Flavonoids Restore Platinum Drug Sensitivity to Ovarian Carcinoma Cells in a Phospho-ERK1/2-Dependent Fashion

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. In-Vitro Experiments
2.3. Differentiation of 3T3-L1 Cells
2.4. Trypan Blue Exclusion Assay
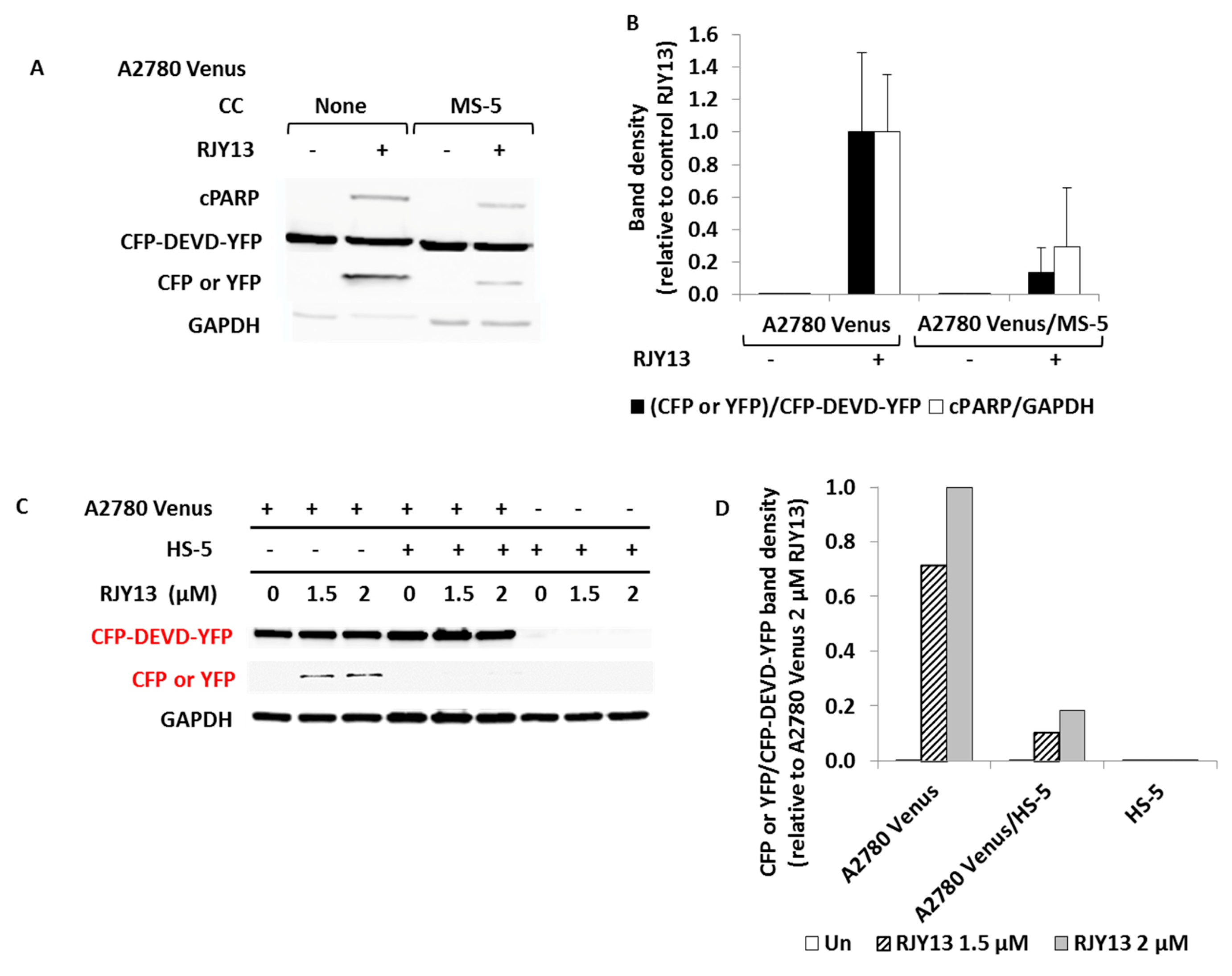
2.5. Construction of A2780/CFP-YFP
2.6. Immunoblotting
2.7. PathScan® Cleaved PARP Assay
3. Results
3.1. Co-culture with MSCs Confers Drug Resistance to OC Cells
3.2. Human MSCs Confer Platinum Drug Resistance to OC Cells
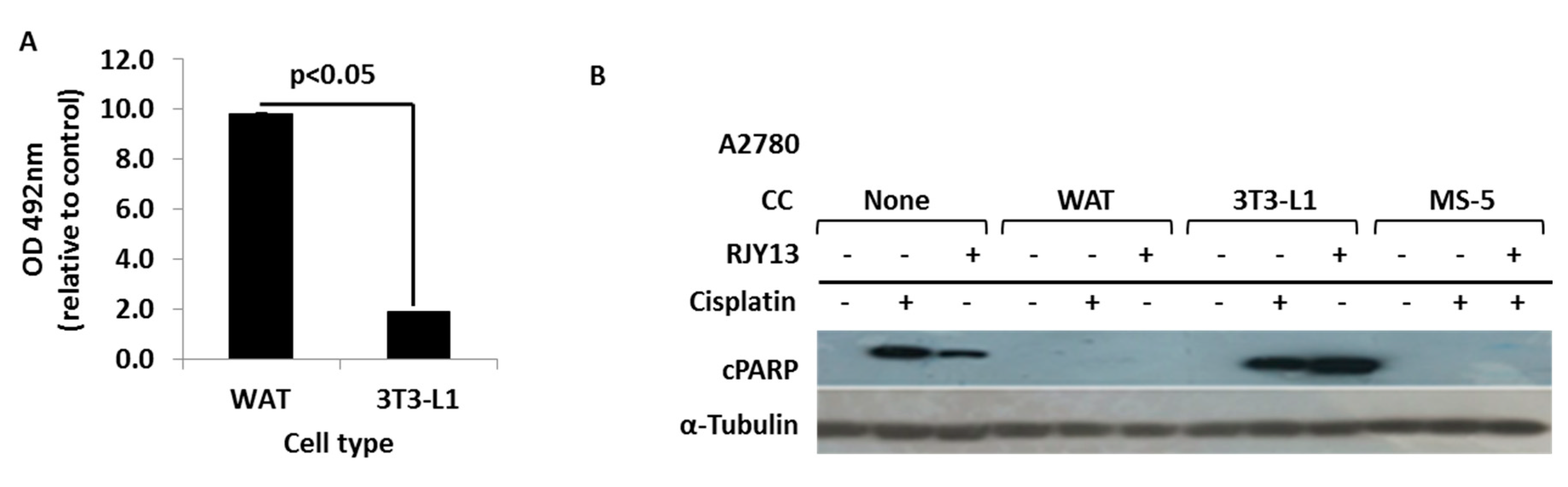
3.3. Co-Culture with Differentiated Murine Adipocyte Progenitor (3T3-L1) Cells Confers Drug Resistance to OC Cells
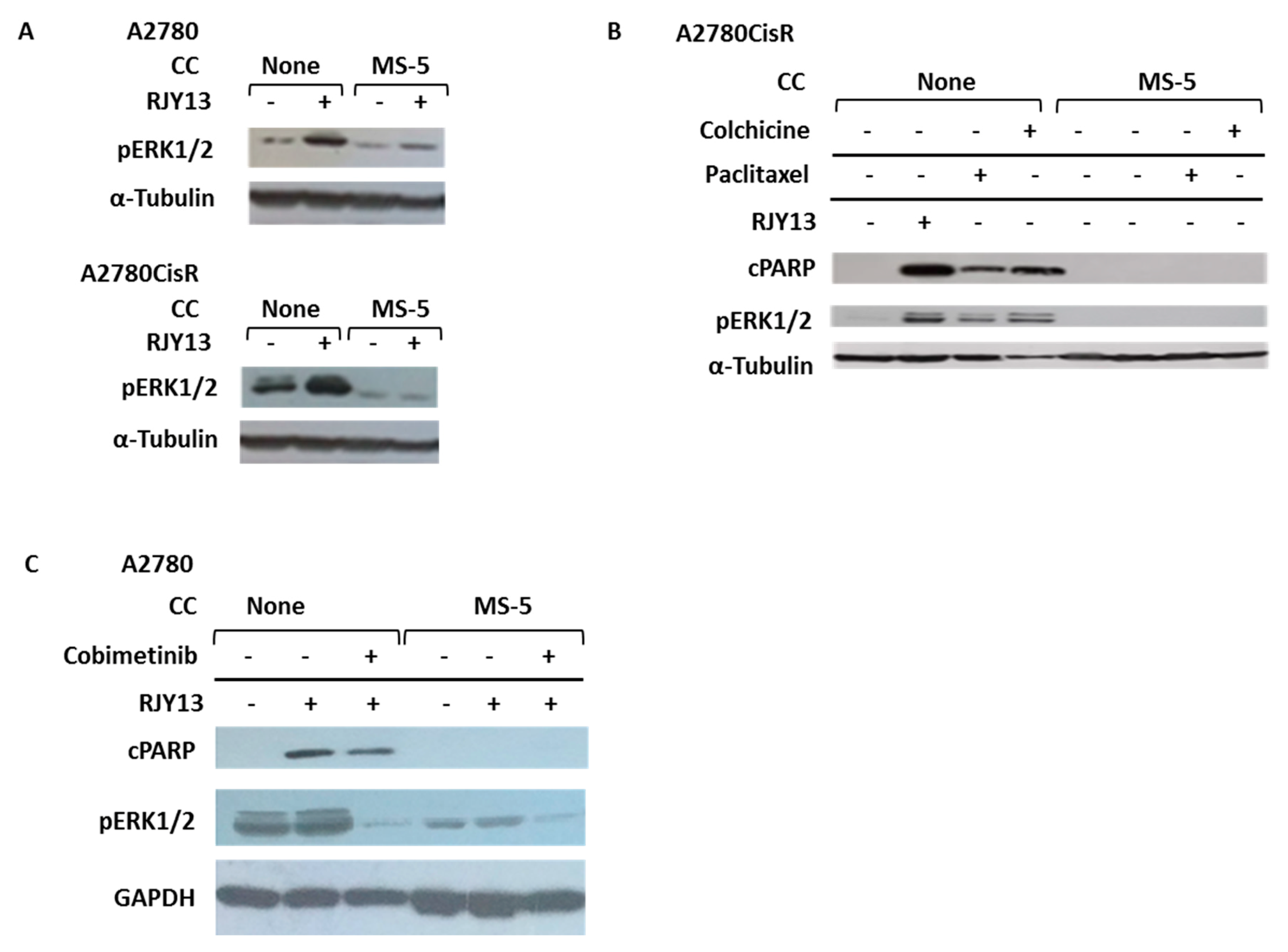
3.4. Co-Culture with MS-5 Cells Blocks Platinum-Induced Phosphorylation of ERK1/2 in OC Cells
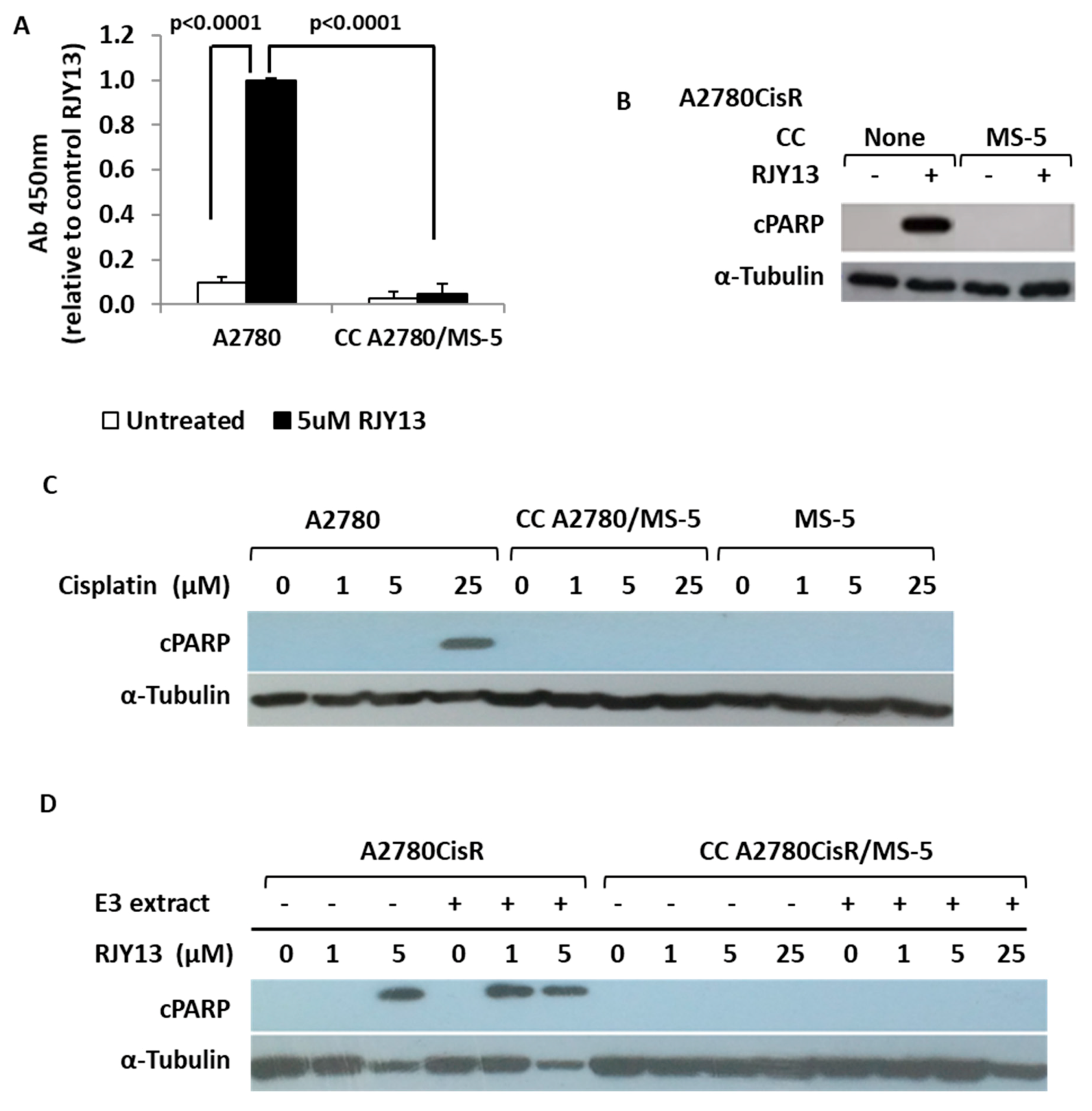
3.5. Flavonoids Restore Platinum Drug Sensitivity to OC Cells Co-Cultured with MS-5 Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Slaughter, K.; Holman, L.L.; Thomas, E.L.; Gunderson, C.C.; Lauer, J.K.; Ding, K.; McMeekin, D.S.; Moore, K.N. Primary and acquired platinum-resistance among women with high grade serous ovarian cancer. Gynecol. Oncol. 2016, 142, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.A.; Howell, S.B. Cellular pharmacology of cisplatin: Perspectives on mechanisms of acquired resistance. Cancer Cells 1990, 2, 35–43. [Google Scholar] [PubMed]
- Ricci, M.S.; Zong, W.-X. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncology 2006, 11, 342–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, B.; Castells, M.; Delord, J.-P.; Couderc, B. Ovarian cancer microenvironment: Implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014, 33, 17–39. [Google Scholar] [CrossRef]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.; Penicka, C.V.; Ladányi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Duong, M.N.; Cleret, A.; Matera, E.-L.; Chettab, K.; Mathé, D.; Valsesia-Wittmann, S.; Clemenceau, B.; Dumontet, C. Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Res. 2015, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Ayantunde, A.A.; Parsons, S.L. Pattern and prognostic factors in patients with malignant ascites: A retrospective study. Ann. Oncol. 2007, 18, 945–949. [Google Scholar] [CrossRef]
- Ahmed, N.; Riley, C.; Oliva, K.; Rice, G.; Quinn, M. Ascites induces modulation of alpha6beta1 integrin and urokinase plasminogen activator receptor expression and associated functions in ovarian carcinoma. Br. J. Cancer 2005, 92, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Riley, C.; Oliva, K.; Stutt, E.; Rice, G.; Quinn, M.A. Integrin-linked kinase expression increases with ovarian tumour grade and is sustained by peritoneal tumour fluid. J. Pathol. 2003, 201, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Wels, J.; Kaplan, R.N.; Rafii, S.; Lyden, D. Migratory neighbors and distant invaders: Tumor-associated niche cells. Genes Dev. 2008, 22, 559–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Robert, V.; Grondin, R.; Rancourt, C.; Piché, A. Malignant ascites protect against TRAIL-induced apoptosis by activating the PI3K/Akt pathway in human ovarian carcinoma cells. Int. J. Cancer 2007, 121, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Zuba-Surma, E.; Kucia, M.; Reca, R.; Wojakowski, W.; Ratajczak, J. The pleiotropic effects of the SDF-1–CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia 2006, 20, 1915–1924. [Google Scholar] [CrossRef] [Green Version]
- Scotton, C.J.; Wilson, J.L.; Milliken, D.; Stamp, G.; Balkwill, F.R. Epithelial cancer cell migration: A role for chemokine receptors? Cancer Res. 2001, 61, 4961–4965. [Google Scholar]
- Jiang, Y.-P.; Wu, X.-H.; Xing, H.-Y.; Du, X.-Y. Role of CXCL12 in metastasis of human ovarian cancer. Chin. Med. J. 2007, 120, 1251–1255. [Google Scholar] [CrossRef]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2011, 18, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Vathipadiekal, V.; Saxena, D.; Mok, S.C.; Hauschka, P.V.; Ozbun, L.; Birrer, M.J. Identification of a Potential Ovarian Cancer Stem Cell Gene Expression Profile from Advanced Stage Papillary Serous Ovarian Cancer. PLoS ONE 2012, 7, e29079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.-M.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.T.; Chen, X.; Trope, C.G.; Davidson, B.; Shih, I.-M.; Wang, T.-L. Notch3 Overexpression Is Related to the Recurrence of Ovarian Cancer and Confers Resistance to Carboplatin. Am. J. Pathol. 2010, 177, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Goncharenko-Khaider, N.; Matte, I.; Lane, D.; Rancourt, C.; Piche, A. Ovarian cancer ascites increase Mcl-1 expression in tumor cells through ERK1/2-Elk-1 signaling to attenuate TRAIL-induced apoptosis. Mol. Cancer 2012, 11, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death - apoptosis, autophagy and senescence. FEBS J. 2009, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.-Y.; Wu, G.S. Bim Protein Degradation Contributes to Cisplatin Resistance. J. Biol. Chem. 2011, 286, 22384–22392. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Choi, C.H.; Oh, H.L.; Kim, Y.K. Role of ERK Activation in Cisplatin-Induced Apoptosis in A172 Human Glioma Cells. NeuroToxicology 2004, 25, 915–924. [Google Scholar] [CrossRef]
- Hertog, M.G. Epidemiological evidence on potential health properties of flavonoids. Proc. Nutr. Soc. 1996, 55, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Atrahimovich, D.; Vaya, J.; Tavori, H.; Khateb, S. Glabridin Protects Paraoxonase 1 from Linoleic Acid Hydroperoxide Inhibition via Specific Interaction: A Fluorescence-Quenching Study. J. Agric. Food Chem. 2012, 60, 3679–3685. [Google Scholar] [CrossRef]
- Atrahimovich, D.; Vaya, J.; Khatib, S. The effects and mechanism of flavonoid-rePON1 interactions. Structure-activity relationship study. Bioorg. Med. Chem. 2013, 21, 3348–3355. [Google Scholar] [CrossRef]
- Duthie, G.G.; Duthie, S.J.; Kyle, J.A.M. Plant polyphenols in cancer and heart disease: Implications as nutritional antioxidants. Nutr. Res. Rev. 2000, 13, 79–106. [Google Scholar] [CrossRef] [Green Version]
- Ramos, S. Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Mol. Nutr. Food Res. 2008, 52, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Yu, L.; You, R.; Yang, Y.; Liao, J.; Chen, D.; Yu, L. Association among Dietary Flavonoids, Flavonoid Subclasses and Ovarian Cancer Risk: A Meta-Analysis. PLoS ONE 2016, 11, e0151134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sak, K. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogn. Rev. 2014, 8, 122–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lee, I.-M.; Zhang, S.M.; Blumberg, J.B.; Buring, J.; Sesso, H.D. Dietary intake of selected flavonols, flavones, and flavonoid-rich foods and risk of cancer in middle-aged and older women. Am. J. Clin. Nutr. 2009, 89, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Youl, E.; Bardy, G.; Magous, R.; Cros, G.; Sejalon, F.; Virsolvy, A.; Richard, S.; Quignard, J.F.; Gross, R.; Petit, P.; et al. Quercetin potentiates insulin secretion and protects INS-1 pancreatic beta-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol. 2010, 161, 799–814. [Google Scholar] [CrossRef] [Green Version]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Lee, S.H.; Son, J.H.; Lee, J.M.; Kang, M.J.; Kim, B.H.; Lee, J.-S.; Ryu, J.K.; Kim, Y.-T. Fisetin Reduces Cell Viability Through Up-Regulation of Phosphorylation of ERK1/2 in Cholangiocarcinoma Cells. Anticancer. Res. 2016, 36, 6109–6116. [Google Scholar] [CrossRef]
- Pal, H.C.; Diamond, A.C.; Strickland, L.R.; Kappes, J.C.; Katiyar, S.K.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin, a dietary flavonoid, augments the anti-invasive and anti-metastatic potential of sorafenib in melanoma. Oncotarget 2016, 7, 1227–1241. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Chen, S.; Zhao, Y.; Ye, X. Fisetin inhibits the proliferation of gastric cancer cells and induces apoptosis through suppression of ERK 1/2 activation. Oncol. Lett. 2018, 15, 8442–8446. [Google Scholar] [CrossRef]
- Youns, M.; Abdel Halim Hegazy, W. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Hyun, J.W. Fisetin induces apoptosis in human nonsmall lung cancer cells via a mitochondria-mediated pathway. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Najajreh, Y.; Khamaisie, H.; Ruimi, N.; Khatib, S.; Katzhendler, J.; Ruthardt, M.; Mahajna, J. Oleylamine-carbonyl-valinol inhibits auto-phosphorylation activity of native and T315I mutated Bcr-Abl, and exhibits selectivity towards oncogenic Bcr-Abl in SupB15 ALL cell lines. Mol. Biol. Rep. 2013, 40, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Ratzon, E.; Najajreh, Y.; Salem, R.; Khamaisie, H.; Ruthardt, M.; Mahajna, J. Platinum (IV)-fatty acid conjugates overcome inherently and acquired Cisplatin resistant cancer cell lines: An in-vitro study. BMC Cancer 2016, 16, 140. [Google Scholar] [CrossRef] [Green Version]
- Oliver, F.J.; De La Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; De Murcia, G.; Murcia, J.M. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef] [Green Version]
- Ben-Arye, E.; Lavie, O.; Samuels, N.; Khamaisie, H.; Schiff, E.; Raz, O.G.; Mahajna, J. Safety of herbal medicine use during chemotherapy in patients with ovarian cancer: A “bedside-to-bench” approach. Med Oncol. 2017, 34. [Google Scholar] [CrossRef]
- Albeck, J.G.; Burke, J.M.; Aldridge, B.B.; Zhang, M.; Lauffenburger, D.A.; Sorger, P.K. Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol. Cell 2008, 30, 11–25. [Google Scholar] [CrossRef] [Green Version]
- Student, A.K.; Hsu, R.Y.; Lane, M.D. Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 preadipocytes. J. Biol. Chem. 1980, 255, 4745–4750. [Google Scholar]
- Musib, L.; Choo, E.; Deng, Y.; Eppler, S.; Rooney, I.; Chan, I.T.; Dresser, M.J. Absolute Bioavailability and Effect of Formulation Change, Food, or Elevated pH with Rabeprazole on Cobimetinib Absorption in Healthy Subjects. Mol. Pharm. 2013, 10, 4046–4054. [Google Scholar] [CrossRef] [PubMed]
- Norouzi-Barough, L.; Sarookhani, M.R.; Sharifi, M.; Moghbelinejad, S.; Jangjoo, S.; Salehi, R. Molecular mechanisms of drug resistance in ovarian cancer. J. Cell. Phys. 2018, 233, 4546–4562. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.M.; Lippard, S.J. Cisplatin: From DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [Google Scholar] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Shi, H.; Ren, F.; Wang, J.-L.; Wu, Q.; Li, X.; Zhang, R. Inhibition of the IGF signaling pathway reverses cisplatin resistance in ovarian cancer cells. BMC Cancer 2017, 17, 851. [Google Scholar] [CrossRef] [PubMed]
- Touboul, C.; Lis, R.; Al Farsi, H.; Raynaud, C.; Warfa, M.; Althawadi, H.; Mery, E.; Mirshahi, M.; Tabrizi, A.R. Mesenchymal stem cells enhance ovarian cancer cell infiltration through IL6 secretion in an amniochorionic membrane based 3D model. J. Transl. Med. 2013, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Sun, R.; Origuchi, M.; Kanehira, M.; Takahata, T.; Itoh, J.; Umezawa, A.; Kijima, H.; Fukuda, S.; Saijo, Y. Mesenchymal Stromal Cells Promote Tumor Growth through the Enhancement of Neovascularization. Mol. Med. 2011, 17, 579–587. [Google Scholar] [CrossRef]
- Greenaway, J.; Moorehead, R.; Shaw, P.; Petrik, J. Epithelial–stromal interaction increases cell proliferation, survival and tumorigenicity in a mouse model of human epithelial ovarian cancer. Gynecol. Oncol. 2008, 108, 385–394. [Google Scholar] [CrossRef]
- Zhu, W.; Xu, W.; Jiang, R.; Qian, H.; Chen, M.; Hu, J.; Cao, W.; Han, C.; Chen, Y. Mesenchymal stem cells derived from bone marrow favor tumor cell growth in vivo. Exp. Mol. Pathol. 2006, 80, 267–274. [Google Scholar] [CrossRef]
- Muehlberg, F.; Song, Y.-H.; Krohn, A.; Pinilla, S.P.; Droll, L.H.; Leng, X.; Seidensticker, M.; Ricke, J.; Altman, A.M.; Devarajan, E.; et al. Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis 2009, 30, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, K.; Kitadai, Y.; Tanaka, M.; Sumida, T.; Kodama, M.; Higashi, Y.; Tanaka, S.; Yasui, W.; Chayama, K. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int. J. Cancer 2010, 127, 2323–2333. [Google Scholar] [CrossRef]
- Lis, R.; Touboul, C.; Halabi, N.; Madduri, A.S.; Querleu, D.; Mezey, J.G.; Malek, J.; Suhre, K.; Tabrizi, A.R. Mesenchymal cell interaction with ovarian cancer cells induces a background dependent pro-metastatic transcriptomic profile. J. Transl. Med. 2014, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Lis, R.; Touboul, C.; Mirshahi, P.; Ali, F.; Mathew, S.; Nolan, D.J.; Maleki, M.; Abdalla, S.A.; Raynaud, C.; Querleu, D.; et al. Tumor associated mesenchymal stem cells protects ovarian cancer cells from hyperthermia through CXCL12. Int. J. Cancer 2010, 128, 715–725. [Google Scholar] [CrossRef]
- Lis, R.; Touboul, C.; Raynaud, C.; Malek, J.A.; Suhre, K.; Mirshahi, M.; Rafii, A. Mesenchymal Cell Interaction with Ovarian Cancer Cells Triggers Pro-Metastatic Properties. PLoS ONE 2012, 7, e38340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun. 2011, 411, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Lehuédé, C.; Li, X.; Dauvillier, S.; Vaysse, C.; Franchet, C.; Clement, E.; Esteve, D.; Longué, M.; Chaltiel, L.; Le Gonidec, S.; et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res. 2019, 21, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojima, K.; Oe, M.; Nakajima, I.; Muroya, S.; Nishimura, T. Dynamics of protein secretion during adipocyte differentiation. FEBS Open Biol. 2016, 6, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Villedieu, M.; Briand, M.; Duval, M.; Heron, J.F.; Gauduchon, P.; Poulain, L. Anticancer and chemosensitizing effects of 2,3-DCPE in ovarian carcinoma cell lines: Link with ERK activation and modulation of p21WAF1/CIP1, Bcl-2 and Bcl-xL expression. Gynecol. Oncol. 2007, 105, 373–384. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, C.; Tantini, B.; Fattori, M.; Stanic’, I.; Pignatti, C.; Clo, C.; Guarnieri, C.; Caldarera, C.M.; Mackintosh, C.; E Pegg, A.; et al. Caspase activation in etoposide-treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett. 2002, 527, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef] [Green Version]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef]
- Chen, J.-R.; Plotkin, L.I.; Aguirre, J.I.; Han, L.; Jilka, R.L.; Kousteni, S.; Bellido, T.; Manolagas, S.C. TransientVersusSustained Phosphorylation and Nuclear Accumulation of ERKs Underlie Anti-VersusPro-apoptotic Effects of Estrogens. J. Biol. Chem. 2005, 280, 4632–4638. [Google Scholar] [CrossRef] [Green Version]
- Zheng, A.; Kallio, A.; Harkonen, P. Tamoxifen-induced rapid death of MCF-7 breast cancer cells is mediated via extracellularly signal-regulated kinase signaling and can be abrogated by estrogen. Endocrinology 2007, 148, 2764–2777. [Google Scholar] [CrossRef] [PubMed]
- Mizrak, S.C.; Renault-Mihara, F.; Parraga, M.; Bogerd, J.; Van De Kant, H.J.G.; López-Casas, P.P.; Paz, M.; Del Mazo, J.; De Rooij, D.G. Phosphoprotein enriched in astrocytes-15 is expressed in mouse testis and protects spermatocytes from apoptosis. Reproduction 2007, 133, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, Y.; Kawai, Y.; Kohda, Y.; Gemba, M. Involvement of activation of NADPH oxidase and extracellular signal-regulated kinase (ERK) in renal cell injury induced by zinc. J. Toxicol. Sci. 2005, 30, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Kohda, Y.; Hiramatsu, J.; Gemba, M. Involvement of MEK/ERK pathway in cephaloridine-induced injury in rat renal cortical slices. Toxicol. Lett. 2003, 143, 185–194. [Google Scholar] [CrossRef]
- Fontanella, R.; Pelagalli, A.; Nardelli, A.; D’Alterio, C.; Ierano’, C.; Cerchia, L.; Lucarelli, E.; Scala, S.; Zannetti, A. A novel antagonist of CXCR4 prevents bone marrow-derived mesenchymal stem cell-mediated osteosarcoma and hepatocellular carcinoma cell migration and invasion. Cancer Lett. 2016, 370, 100–107. [Google Scholar] [CrossRef]
- Georgiou, K.R.; Foster, B.K.; Xian, C.J. Damage and recovery of the bone marrow microenvironment induced by cancer chemotherapy-potential regulatory role of chemokine CXCL12/receptor CXCR4 signalling. Curr. Mol. Med. 2010, 10, 440–453. [Google Scholar] [CrossRef]
- Shankar, V.; Hori, H.; Kihira, K.; Lei, Q.; Toyoda, H.; Iwamoto, S.; Komada, Y. Mesenchymal Stromal Cell Secretome Up-Regulates 47 kDa CXCR4 Expression, and Induce Invasiveness in Neuroblastoma Cell Lines. PLoS ONE 2015, 10, e0120069. [Google Scholar] [CrossRef]
- Song, C.; Li, G. CXCR4 and matrix metalloproteinase-2 are involved in mesenchymal stromal cell homing and engraftment to tumors. Cytotherapy 2011, 13, 549–561. [Google Scholar] [CrossRef]
- Wormann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.-J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; Van Amersfoort, M.; et al. Mesenchymal Stem Cells Induce Resistance to Chemotherapy through the Release of Platinum-Induced Fatty Acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Wang, J.; Chen, M.; Chen, F.; Tu, W. Exosomal microRNA146a derived from mesenchymal stem cells increases the sensitivity of ovarian cancer cells to docetaxel and taxane via a LAMC2mediated PI3K/Akt axis. Int. J. Mol. Med. 2020, 46, 609–620. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koren Carmi, Y.; Mahmoud, H.; Khamaisi, H.; Adawi, R.; Gopas, J.; Mahajna, J. Flavonoids Restore Platinum Drug Sensitivity to Ovarian Carcinoma Cells in a Phospho-ERK1/2-Dependent Fashion. Int. J. Mol. Sci. 2020, 21, 6533. https://doi.org/10.3390/ijms21186533
Koren Carmi Y, Mahmoud H, Khamaisi H, Adawi R, Gopas J, Mahajna J. Flavonoids Restore Platinum Drug Sensitivity to Ovarian Carcinoma Cells in a Phospho-ERK1/2-Dependent Fashion. International Journal of Molecular Sciences. 2020; 21(18):6533. https://doi.org/10.3390/ijms21186533
Chicago/Turabian StyleKoren Carmi, Yifat, Hatem Mahmoud, Hazem Khamaisi, Rina Adawi, Jacob Gopas, and Jamal Mahajna. 2020. "Flavonoids Restore Platinum Drug Sensitivity to Ovarian Carcinoma Cells in a Phospho-ERK1/2-Dependent Fashion" International Journal of Molecular Sciences 21, no. 18: 6533. https://doi.org/10.3390/ijms21186533
APA StyleKoren Carmi, Y., Mahmoud, H., Khamaisi, H., Adawi, R., Gopas, J., & Mahajna, J. (2020). Flavonoids Restore Platinum Drug Sensitivity to Ovarian Carcinoma Cells in a Phospho-ERK1/2-Dependent Fashion. International Journal of Molecular Sciences, 21(18), 6533. https://doi.org/10.3390/ijms21186533