Fructose and Uric Acid: Major Mediators of Cardiovascular Disease Risk Starting at Pediatric Age

 ,
,  , and
, and 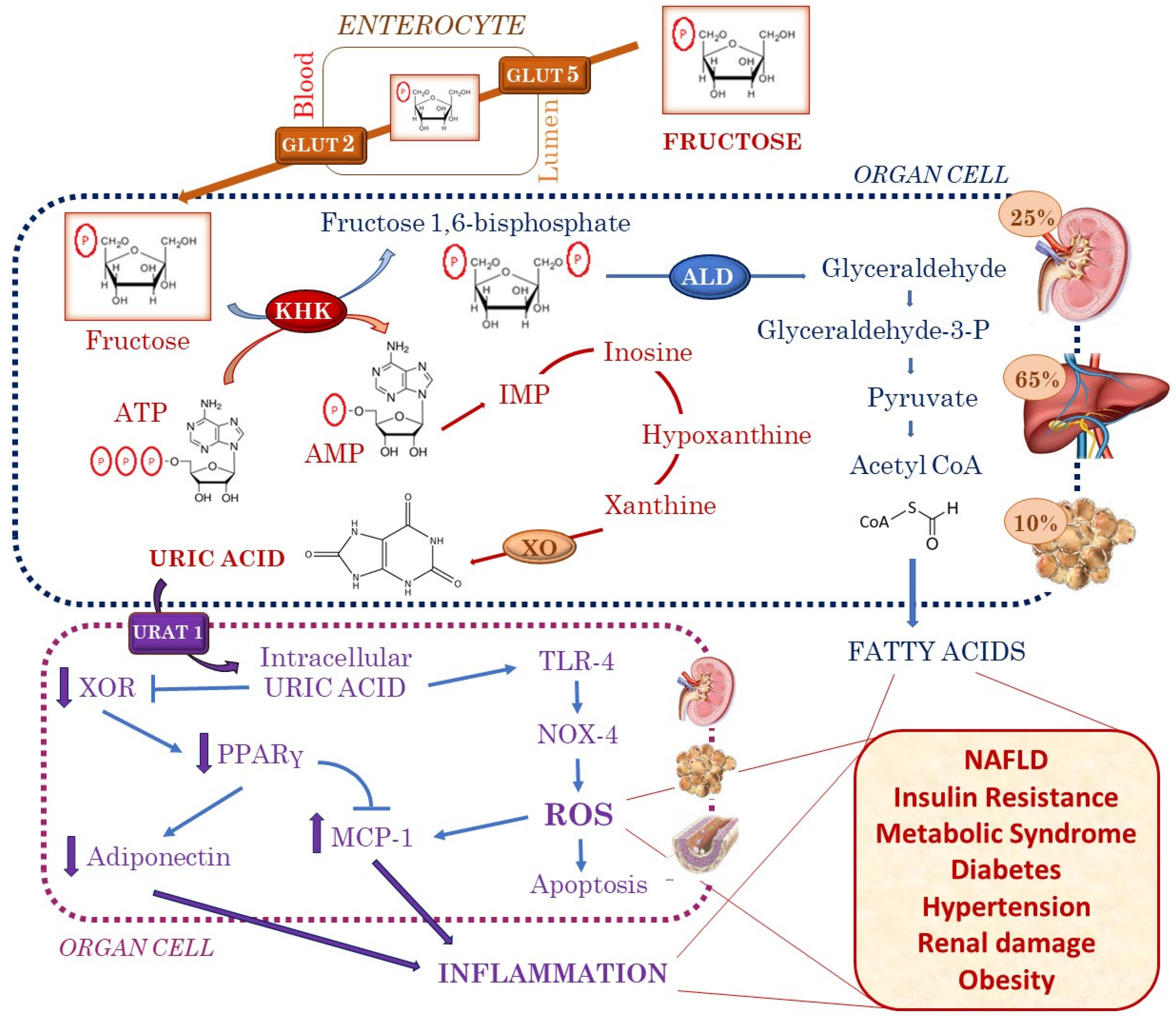{kind=link}
Abstract
1. Introduction
2. Fructose Is Associated with Increased Uric Acid Production
3. Insulin Resistance and Metabolic Syndrome
4. Obesity
5. Diabetes
6. Hypertension
7. Renal Damage
8. NAFLD
9. Why Are These Pathogenetic Mechanisms Extraordinarily Important in Children?
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228S–1235S. [Google Scholar] [CrossRef]
- Malik, V.S.; Hu, F.B. Fructose and cardiometabolic health: What the evidence from sugar-sweetened beverages tells us. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B.; Malik, V.S. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: Epidemiologic evidence. Physiol. Behav. 2010, 100, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Pan, A.; Willett, W.C.; Hu, F.B. Sugar sweetened beverages and weight gain in children and adults: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2013, 98, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- Luger, M.; Lafontan, M.; Bes-Rastrollo, M.; Winzer, E.; Yumuk, V.; Farpour-Lambert, N. Sugar-sweetened beverages and weight gain in children and adults: A systematic review from 2013 to 2015 and a comparison with previous studies. Obes. Facts 2017, 10, 674–693. [Google Scholar] [CrossRef]
- Van den Berghe, G. Fructose: Metabolism and short-term effects on carbohydrate and purine metabolic pathways. Prog. Biochem. Pharm. 1986, 21, 1–32. [Google Scholar]
- Drewnowski, A.; Bellisle, F. Liquid calories, sugar, and body weight. Am. J. Clin. Nutr. 2007, 85, 651–661. [Google Scholar] [CrossRef]
- Basciano, H.; Federico, L.; Adeli, K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metab. (Lond.) 2005, 2, 5. [Google Scholar] [CrossRef]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef]
- Lin, W.T.; Chan, T.F.; Huang, H.L.; Lee, C.Y.; Tsai, S.; Wu, P.W.; Yang, Y.C.; Wang, T.N.; Lee, C.H. Fructose-rich beverage intake and central adiposity, uric acid, and pediatric insulin resistance. J. Pediatr. 2016, 171, 90–96. [Google Scholar] [CrossRef]
- Galderisi, A.; Giannini, C.; Van Name, M.; Caprio, S. Fructose consumption contributes to hyperinsulinemia in adolescents with obesity through a GLP-1-mediated mechanism. J. Clin. Endocrinol. Metab. 2019, 104, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Matikainen, N.; Söderlund, S.; Björnson, E.; Bogl, L.H.; Pietiläinen, K.H.; Hakkarainen, A.; Lundbom, N.; Eliasson, B.; Räsänen, S.M.; Rivellese, A.; et al. Fructose intervention for 12 weeks does not impair glycemic control or incretin hormone responses during oral glucose or mixed meal tests in obese men. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef]
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.Y.; Johnson, R.J.; Scarpace, P.J. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1370–R1375. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Keim, N.L.; et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. (Lond.) 2012, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andrés-Hernando, A.; Ishimoto, T.; Sánchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef]
- Milanesi, S.; Verzola, D.; Cappadona, F.; Bonino, B.; Murugavel, A.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid and angiotensin II additively promote inflammation and oxidative stress in human proximal tubule cells by activation of toll-like receptor 4. J. Cell. Physiol. 2019, 234, 10868–10876. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.A.; Sanchez-Lozada, L.G.; Johnson, R.J.; Kang, D.H. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef]
- Quiñones Galvan, A.; Natali, A.; Baldi, S.; Frascerra, S.; Sanna, G.; Ciociaro, D.; Ferrannini, E. Effect of insulin on uric acid excretion in humans. Am. J. Physiol. 1995, 268, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Muscelli, E.; Natali, A.; Bianchi, S.; Bigazzi, R.; Galvan, A.Q.; Sironi, A.M.; Frascerra, S.; Ciociaro, D.; Ferrannini, E. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am. J. Hypertens. 1996, 9, 746–752. [Google Scholar] [CrossRef]
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011, 60, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Nittari, G.; Scuri, S.; Petrelli, F.; Pirillo, I.; di Luca, N.M.; Grappasonni, I. Fighting obesity in children from European World Health Organization member states. Epidemiological data, medical-social aspects, and prevention programs. Clin. Ther. 2019, 170, e223–e230. [Google Scholar] [CrossRef]
- Freedman, D.S.; Khan, L.K.; Dietz, W.H.; Srinivasan, S.R.; Berenson, G.S. Relationship of childhood obesity to coronary heart disease risk factors in adulthood: The Bogalusa Heart Study. Pediatrics 2001, 108, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Shoham, D.A.; Durazo-Arvizu, R.; Kramer, H.; Luke, A.; Vupputuri, S.; Kshirsagar, A.; Cooper, R.S. Sugary soda consumption and albuminuria: Results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS ONE 2008, 3, e3431. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Kit, B.K.; Carroll, M.D.; Park, S. Consumption of sugar drinks in the United States, 2005–2008. NCHS Data Brief 2011, 71, 1–8. [Google Scholar]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-sweetened beverage consumption among U.S. Adults, 2011–2014. NCHS Data Brief 2017, 270, 1–8. [Google Scholar]
- Ng, S.W.; Ni Mhurchu, C.; Jebb, S.A.; Popkin, B.M. Patterns and trends of beverage consumption among children and adults in Great Britain, 1986–2009. Br. J. Nutr. 2012, 108, 536–551. [Google Scholar] [CrossRef]
- Han, E.; Kim, T.H.; Powell, L.M. Beverage consumption and individual-level associations in South Korea. BMC Public Health 2013, 13, 195. [Google Scholar] [CrossRef]
- Lin, W.T.; Huang, H.L.; Huang, M.C.; Chan, T.F.; Ciou, S.Y.; Lee, C.Y.; Chiu, Y.W.; Duh, T.H.; Lin, P.L.; Wang, T.N.; et al. Effects on uric acid, body mass index and blood pressure in adolescents of consuming beverages sweetened with high-fructose corn syrup. Int. J. Obes. (Lond.) 2013, 37, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.E. Does consumption of high-fructose corn syrup beverages cause obesity in children? Pediatr. Obes. 2013, 8, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Khitan, Z.; Kim, D.H. Fructose: A key factor in the development of metabolic syndrome and hypertension. J. Nutr. Metab. 2013, 2013, 682673. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Feldman, H.A.; Chomitz, V.R.; Antonelli, T.A.; Gortmaker, S.L.; Osganian, S.K.; Ludwig, D.S. A randomized trial of sugar-sweetened beverages and adolescent body weight. N. Engl. J. Med. 2012, 367, 1407–1416. [Google Scholar] [CrossRef]
- de Ruyter, J.C.; Olthof, M.R.; Seidell, J.C.; Katan, M.B. A trial of sugar-free or sugar-sweetened beverages and body weight in children. N. Engl. J. Med. 2012, 367, 1397–1406. [Google Scholar] [CrossRef]
- Kelishadi, R.; Roufarshbaf, M.; Soheili, S.; Payghambarzadeh, F.; Masjedi, M. Association of childhood obesity and the immune system: A systematic review of reviews. Child. Obes. 2017, 13, 332–346. [Google Scholar] [CrossRef]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Umano, G.R.; Pistone, C.; Tondina, E.; Moiraghi, A.; Lauretta, D.; Miraglia Del Giudice, E.; Brambilla, I. Pediatric obesity and the immune system. Front. Pediatr. 2019, 7, 487. [Google Scholar] [CrossRef]
- Harlan, W.R.; Cornoni-Huntley, J.; Leaverton, P.E. Physiologic determinants of serum urate levels in adolescence. Pediatrics 1979, 63, 569–575. [Google Scholar]
- Rocha, E.P.A.A.; Vogel, M.; Stanik, J.; Pietzner, D.; Willenberg, A.; Körner, A.; Kiess, W. Serum uric acid levels as an indicator for metabolically unhealthy obesity in children and adolescents. Horm. Res. Paediatr. 2018, 90, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, D.; Unwin, R. The pathophysiology of hyperuricaemia and its possible relationship to cardiovascular disease, morbidity and mortality. BMC Nephrol. 2013, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Genoni, G.; Menegon, V.; Secco, G.G.; Sonzini, M.; Martelli, M.; Castagno, M.; Ricotti, R.; Monzani, A.; Aronici, M.; Grossini, E.; et al. Insulin resistance, serum uric acid and metabolic syndrome are linked to cardiovascular dysfunction in pediatric obesity. Int. J. Cardiol. 2017, 249, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Romaguera, D.; Norat, T.; Wark, P.A.; Vergnaud, A.C.; Schulze, M.B.; van Woudenbergh, G.J.; Drogan, D.; Amiano, P.; Molina-Montes, E.; Sánchez, M.J.; et al. Consumption of sweet beverages and type 2 diabetes incidence in European adults: Results from EPIC-InterAct. Diabetologia 2013, 56, 1520–1530. [Google Scholar]
- Muraki, I.; Imamura, F.; Manson, J.E.; Hu, F.B.; Willett, W.C.; van Dam, R.M.; Sun, Q. Fruit consumption and risk of type 2 diabetes: Results from three prospective longitudinal cohort studies. BMJ 2013, 347, f5001. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Schwarz, J.M.; Havel, P.J. Adverse metabolic effects of dietary fructose: Results from the recent epidemiological, clinical, and mechanistic studies. Curr. Opin. Lipidol. 2013, 24, 198–206. [Google Scholar] [CrossRef]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef]
- Choi, H.K.; Ford, E.S. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am. J. Med. 2007, 120, 442–447. [Google Scholar] [CrossRef]
- Viazzi, F.; Leoncini, G.; Vercelli, M.; Deferrari, G.; Pontremoli, R. Serum uric acid levels predict new-onset type 2 diabetes in hospitalized patients with primary hypertension: The MAGIC study. Diabetes Care 2011, 34, 126–128. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; et al. High serum uric acid and increased risk of type 2 diabetes: A systemic review and meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e56864. [Google Scholar] [CrossRef]
- Bjornstad, P.; Laffel, L.; Lynch, J.; El Ghormli, L.; Weinstock, R.S.; Tollefsen, S.E.; Nadeau, K.J.; TODAY Study Group. Elevated serum uric acid is associated with greater risk for hypertension and diabetic kidney diseases in obese adolescents with type 2 diabetes: An observational analysis from the treatment options for type 2 diabetes in adolescents and youth (TODAY) study. Diabetes Care 2019, 42, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Jayalath, V.H.; de Souza, R.J.; Ha, V.; Mirrahimi, A.; Blanco-Mejia, S.; Di Buono, M.; Jenkins, A.L.; Leiter, L.A.; Wolever, T.M.; Beyene, J.; et al. Sugar-sweetened beverage consumption and incident hypertension: A systematic review and meta-analysis of prospective cohorts. Am. J. Clin. Nutr. 2015, 102, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Cabral, P.D.; Hong, N.J.; Hye Khan, M.A.; Ortiz, P.A.; Beierwaltes, W.H.; Imig, J.D.; Garvin, J.L. Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to angiotensin II. Hypertension 2014, 63, e68–e73. [Google Scholar] [CrossRef]
- Xu, C.; Lu, A.; Lu, X.; Zhang, L.; Fang, H.; Zhou, L.; Yang, T. Activation of renal (pro)renin receptor contributes to high fructose-induced salt sensitivity. Hypertension 2017, 69, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Yokota, R.; Ronchi, F.A.; Fernandes, F.B.; Jara, Z.P.; Rosa, R.M.; Leite, A.P.O.; Fiorino, P.; Farah, V.; do Nascimento, N.R.F.; Fonteles, M.C.; et al. Intra-renal angiotensin levels are increased in high-fructose fed rats in the extracorporeal renal perfusion model. Front. Physiol. 2018, 9, 1433. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.; Gardiner, S.M.; Elmes, M.; Gardner, D.S. Excess maternal salt or fructose intake programmes sex-specific, stress- and fructose-sensitive hypertension in the offspring. Br. J. Nutr. 2016, 115, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Gordish, K.L.; Kassem, K.M.; Ortiz, P.A.; Beierwaltes, W.H. Moderate (20%) fructose-enriched diet stimulates salt-sensitive hypertension with increased salt retention and decreased renal nitric oxide. Physiol. Rep. 2017, 5, e13162. [Google Scholar] [CrossRef]
- Johnson, R.J.; Segal, M.S.; Srinivas, T.; Ejaz, A.; Mu, W.; Roncal, C.; Sánchez-Lozada, L.G.; Gersch, M.; Rodriguez-Iturbe, B.; Kang, D.H.; et al. Essential hypertension, progressive renal disease, and uric acid: A pathogenetic link? J. Am. Soc. Nephrol. 2015, 16, 1909–1919. [Google Scholar] [CrossRef]
- Kanbay, M.; Jensen, T.; Solak, Y.; Le, M.; Roncal-Jimenez, C.; Rivard, C.; Lanaspa, M.A.; Nakagawa, T.; Johnson, R.J. Uric acid in metabolic syndrome: From an innocent bystander to a central player. Eur. J. Intern. Med. 2016, 29, 3–8. [Google Scholar] [CrossRef]
- Li, N.; Zhang, S.; Li, W.; Wang, L.; Liu, H.; Li, W.; Zhang, T.; Liu, G.; Du, Y.; Leng, J. Prevalence of hyperuricemia and its related risk factors among preschool children from China. Sci. Rep. 2017, 7, 9448. [Google Scholar] [CrossRef]
- Viazzi, F.; Antolini, L.; Giussani, M.; Brambilla, P.; Galbiati, S.; Mastriani, S.; Stella, A.; Pontremoli, R.; Valsecchi, M.G.; Genovesi, S. Serum uric acid and blood pressure in children at cardiovascular risk. Pediatrics 2013, 132, e93–e99. [Google Scholar] [CrossRef] [PubMed]
- Viazzi, F.; Rebora, P.; Giussani, M.; Orlando, A.; Stella, A.; Antolini, L.; Valsecchi, M.G.; Pontremoli, R.; Genovesi, S. Increased serum uric acid levels blunt the antihypertensive efficacy of lifestyle modifications in children at cardiovascular risk. Hypertension 2016, 67, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Feig, D.I.; Soletsky, B.; Johnson, R.J. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: A randomized trial. JAMA 2008, 300, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Soletsky, B.; Feig, D.I. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension 2012, 60, 1148–1156. [Google Scholar] [CrossRef]
- Kubota, M. Hyperuricemia in children and adolescents: Present knowledge and future directions. J. Nutr. Metab. 2019, 2019, 3480718. [Google Scholar] [CrossRef]
- Johnson, R.J.; Sanchez-Lozada, L.G.; Nakagawa, T. The effect of fructose on renal biology and disease. J. Am. Soc. Nephrol. 2010, 21, 2036–2039. [Google Scholar] [CrossRef]
- Nakayama, T.; Kosugi, T.; Connor, T.; Gersch, M.; Sanchez-Lozada, L.G.; Roncal, C.; Perez-Pozo, S.E.; Johnson, R.J.; Nakagawa, T. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am. J. Physiol. Ren. Physiol. 2010, 298, F712–F720. [Google Scholar] [CrossRef]
- Gersch, M.S.; Mu, W.; Cirillo, P.; Reungjui, S.; Zhang, L.; Roncal, C.; Sautin, Y.Y.; Johnson, R.J.; Nakagawa, T. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2007, 293, F1256–F1261. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Ishimoto, T.; Cicerchi, C.; Tamura, Y.; Roncal-Jimenez, C.A.; Chen, W.; Tanabe, K.; Andres-Hernando, A.; Orlicky, D.J.; Finol, E.; et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. J. Am. Soc. Nephrol. 2014, 25, 2526–2538. [Google Scholar] [CrossRef]
- Roncal Jimenez, C.A.; Ishimoto, T.; Lanaspa, M.; Rivard, C.J.; Nakagawa, T.; Ejaz, A.A.; Cicerchi, C.; Inaba, S.; Le, M.; Miyazaki, M.; et al. Fructokinase activity mediates dehydration induced renal injury. Kidney Int. 2014, 86, 294–302. [Google Scholar] [CrossRef]
- Roncal-Jimenez, C.A.; Ishimoto, T.; Lanaspa, M.A.; Milagres, T.; Hernando, A.A.; Jensen, T.; Miyazaki, M.; Doke, T.; Hayasaki, T.; Nakagawa, T.; et al. Aging-associated renal disease in mice is fructokinase dependent. Am. J. Physiol. Ren. Physiol. 2016, 311, F722–F730. [Google Scholar] [CrossRef] [PubMed]
- Rodenbach, K.E.; Schneider, M.F.; Furth, S.L.; Moxey-Mims, M.M.; Mitsnefes, M.M.; Weaver, D.J.; Warady, B.A.; Schwartz, G.J. Hyperuricemia and progression of CKD in children and adolescents: The chronic kidney disease in children (CKiD) cohort study. Am. J. Kidney Dis. 2015, 66, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Viazzi, F.; Leoncini, G.; Ratto, E.; Falqui, V.; Parodi, A.; Conti, N.; Derchi, L.E.; Tomolillo, C.; Deferrari, G.; Pontremoli, R. Mild hyperuricemia and subclinical renal damage in untreated primary hypertension. Am. J. Hypertens. 2007, 20, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE 2014, 16, e115210. [Google Scholar] [CrossRef]
- Russo, E.; Drovandi, S.; Salvidio, G.; Verzola, D.; Esposito, P.; Garibotto, G.; Viazzi, F. Increased serum uric acid levels are associated to renal arteriolopathy and predict poor outcome in IgA nephropathy. Nutr. Metab. Cardiovasc. Dis. 2020. under review. [Google Scholar]
- Kohagura, K.; Kochi, M.; Miyagi, T.; Kinjyo, T.; Maehara, Y.; Nagahama, K.; Sakima, A.; Iseki, K.; Ohya, Y. An association between uric acid levels and renal arteriolopathy in chronic kidney disease: A biopsy-based study. Hypertens. Res. 2013, 36, 43–49. [Google Scholar] [CrossRef]
- Zhu, B.; Yu, D.R.; Lv, J.C.; Lin, Y.; Li, Q.; Yin, J.Z.; Du, Y.Y.; Tang, X.L.; Mao, L.C.; Li, Q.F.; et al. Uric acid as a predictor of immunoglobulin a nephropathy progression: A cohort study of 1965 cases. Am. J. Nephrol. 2018, 48, 127–136. [Google Scholar] [CrossRef]
- Kang, D.H. Hyperuricemia and progression of chronic kidney disease: Role of phenotype transition of renal tubular and endothelial cells. Contrib. Nephrol. 2018, 192, 48–55. [Google Scholar] [CrossRef]
- Bonino, B.; Leoncini, G.; Russo, E.; Pontremoli, R.; Viazzi, F. Uric acid in CKD: Has the jury come to the verdict? J. Nephrol. 2020, in press. [Google Scholar] [CrossRef]
- Calzadilla Bertot, L.; Adams, L.A. The natural course of non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef]
- Mosca, A.; Della Corte, C.; Sartorelli, M.R.; Ferretti, F.; Nicita, F.; Vania, A.; Nobili, V. Beverage consumption and paediatric NAFLD. Eat. Weight Disord. 2016, 21, 581–588. [Google Scholar] [CrossRef]
- Marzuillo, P.; Grandone, A.; Perrone, L.; Miraglia Del Giudice, E. Understanding the pathophysiological mechanisms in the pediatric non-alcoholic fatty liver disease: The role of genetics. World J. Hepatol. 2015, 7, 1439–1443. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: A follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Di Sessa, A.; Umano, G.R.; Miraglia Del Giudice, E.; Santoro, N. From the liver to the heart: Cardiac dysfunction in obese children with non-alcoholic fatty liver disease. World J. Hepatol. 2017, 9, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Koot, B.G.; de Groot, E.; van der Baan-Slootweg, O.H.; Bohte, A.E.; Nederveen, A.J.; Jansen, P.L.; Stoker, J.; Benninga, M.A. Nonalcoholic fatty liver disease and cardiovascular risk in children with obesity. Obesity (Silver Spring) 2015, 23, 1239–1943. [Google Scholar] [CrossRef]
- Madan, S.A.; John, F.; Pyrsopoulos, N.; Pitchumoni, C.S. Nonalcoholic fatty liver disease and carotid artery atherosclerosis in children and adults: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1237–1248. [Google Scholar] [CrossRef]
- Singh, G.K.; Vitola, B.E.; Holland, M.R.; Sekarski, T.; Patterson, B.W.; Magkos, F.; Klein, S. Alterations in ventricular structure and function in obese adolescents with nonalcoholic fatty liver disease. J. Pediatr. 2013, 162, 1160–1168.e1. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Perla, F.M.; Roggini, M.; Andreoli, G.; D’Avanzo, M.; Chiesa, C. A systematic review of NAFLD-associated extrahepatic disorders in youths. J. Clin. Med. 2019, 8, 868. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Jin, R.; Willment, A.; Patel, S.S.; Sun, X.; Song, M.; Mannery, Y.O.; Kosters, A.; McClain, C.J.; Vos, M.B. Fructos induced endotoxemia in pediatric nonalcoholic Fatty liver disease. Int. J. Hepatol. 2014, 2014, 560620. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; et al. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasomedependent mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Elshaghabee, F.M.; Bockelmann, W.; Meske, D.; de Vrese, M.; Walte, H.G.; Schrezenmeir, J.; Heller, K.J. Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front. Microbiol. 2016, 7, 47. [Google Scholar] [CrossRef]
- Elshaghabee, F.M.F.; Rokana, N.; Panwar, H.; Heller, K.J.; Schrezenmeir, J. Probiotics for dietary management of non-alcoholic fatty liver disease. Environ. Chem. Lett. 2019, 17, 1553–1563. [Google Scholar] [CrossRef]
- Liu, Z.; Que, S.; Zhou, L.; Zheng, S. Dose-response relationship of serum uric acid with metabolic syndrome and non-alcoholic fatty liver disease incidence: A meta-analysis of prospective studies. Sci. Rep. 2015, 5, 14325. [Google Scholar] [CrossRef]
- Zhou, Y.; Wei, F.; Fan, Y. High serum uric acid and risk of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Clin. Biochem. 2016, 49, 636–642. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; Cha, B.S. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- García-Arroyo, F.E.; Monroy-Sánchez, F.; Muñoz-Jiménez, I.; Gonzaga, G.; Andrés-Hernando, A.; Zazueta, C.; Juárez-Rojas, J.G.; Lanaspa, M.A.; Johnson, R.J.; Sánchez-Lozada, L.G. Allopurinol prevents the lipogenic response induced by an acute oral fructose challenge in short-term fructose fed rats. Biomolecules 2019, 9, 601. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Seno, Y.; Kushiyama, A.; Sakoda, H.; Fujishiro, M.; Katasako, A.; Mori, K.; Matsunaga, Y.; Fukushima, T.; Kanaoka, R.; et al. The xanthine oxidase inhibitor febuxostat suppresses development of nonalcoholic steatohepatitis in a rodent model. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G42–G51. [Google Scholar] [CrossRef]
- Takir, M.; Kostek, O.; Ozkok, A.; Elcioglu, O.C.; Bakan, A.; Erek, A.; Mutlu, H.H.; Telci, O.; Semerci, A.; Odabas, A.R.; et al. Lowering uric acid with allopurinol improves insulin resistance and systemic inflammation in asymptomatic hyperuricemia. J. Investig. Med. 2015, 63, 924–929. [Google Scholar] [CrossRef]
- Meng, J.; Li, Y.; Yuan, X.; Lu, Y. Effects of febuxostat on insulin resistance and expression of high-sensitivity C-reactive protein in patients with primary gout. Rheumatol. Int. 2017, 37, 299–303. [Google Scholar] [CrossRef]
- Pisano, A.; Cernaro, V.; Gembillo, G.; D’Arrigo, G.; Buemi, M.; Bolignano, D. Xanthine oxidase inhibitors for improving renal function in chronic kidney disease patients: An updated systematic review and meta-analysis. Int. J. Mol. Sci. 2017, 18, E2283. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, E.; Leoncini, G.; Esposito, P.; Garibotto, G.; Pontremoli, R.; Viazzi, F. Fructose and Uric Acid: Major Mediators of Cardiovascular Disease Risk Starting at Pediatric Age. Int. J. Mol. Sci. 2020, 21, 4479. https://doi.org/10.3390/ijms21124479
Russo E, Leoncini G, Esposito P, Garibotto G, Pontremoli R, Viazzi F. Fructose and Uric Acid: Major Mediators of Cardiovascular Disease Risk Starting at Pediatric Age. International Journal of Molecular Sciences. 2020; 21(12):4479. https://doi.org/10.3390/ijms21124479
Chicago/Turabian StyleRusso, Elisa, Giovanna Leoncini, Pasquale Esposito, Giacomo Garibotto, Roberto Pontremoli, and Francesca Viazzi. 2020. "Fructose and Uric Acid: Major Mediators of Cardiovascular Disease Risk Starting at Pediatric Age" International Journal of Molecular Sciences 21, no. 12: 4479. https://doi.org/10.3390/ijms21124479
APA StyleRusso, E., Leoncini, G., Esposito, P., Garibotto, G., Pontremoli, R., & Viazzi, F. (2020). Fructose and Uric Acid: Major Mediators of Cardiovascular Disease Risk Starting at Pediatric Age. International Journal of Molecular Sciences, 21(12), 4479. https://doi.org/10.3390/ijms21124479