The Product of Matrix Metalloproteinase Cleavage of Doxorubicin Conjugate for Anticancer Drug Delivery: Calorimetric, Spectroscopic, and Molecular Dynamics Studies on Peptide–Doxorubicin Binding to DNA

 , , , , , ,
, , , , , ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
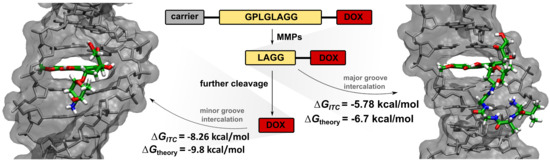
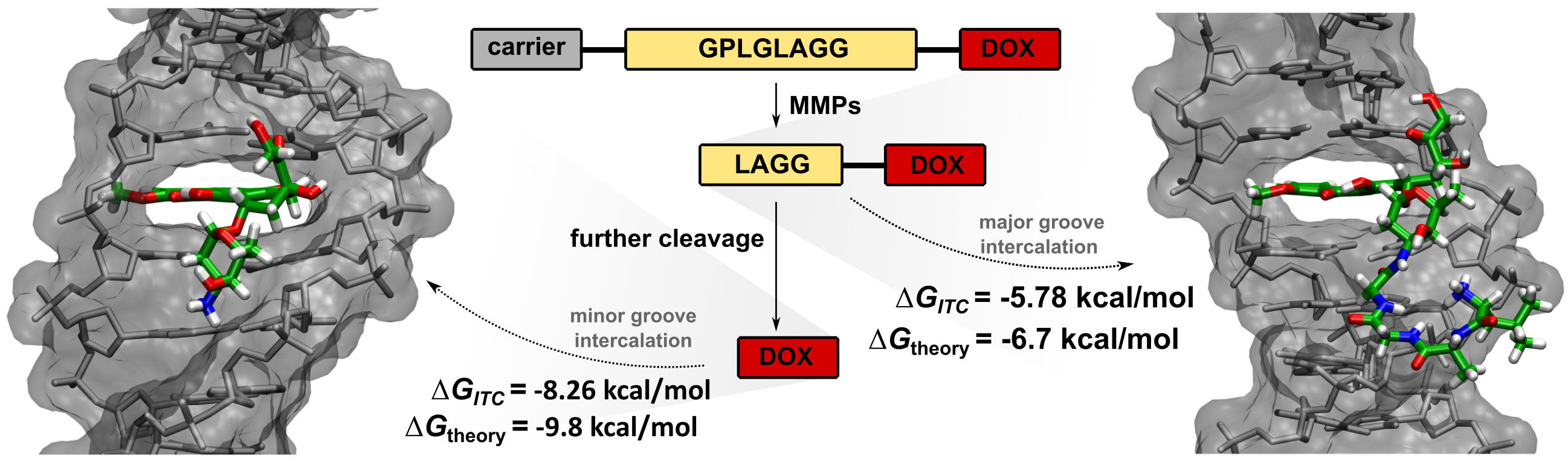
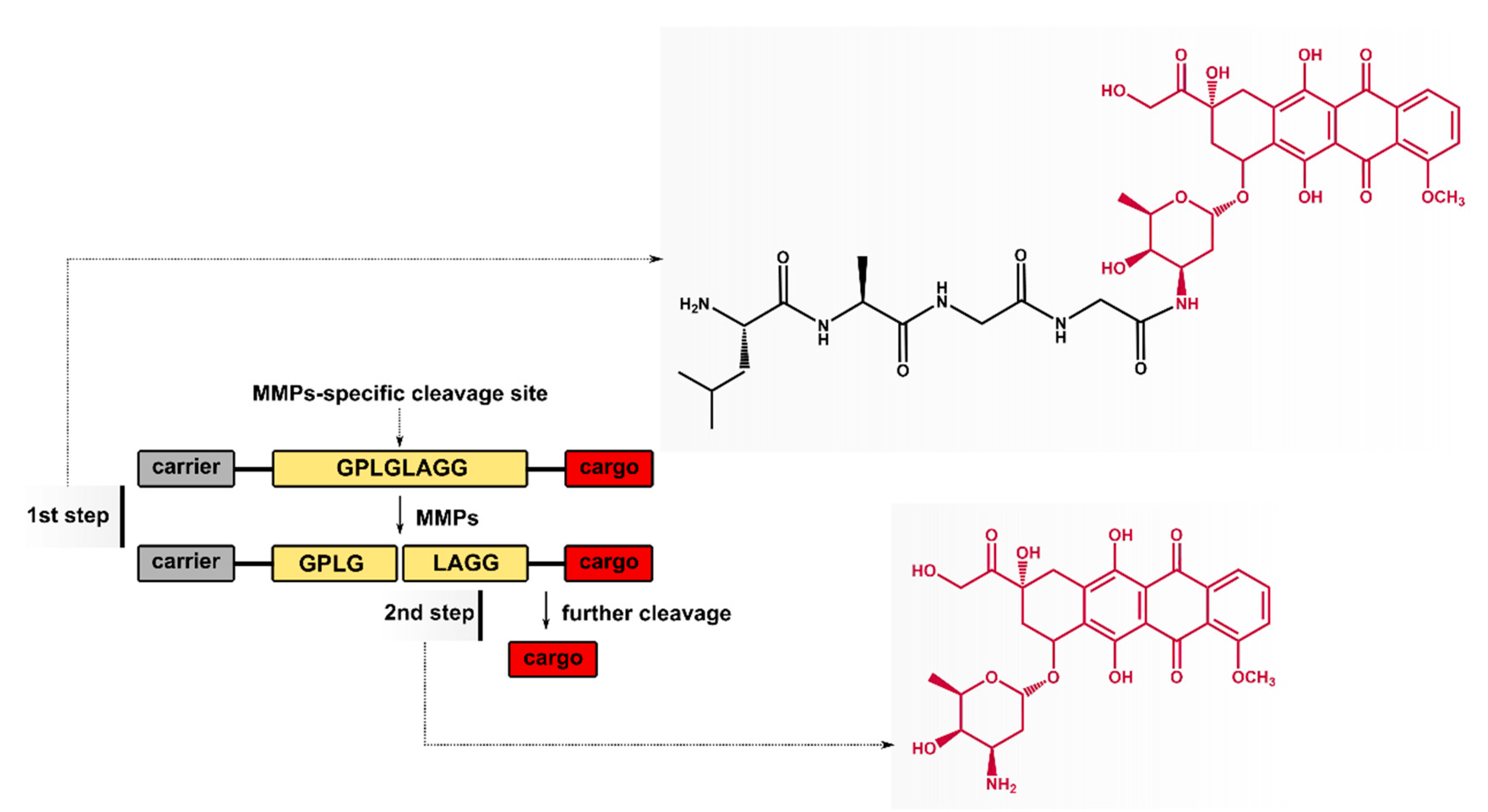
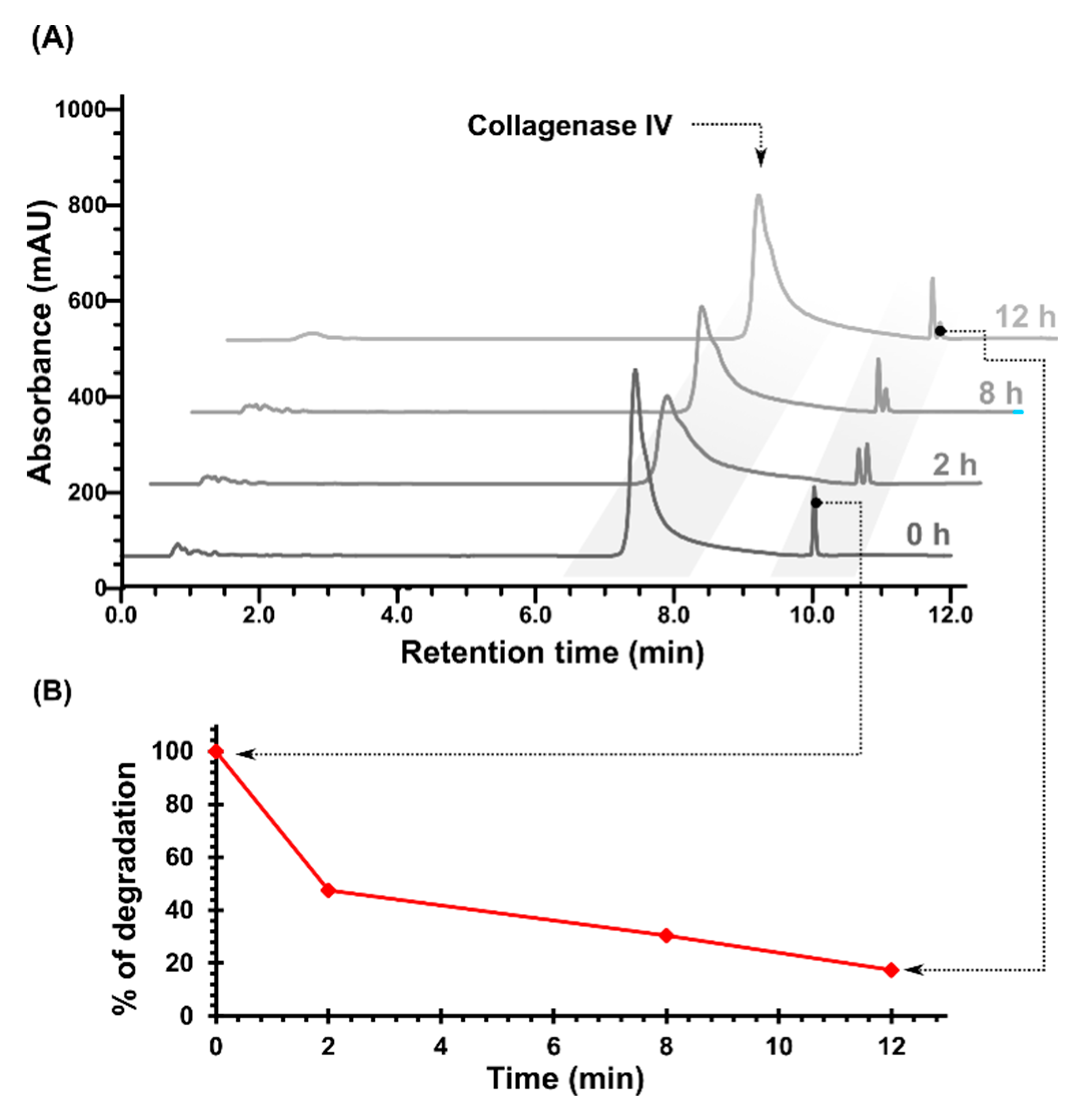
2.1. Cleavage of Peptide by MMPs
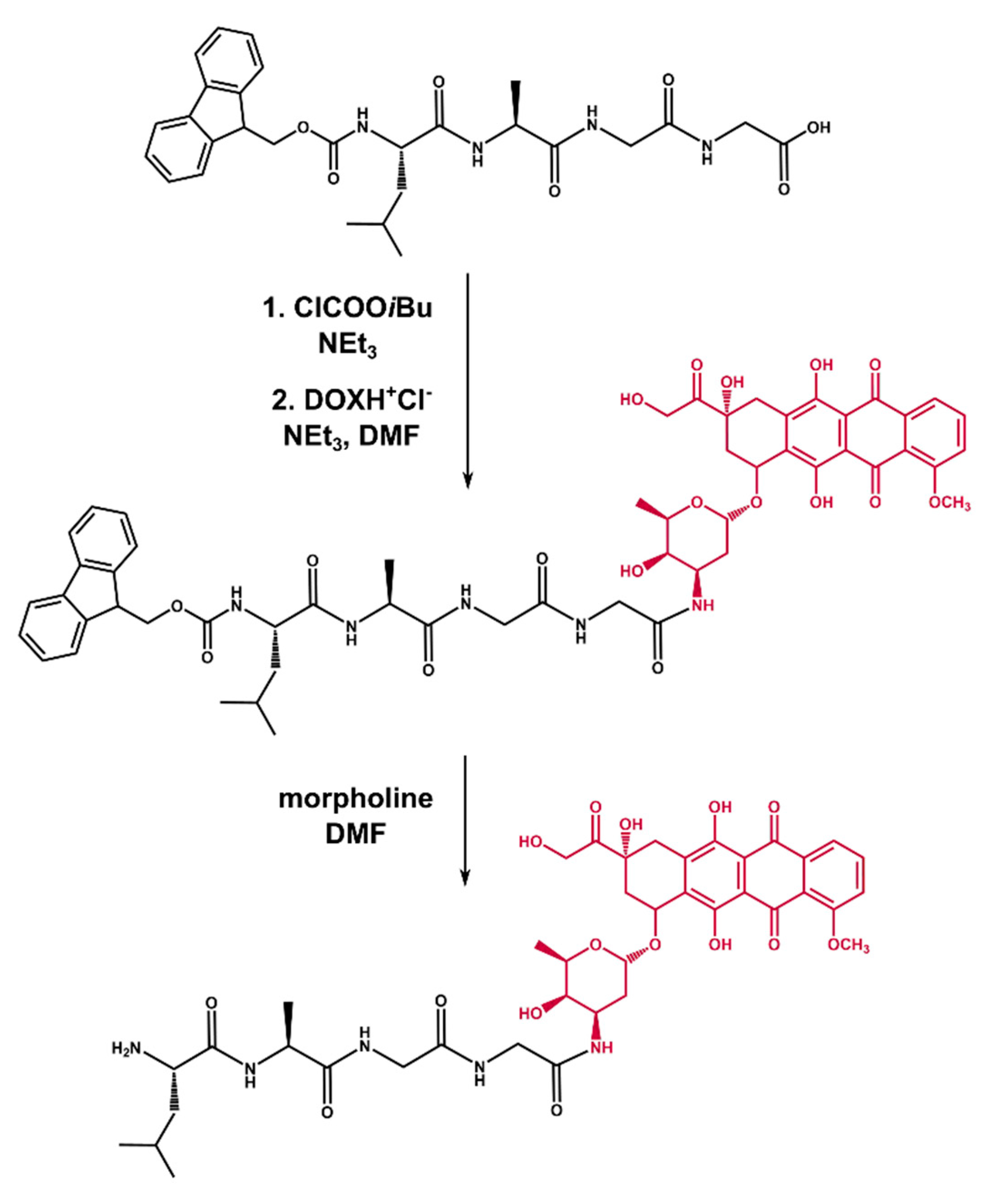
2.2. Synthesis of Tetrapeptide–DOX
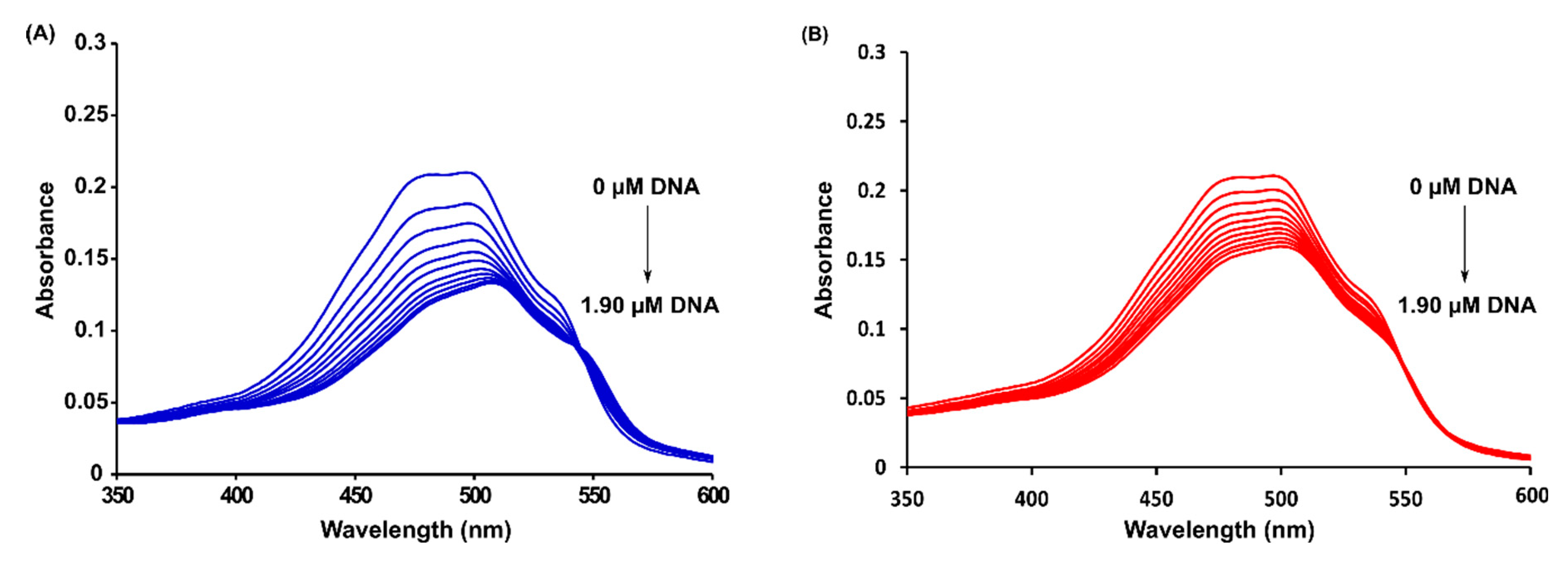
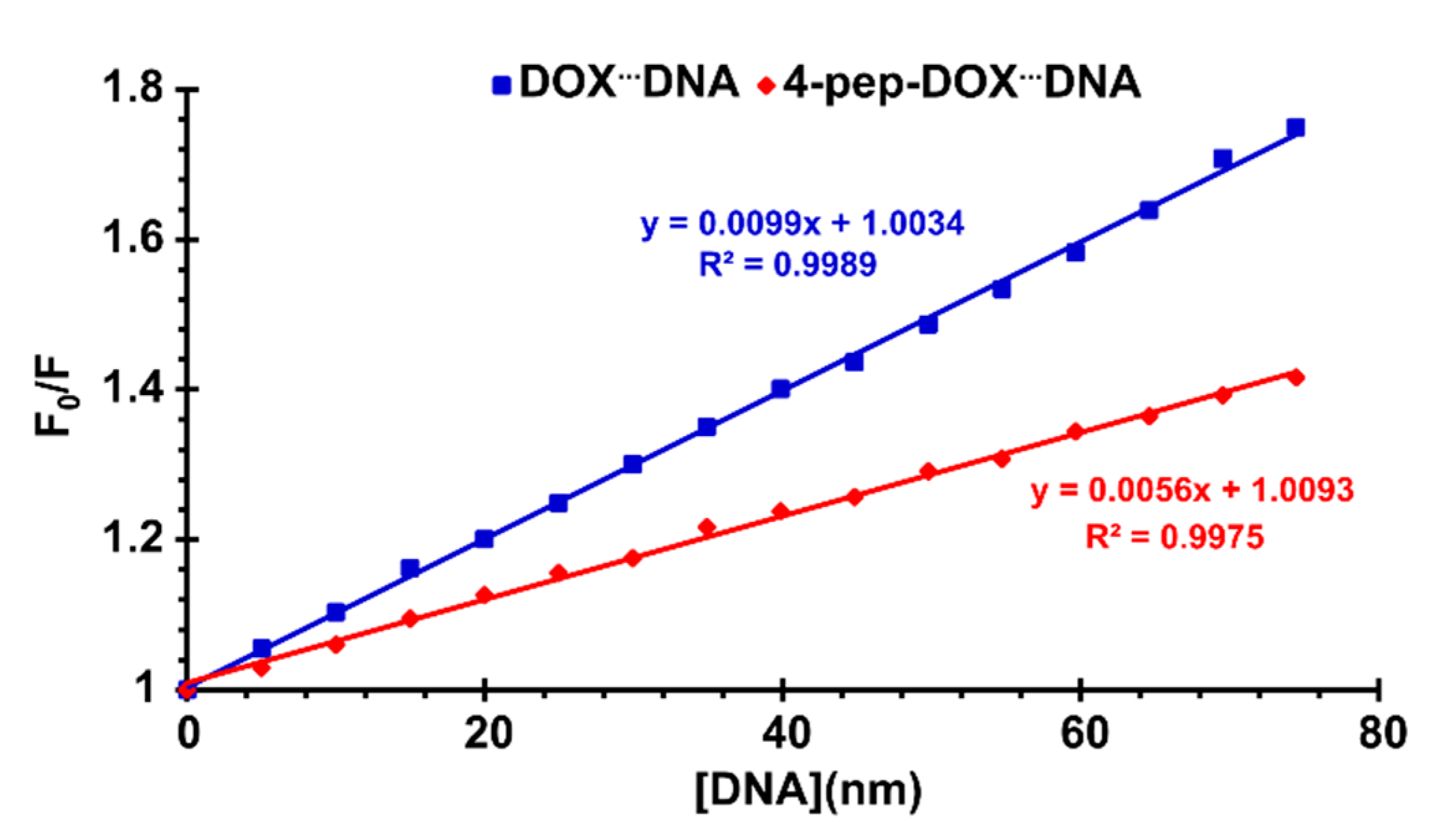
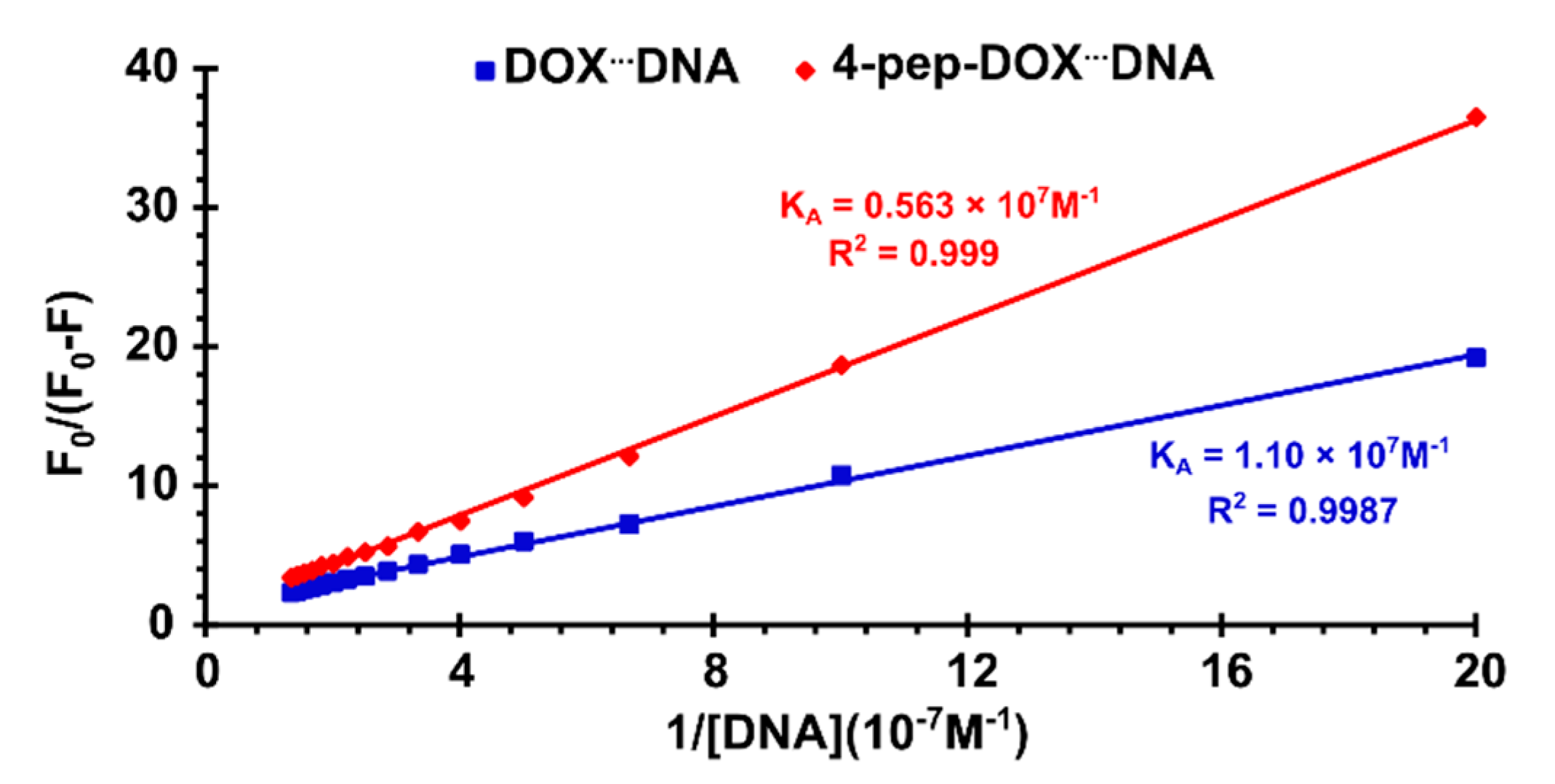
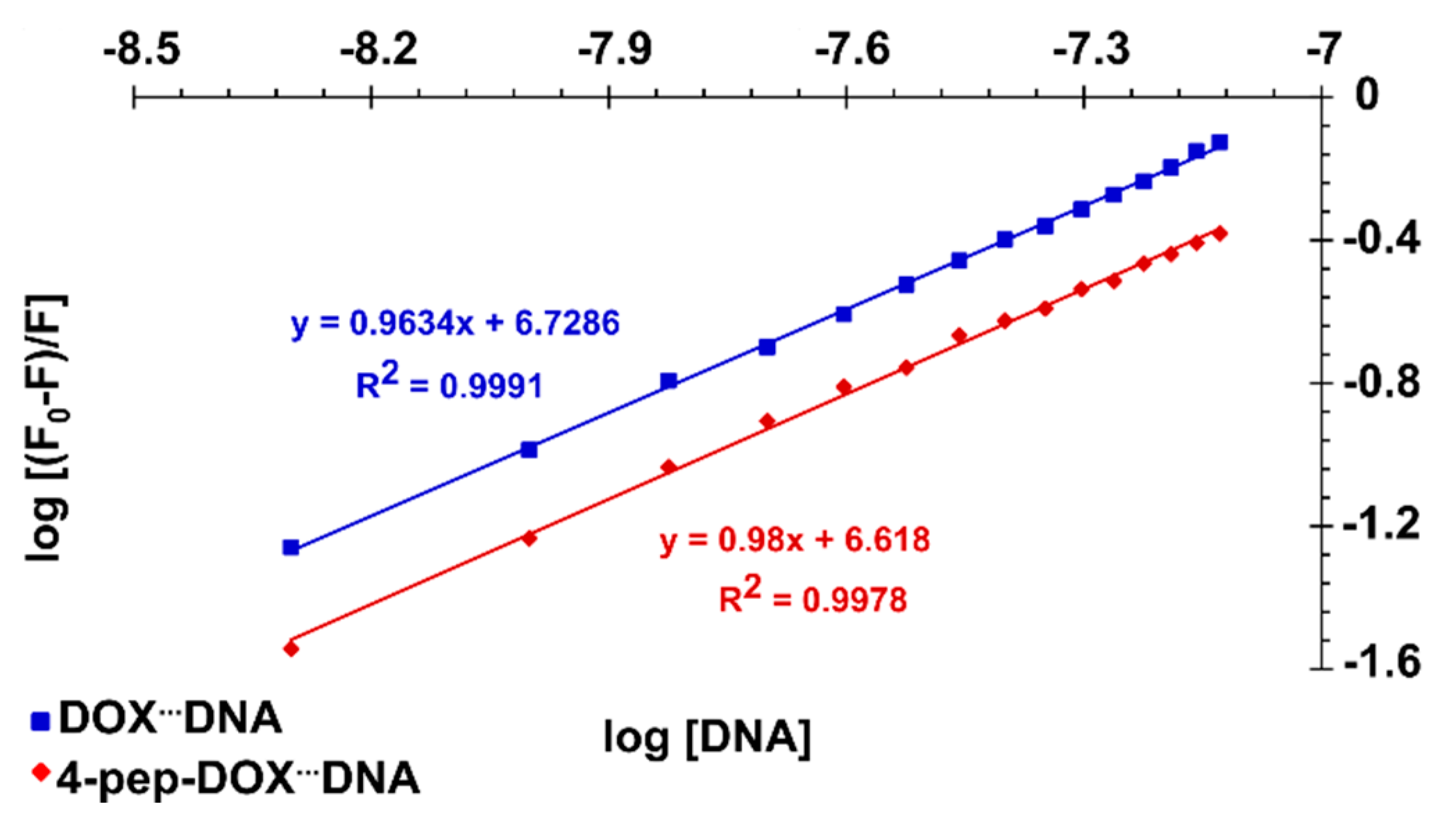
2.3. Spectrophotometric Titrations
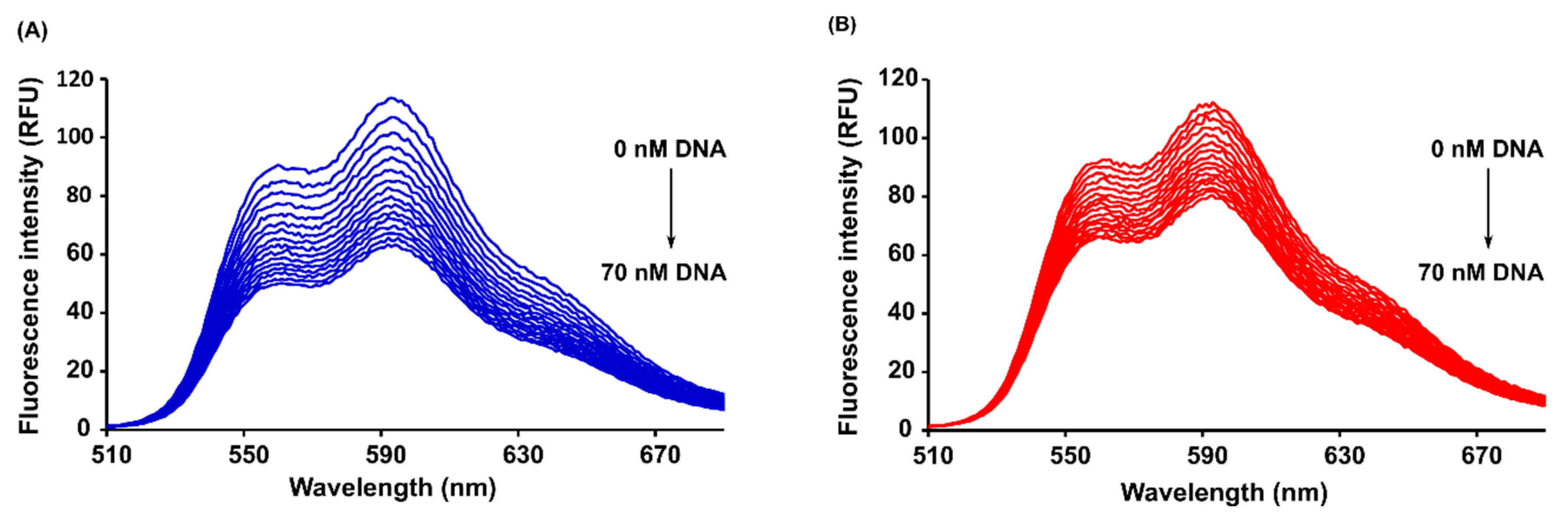
2.4. Steady-State and Time-Resolved Spectroscopy
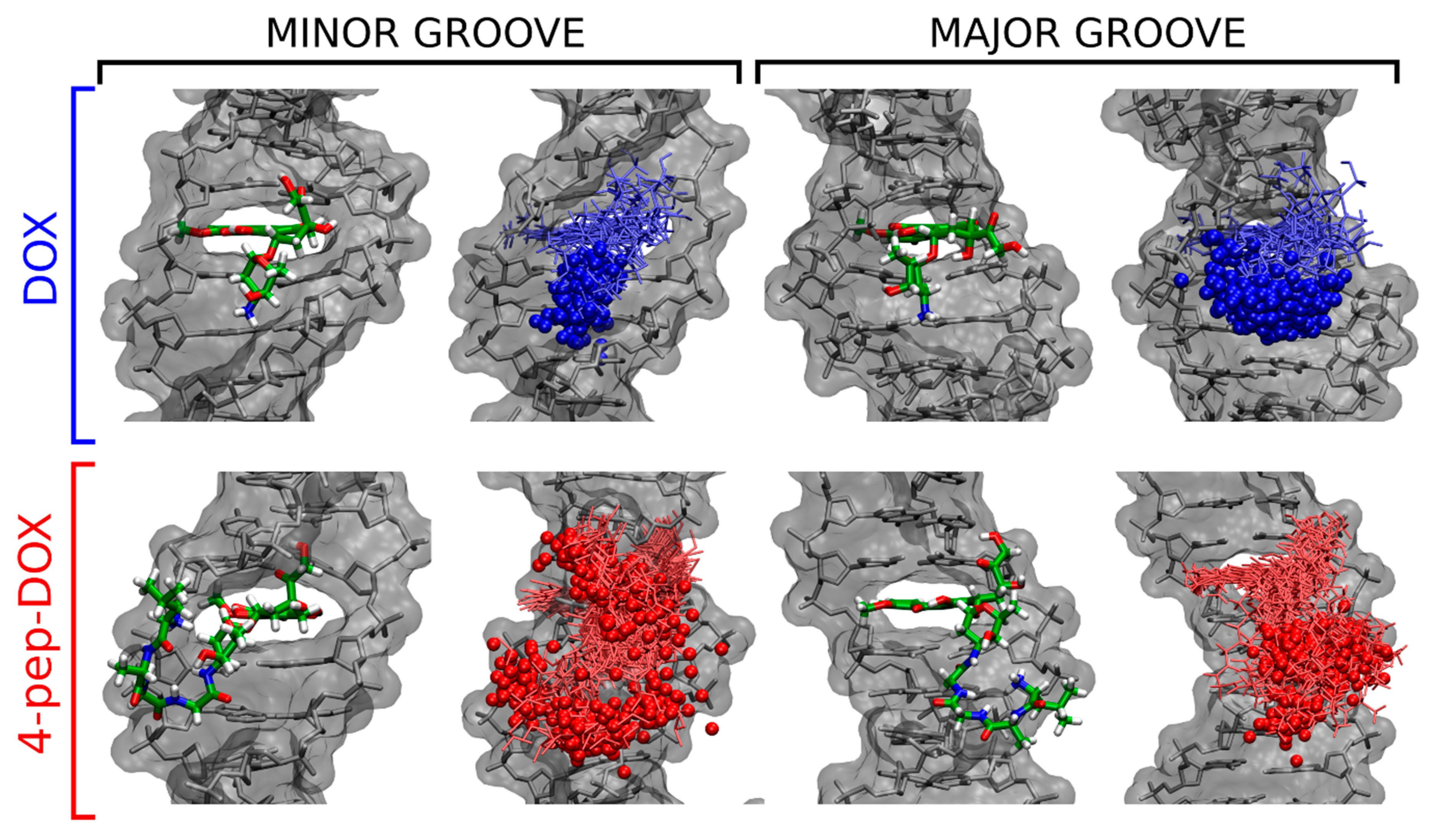
2.5. Isothermal Titration Calorimetry
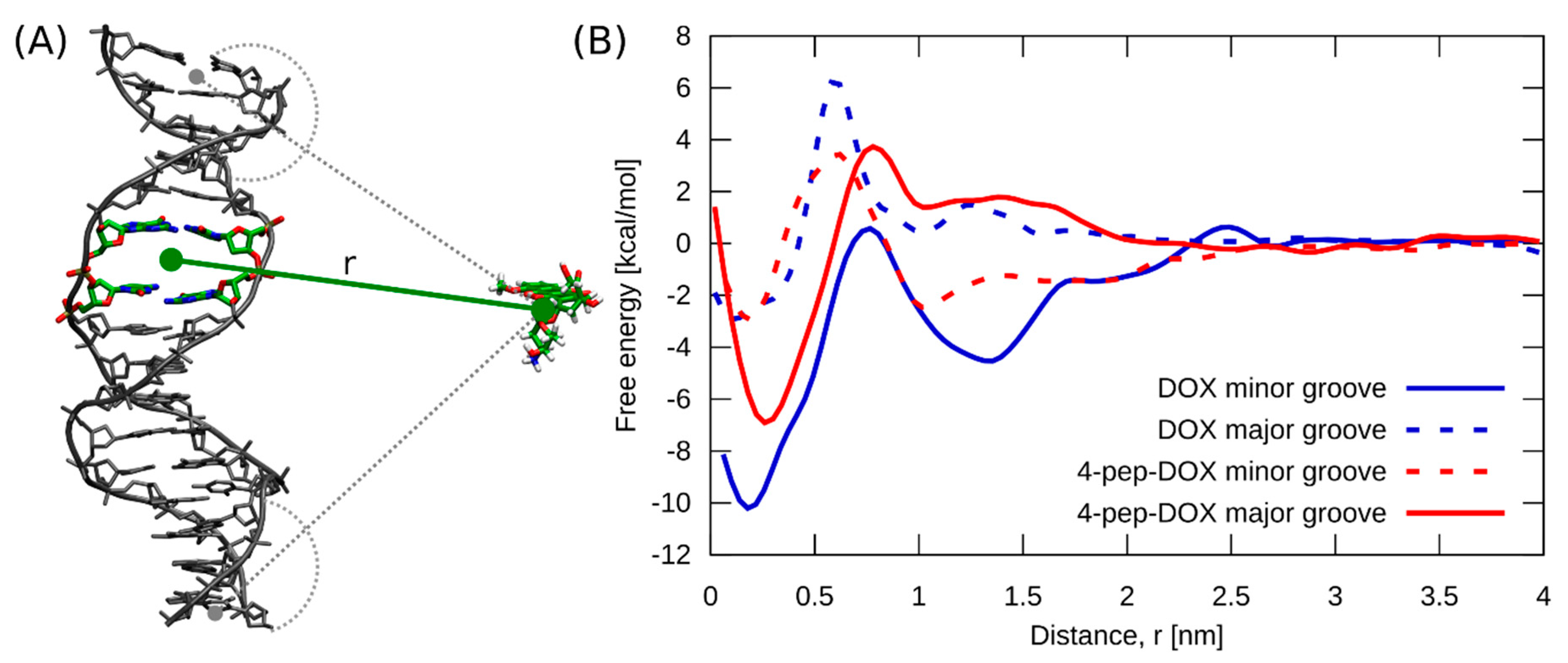
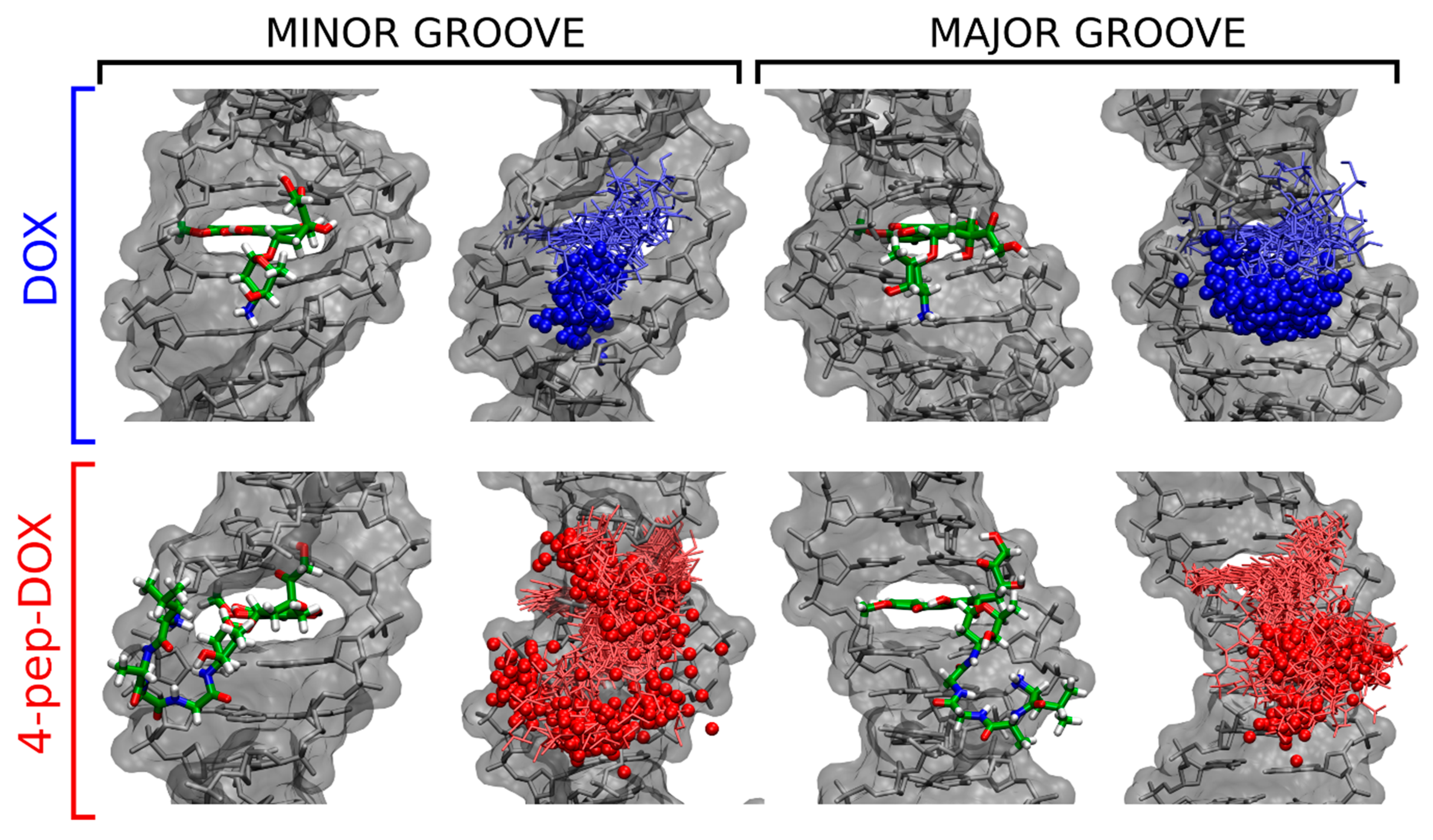
2.6. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Assembly of dsDNA
3.2.2. Enzymatic Cleavage of Peptide by MMPs
3.2.3. Synthesis of Fmoc–Leu–Ala–Gly–Gly–DOX
3.2.4. Synthesis of Leu–Ala–Gly–Gly–DOX
3.2.5. Spectrophotometric Titrations
3.2.6. Steady-State and Time-Resolved Fluorescence Spectroscopy
3.2.7. Isothermal Titration Calorimetry
3.2.8. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DOX | Doxorubicin |
| MMPs | Matrix metalloproteinases |
| MD | Molecular dynamics |
| ITC | Isothermal titration calorimetry |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavan, H.; Azadi, A.; Veisani, Y. Return to work in cancer patients: A systematic review and meta-analysis. Indian J. Palliat. Care 2019, 25, 147–152. [Google Scholar] [PubMed]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharm. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Petrioli, R.; Fiaschi, A.I.; Francini, E.; Pascucci, A.; Francini, G. The role of doxorubicin and epirubicin in the treatment of patients with metastatic hormone-refractory prostate cancer. Cancer Treat. Rev. 2008, 34, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Arcamone, F. Doxorubicin: Anticancer Antibiotics; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacology developments in antitumor activity and cardiotoxicity. Pharm. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binaschi, M.; Capranico, G.; Dal Bo, L.; Zunino, F. Relationship between lethal effects and topoisomerase II-mediated double-stranded DNA breaks produced by anthracyclines with different sequence specificity. Mol. Pharm. 1997, 51, 1053–1059. [Google Scholar] [CrossRef]
- Feinstein, E.; Canaani, E.; Weiner, L.M. Dependence of nucleic acid degradation on in situ free-radical production by adriamycin. Biochemistry 1993, 32, 13156–13161. [Google Scholar] [CrossRef]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Cutts, S.M.; Parsons, P.G.; Sturm, R.A.; Phillips, D.R. Adriamycin-induced DNA adducts inhibit the DNA interactions of transcription factors and RNA polymerase. J. Biol. Chem. 1996, 271, 5422–5429. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Bally, M.B.; Nayar, R.; Masin, D.; Cullis, P.R.; Mayer, L.D. Studies on the myelosuppressive activity of doxorubicin entrapped in liposomes. Cancer Chemother. Pharm. 1990, 27, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.; Cardoso, S.; Correia, S.; Oliveira, P.; Santos, M.; Moreira, P. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Singh, S.; Wlillard, J.; Singh, R. Drug delivery approaches for breast cancer. Int. J. Nanomed. 2017, 12, 6205–6218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudra, A.; Li, J.; Shakur, R.; Bhagchandani, S.; Langer, R. Trends in therapeutic conjugates: Bench to clinic. Bioconjug. Chem. 2020, 31, 462–473. [Google Scholar] [CrossRef]
- Janssen, M.; Mihov, G.; Welting, T.; Thies, J.; Emans, P. Drugs and polymers for delivery systems in OA joints: Clinical needs and opportunities. Polymers 2014, 6, 799–819. [Google Scholar] [CrossRef]
- Milton Harris, J.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Yoo, H.S.; Park, T.G. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. J. Control. Release 2001, 70, 63–70. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 1–20. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Vihinen, P.; Kähäri, V.M. Matrix metalloproteinases in cancer: Prognostic markers and therapeutic targets. Int. J. Cancer 2002, 99, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Xu, Z.; Chen, Y. Doxorubicin conjugated with a trastuzumab epitope and an MMP-2 sensitive peptide linker for the treatment of HER2-positive breast cancer. Drug Deliv. 2018, 25, 448–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Wang, T.; Perche, F.; Taigind, A.; Torchilin, V.P. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc. Natl. Acad. Sci. USA 2013, 110, 17047–17052. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yuan, Z.F.; Wang, Y.; Chen, W.H.; Luo, G.F.; Cheng, S.X.; Zhuo, R.X.; Zhang, X.Z. Multifunctional envelope-type mesoporous silica nanoparticles for tumor-triggered targeting drug delivery. J. Am. Chem. Soc. 2013, 135, 5068–5073. [Google Scholar] [CrossRef]
- Bacinello, D.; Garanger, E.; Taton, D.; Tam, K.C.; Lecommandoux, S. Enzyme-degradable self-assembled nanostructures from polymer-peptide hybrids. Biomacromolecules 2014, 15, 1882–1888. [Google Scholar] [CrossRef]
- Lee, G.Y.; Park, K.; Kim, S.Y.; Byun, Y. MMPs-specific PEGylated peptide-DOX conjugate micelles that can contain free doxorubicin. Eur. J. Pharm. Biopharm. 2007, 67, 646–654. [Google Scholar] [CrossRef]
- Kratz, F.; Müller, I.A.; Ryppa, C.; Warnecke, A. Prodrug strategies in anticancer chemotherapy. ChemMedChem 2008, 3, 20–53. [Google Scholar] [CrossRef]
- Mansour, A.M.; Drevs, J.; Esser, N.; Hamada, F.M.; Badary, O.A.; Unger, C.; Fichtner, I.; Kratz, F. A new approach for the treatment of malignant melanoma: Enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer Res. 2003, 63, 4062–4066. [Google Scholar]
- Guarnieri, D.; Biondi, M.; Yu, H.; Belli, V.; Falanga, A.P.; Cantisani, M.; Galdiero, S.; Netti, P.A. Tumor-activated prodrug (TAP)-conjugated nanoparticles with cleavable domains for safe doxorubicin delivery. Biotechnol. Bioeng. 2015, 112, 601–611. [Google Scholar] [CrossRef]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.X.; Yang, X.Z.; Sun, C.Y.; Mao, C.Q.; Zhu, Y.H.; Wang, J. Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials 2014, 35, 7622–7634. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, K.; KrishnaKumar, V.G.; Gupta, S. Piperazine and DBU: A safer alternative for rapid and efficient Fmoc deprotection in solid phase peptide synthesis. RSC Adv. 2015, 5, 104417–104425. [Google Scholar] [CrossRef]
- Fields, G.B. Methods for removing the Fmoc group. Methods Mol. Biol. 1994, 35, 17–27. [Google Scholar] [PubMed]
- Eftink, M.R.; Ghiron, C.A. Fluorescence quenching studies with proteins. Anal. Biochem. 1981, 114, 199–227. [Google Scholar] [CrossRef]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Xiang, G.; Tong, C.; Lin, H. Nitroaniline isomers interaction with bovine serum albumin and toxicological implications. J. Fluoresc. 2007, 17, 512–521. [Google Scholar] [CrossRef]
- Congdon, R.W.; Muth, G.W.; Splittgerber, A.G. The binding interaction of coomassie blue with proteins. Anal. Biochem. 1993, 213, 407–413. [Google Scholar] [CrossRef]
- Dai, X.; Yue, Z.; Eccleston, M.E.; Swartling, J.; Slater, N.K.H.; Kaminski, C.F. Fluorescence intensity and lifetime imaging of free and micellar-encapsulated doxorubicin in living cells. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Chaires, J.B. A thermodynamic signature for drug-DNA binding mode. Arch. Biochem. Biophys. 2006, 453, 26–31. [Google Scholar] [CrossRef]
- Beauchamp, K.A.; Bowman, G.R.; Lane, T.J.; Maibaum, L.; Haque, I.S.; Pande, V.S. MSMBuilder2: Modelling conformational dynamics on the picosecond to millisecond scale. J. Chem. Theory Comput. 2011, 7, 3412–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, N.Q.; Gao, W.; Xiang, B.; Qi, X.R. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int. J. Nanomed. 2012, 7, 1613–1621. [Google Scholar]
- Munnier, E.; Cohen-Jonathan, S.; Linassier, C.; Douziech-Eyrolles, L.; Marchais, H.; Soucé, M.; Hervé, K.; Dubois, P.; Chourpa, I. Novel method of doxorubicin-SPION reversible association for magnetic drug targeting. Int. J. Pharm. 2008, 363, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Panuszko, A.; Bruździak, P.; Zielkiewicz, J.; Wyrzykowski, D.; Stangret, J. Effects of urea and trimethylamine-N-oxide on the properties of water and the secondary structure of hen egg white lysozyme. J. Phys. Chem. B 2009, 113, 14797–14809. [Google Scholar] [CrossRef] [PubMed]
- Frederick, C.A.; Dean Williams, L.; Ughetto, G.; van der Marel, G.A.; van Boom, H.-J.; Rich, A.; Wang, A.H.-J. Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin. Biochemisty 1990, 29, 2538–2549. [Google Scholar] [CrossRef]
- Lu, X.-J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualisation of three-dimensional nucleic acid structures. Nucl. Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balaceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2015, 13, 55–58. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Software for Calculation; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995, 16, 1339–1350. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | DOX…dsDNA | 4-pep–DOX…dsDNA |
|---|---|---|
| logKITC | 6.03 (±0.04) | 4.24 (±0.09) |
| ΔHITC [kcal/mol] | −9.52 (±0.17) | −5.84 (±1.27) |
| TΔSITC [kcal/mol] | −1.3 | −0.05 |
| ΔGITC [kcal/mol] | −8.26 (±0.05) | −5.78 (±0.12) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butowska, K.; Żamojć, K.; Kogut, M.; Kozak, W.; Wyrzykowski, D.; Wiczk, W.; Czub, J.; Piosik, J.; Rak, J. The Product of Matrix Metalloproteinase Cleavage of Doxorubicin Conjugate for Anticancer Drug Delivery: Calorimetric, Spectroscopic, and Molecular Dynamics Studies on Peptide–Doxorubicin Binding to DNA. Int. J. Mol. Sci. 2020, 21, 6923. https://doi.org/10.3390/ijms21186923
Butowska K, Żamojć K, Kogut M, Kozak W, Wyrzykowski D, Wiczk W, Czub J, Piosik J, Rak J. The Product of Matrix Metalloproteinase Cleavage of Doxorubicin Conjugate for Anticancer Drug Delivery: Calorimetric, Spectroscopic, and Molecular Dynamics Studies on Peptide–Doxorubicin Binding to DNA. International Journal of Molecular Sciences. 2020; 21(18):6923. https://doi.org/10.3390/ijms21186923
Chicago/Turabian StyleButowska, Kamila, Krzysztof Żamojć, Mateusz Kogut, Witold Kozak, Dariusz Wyrzykowski, Wiesław Wiczk, Jacek Czub, Jacek Piosik, and Janusz Rak. 2020. "The Product of Matrix Metalloproteinase Cleavage of Doxorubicin Conjugate for Anticancer Drug Delivery: Calorimetric, Spectroscopic, and Molecular Dynamics Studies on Peptide–Doxorubicin Binding to DNA" International Journal of Molecular Sciences 21, no. 18: 6923. https://doi.org/10.3390/ijms21186923
APA StyleButowska, K., Żamojć, K., Kogut, M., Kozak, W., Wyrzykowski, D., Wiczk, W., Czub, J., Piosik, J., & Rak, J. (2020). The Product of Matrix Metalloproteinase Cleavage of Doxorubicin Conjugate for Anticancer Drug Delivery: Calorimetric, Spectroscopic, and Molecular Dynamics Studies on Peptide–Doxorubicin Binding to DNA. International Journal of Molecular Sciences, 21(18), 6923. https://doi.org/10.3390/ijms21186923