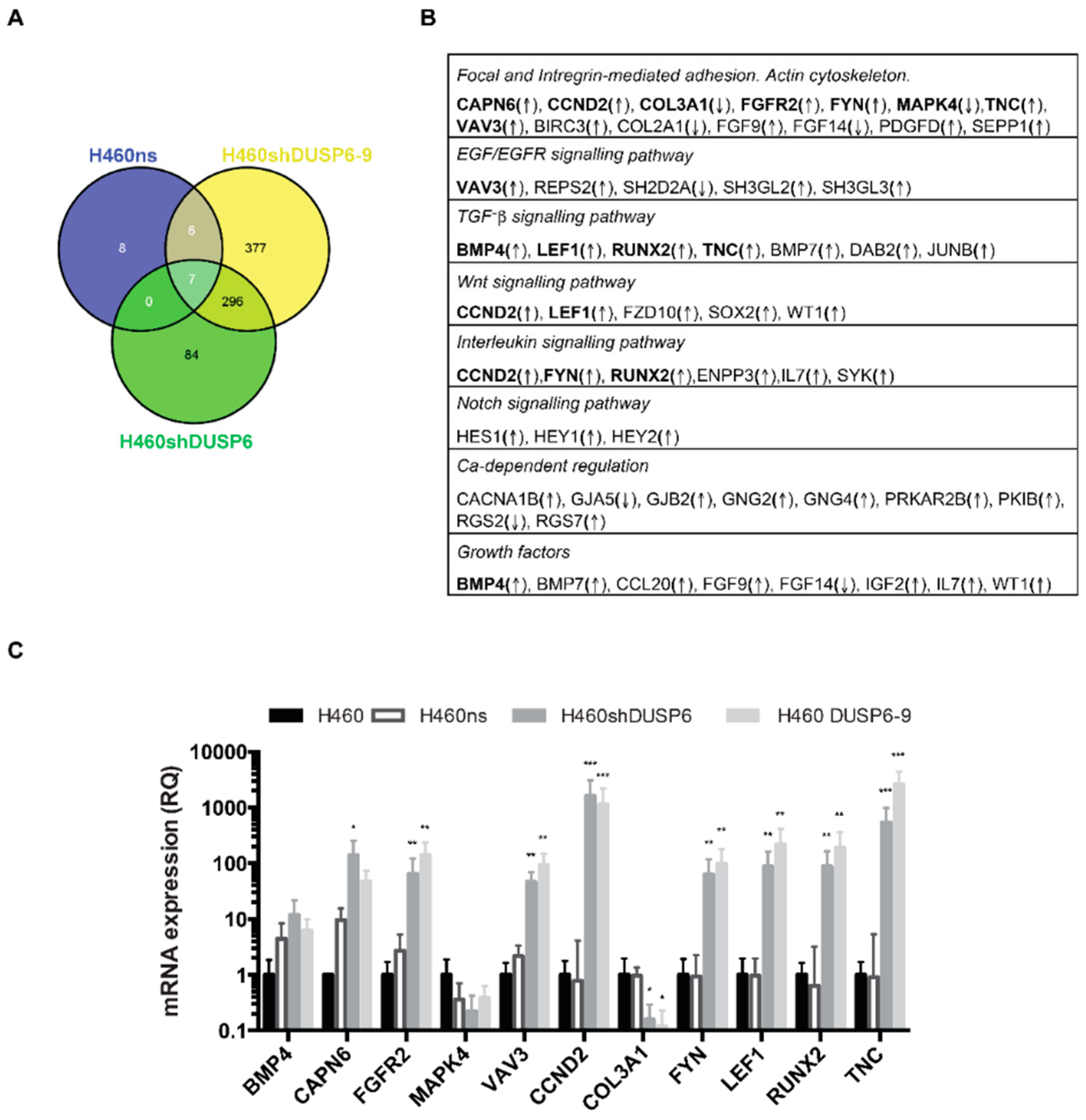
2.3. Gene Expression Profiles of H460 DUSP6 Depleted Cells
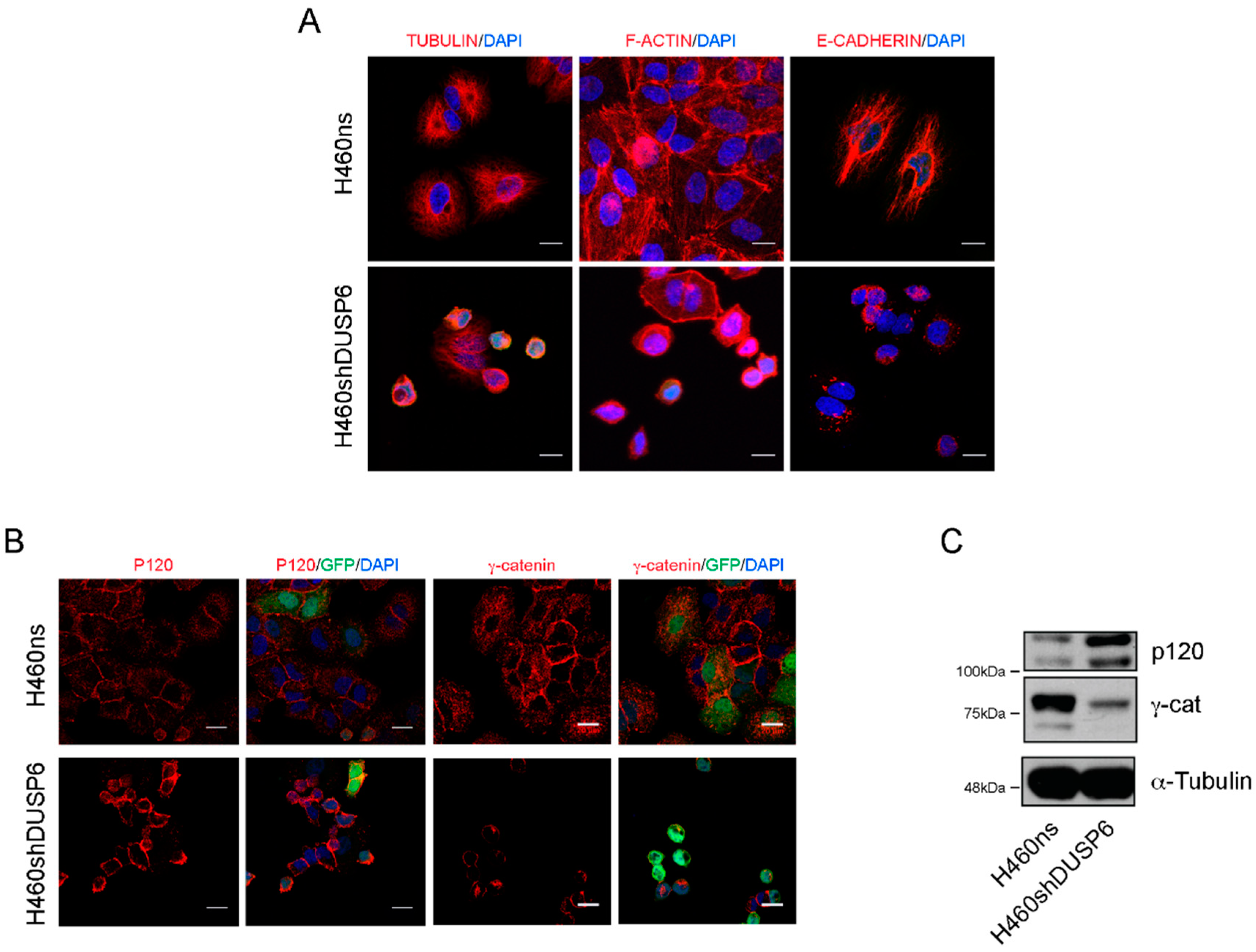
2.4. DUSP6 Depleted Cells Show Changes in the Organization at Actin Cytoskeleton and Cell-Cell Contact
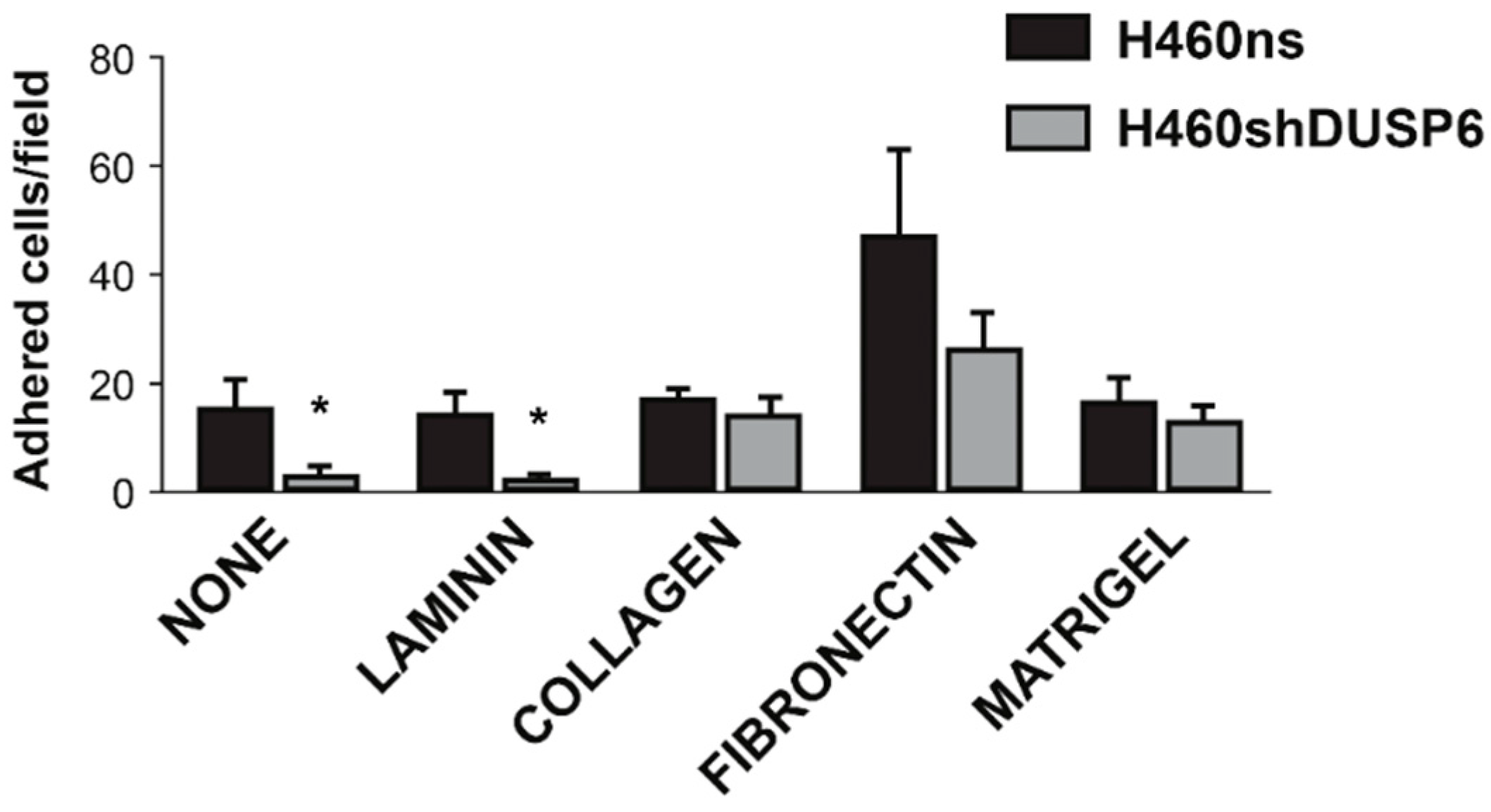
2.5. Focal Adhesion Pathway Is Deregulated in Non-Small Cell Lung Cancer Cells Depleted of DUSP6
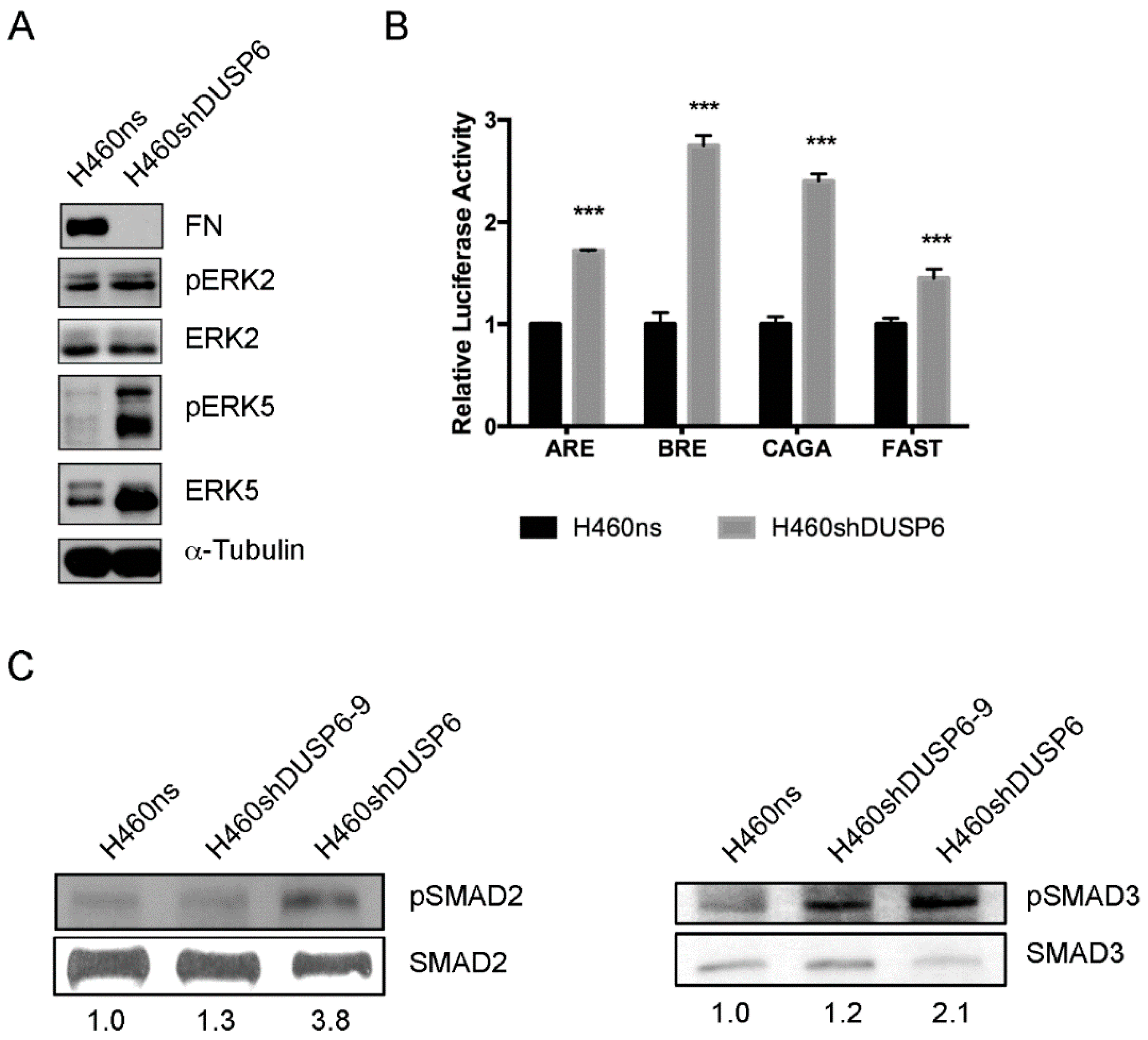
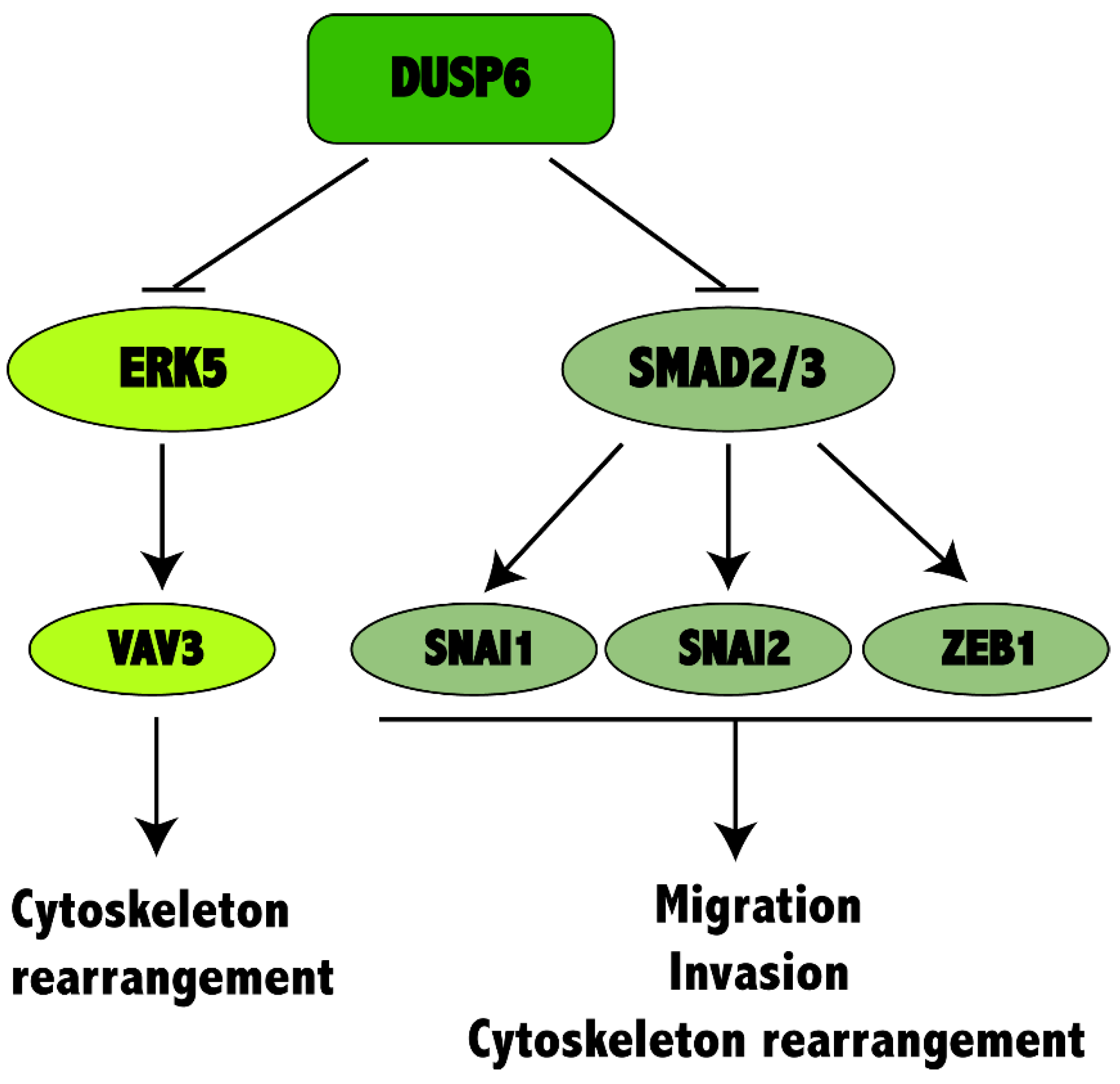
2.6. DUSP6 Depletion Activates MAPK and TGF-β Signaling Pathway in Non-Small Cell Lung Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Compounds and Luciferase Assay
4.2. Lentivirus Production and Cell Infections
4.3. RNA-seq Studies
4.4. Validation of mRNA Changes by RT-q-PCR
4.5. Evaluation of Gene Expression of Epithelial to Mesenchymal Transition Associated Genes by RT-q-PCR
4.6. Western Blots
4.7. Cell Motility Assay
4.8. Immunofluorescence
4.9. Adhesion Assays
4.10. Tumorogenicity in Xenograft Mouse Model
4.11. Statistical Analysis
4.12. “In Silico” Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American Type Culture Collection |
| ECACC | European Collection of Cell Cultures |
| NSCLC | Non-Small Cell Lung Cancer |
| MAPKs | Mitogen-activated Protein Kinases |
| ESCC | Esophageal squamous cell carcinoma |
| NPC | Nasopharyngeal carcinoma |
| ECM | Extracellular matrix |
| GIST | Gastrointestinal stromal tumors |
| FDR | False Discovery rate |
| ROX | (6-Carboxyl-X-Rho-damine |
References
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Zhang, Z.; Stiegler, A.L.; Boggon, T.J.; Kobayashi, S.; Halmos, B. EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 2010, 1, 497–514. [Google Scholar] [PubMed]
- Giurisato, E.; Tournier, C. Can tumour cells proliferate without ERK5? Cell Cycle 2016, 15, 619–620. [Google Scholar] [CrossRef] [PubMed]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Barros, J.C.; Marshall, C.J. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J. Cell Sci. 2005, 118, 1663–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schramp, M.; Ying, O.; Kim, T.Y.; Martin, G.S. ERK5 promotes Src-induced podosome formation by limiting Rho activation. J. Cell Biol. 2008, 181, 1195–1210. [Google Scholar] [CrossRef] [Green Version]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.C.; Chen, H.; Ko, J.M.; Chan, K.W.; Chan, Y.P.; Law, S.; Chua, D.; Kwong, D.L.; Lung, H.L.; Srivastava, G.; et al. Tumor suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell invasion and epithelial-mesenchymal transition (EMT)-associated phenotype. Int. J. Cancer 2012, 130, 83–95. [Google Scholar] [CrossRef]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.; Macdonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y.; et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef]
- Cai, B.; Jiang, X. Revealing Biological Pathways Implicated in Lung Cancer from TCGA Gene Expression Data Using Gene Set Enrichment Analysis. Cancer Inform. 2014, 13, 113–121. [Google Scholar] [CrossRef]
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011, 71, 245–254. [Google Scholar] [CrossRef]
- Spaderna, S.; Schmalhofer, O.; Wahlbuhl, M.; Dimmler, A.; Bauer, K.; Sultan, A.; Hlubek, F.; Jung, A.; Strand, D.; Eger, A.; et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008, 68, 537–544. [Google Scholar] [CrossRef]
- Qi, W.; Yang, Z.; Li, H.; Cui, Y.; Xuan, Y. The role of Tenascin-C and Twist1 in gastric cancer: Cancer progression and prognosis. Acta Pathol. Microbiol. Immunol. Scand. 2019, 127, 64–71. [Google Scholar] [CrossRef]
- Shen, C.; Wang, C.; Yin, Y.; Chen, H.; Yin, X.; Cai, Z.; Chen, Z.; Zhang, B.; Zhou, Z. Tenascin-C expression is significantly associated with the progression and prognosis in gastric GISTs. Medicine 2019, 98, e14045. [Google Scholar] [CrossRef]
- Paron, I.; Berchtold, S.; Voros, J.; Shamarla, M.; Erkan, M.; Hofler, H.; Esposito, I. Tenascin-C enhances pancreatic cancer cell growth and motility and affects cell adhesion through activation of the integrin pathway. PLoS ONE 2011, 6, e21684. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, C.; Qi, W.; Cui, C.; Cui, Y.; Xuan, Y. Tenascin-C as a prognostic determinant of colorectal cancer through induction of epithelial-to-mesenchymal transition and proliferation. Exp. Mol. Pathol. 2018, 105, 216–222. [Google Scholar] [CrossRef]
- He, X.; Chen, S.Y.; Yang, Z.; Zhang, J.; Wang, W.; Liu, M.Y.; Niu, Y.; Wei, X.M.; Li, H.M.; Hu, W.N.; et al. miR-4317 suppresses non-small cell lung cancer (NSCLC) by targeting fibroblast growth factor 9 (FGF9) and cyclin D2 (CCND2). J. Exp. Clin. Cancer Res. 2018, 37, 230. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.; Wu, N.; Wu, L.; Wang, C.; Lin, Y. Overexpression of miR-499-5p inhibits non-small cell lung cancer proliferation and metastasis by targeting VAV3. Sci. Rep. 2016, 6, 23100. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Meng, L.; Sun, H.; Li, Z.; Zhang, X.; Hua, S. MicroRNA-196b Inhibits Cell Growth and Metastasis of Lung Cancer Cells by Targeting Runx2. Cell. Physiol. Biochem. 2017, 43, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Hao, Y.; Huang, S.; Ma, Y.; Li, X.; Li, D.; Mao, Y. MiR-557 works as a tumor suppressor in human lung cancers by negatively regulating LEF1 expression. Tumour Biol. 2017, 39, 1010428317709467. [Google Scholar] [CrossRef]
- Timsah, Z.; Berrout, J.; Suraokar, M.; Behrens, C.; Song, J.; Lee, J.J.; Ivan, C.; Gagea, M.; Shires, M.; Hu, X.; et al. Expression pattern of FGFR2, Grb2 and Plcgamma1 acts as a novel prognostic marker of recurrence recurrence-free survival in lung adenocarcinoma. Am. J. Cancer Res. 2015, 5, 3135–3148. [Google Scholar]
- Kim, J.S.; Kurie, J.M.; Ahn, Y.H. BMP4 depletion by miR-200 inhibits tumorigenesis and metastasis of lung adenocarcinoma cells. Mol. Cancer 2015, 14, 173. [Google Scholar] [CrossRef]
- Xie, Y.G.; Yu, Y.; Hou, L.K.; Wang, X.; Zhang, B.; Cao, X.C. FYN promotes breast cancer progression through epithelial-mesenchymal transition. Oncol. Rep. 2016, 36, 1000–1006. [Google Scholar] [CrossRef]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017, 392, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.N.; Liao, Y.F.; Lu, Y.X.; Wang, Y.; Lu, J.H.; Zeng, Z.L.; Huang, Q.T.; Sheng, H.; Yun, J.P.; Xie, D.; Ju, H.Q.; Xu, R.H. Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 2018, 412, 243–255. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.; Kokkalis, A.; Roumelioti, F.; Gagos, S.; Tzetis, M.; Canovas, B.; et al. Chronic p53-independent p21 expression causesgenomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual Role of the Alternative Reading Frame ARF Protein in Cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Lin, X.L.; Liu, M.; Liu, Y.; Hu, H.; Pan, Y.; Zou, W.; Fan, X.; Hu, X. Transforming growth factor beta1 promotes migration and invasion in HepG2 cells: Epithelialtomesenchymal transition via JAK/STAT3 signaling. Int. J. Mol. Med. 2018, 41, 129–136. [Google Scholar]
- Reiser, J.; Harmison, G.; Kluepfel-Stahl, S.; Brady, R.O.; Karlsson, S.; Schubert, M. Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl. Acad. Sci. USA 1996, 93, 15266–15271. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 1, R10. [Google Scholar]
- Tadesse, M.G.; Ibrahim, J.G.; Vannucci, M.; Gentleman, R. Wavelet thresholding with bayesian false discovery rate control. Biometrics 2005, 61, 25–35. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| SNAI1 | 5′ ACCCACACTGGCGAGAAG 3′ | 5′ GAGAAGGATGTGGGGTCCTT 3′ |
| SNAI2 | 5′ TGGTTGCTTCAA GGACACAT 3′ | 5′ GTTGCAGTGAGGGCAAAGAA 3′ |
| FN1 | 5′ CCCTTACAGTTCAGGGTTCC 3′ | 5′ TTCAAGCCTTCGTTGACAGA 3′ |
| VIM | 5′ GACAATGCGTCTCTGGCACGT 3′ | 5′ TCTTCTGCCTCCTGCAGGTTCTT 3′ |
| ZEB1 | 5′ GCTGACCAGAACAGTGTT 3′ | 5′ CAGAGTCATTCTGATCCTC 3′ |
| GAPDH | 5′ GAGAGACCCTCACTGCTG 3′ | 5′ GATGGTACATGACAAGGTGC 3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moncho-Amor, V.; Pintado-Berninches, L.; Ibañez de Cáceres, I.; Martín-Villar, E.; Quintanilla, M.; Chakravarty, P.; Cortes-Sempere, M.; Fernández-Varas, B.; Rodriguez-Antolín, C.; de Castro, J.; et al. Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2019, 20, 2036. https://doi.org/10.3390/ijms20082036
Moncho-Amor V, Pintado-Berninches L, Ibañez de Cáceres I, Martín-Villar E, Quintanilla M, Chakravarty P, Cortes-Sempere M, Fernández-Varas B, Rodriguez-Antolín C, de Castro J, et al. Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2019; 20(8):2036. https://doi.org/10.3390/ijms20082036
Chicago/Turabian StyleMoncho-Amor, Verónica, Laura Pintado-Berninches, Inmaculada Ibañez de Cáceres, Ester Martín-Villar, Miguel Quintanilla, Probir Chakravarty, María Cortes-Sempere, Beatriz Fernández-Varas, Carlos Rodriguez-Antolín, Javier de Castro, and et al. 2019. "Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 20, no. 8: 2036. https://doi.org/10.3390/ijms20082036
APA StyleMoncho-Amor, V., Pintado-Berninches, L., Ibañez de Cáceres, I., Martín-Villar, E., Quintanilla, M., Chakravarty, P., Cortes-Sempere, M., Fernández-Varas, B., Rodriguez-Antolín, C., de Castro, J., Sastre, L., & Perona, R. (2019). Role of Dusp6 Phosphatase as a Tumor Suppressor in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences, 20(8), 2036. https://doi.org/10.3390/ijms20082036