Genome-Wide Gene Expression Profiles Analysis Reveal Novel Insights into Drought Stress in Foxtail Millet (Setaria italica L.)

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic and Physiological Analyses of Foxtail Millet under Drought Stress and Rehydration
2.2. Overview of Transcriptome Sequencing and Differential Expression Genes Responding to Drought Stress
2.3. Transcriptomics Analysis Revealed Complex Mechanisms Involved in Drought Response in Foxtail Millet
2.4. Drought-Responsive Genes Are Mainly Related to Photosynthesis, Signal Transduction and TFs
2.5. Some of DEGs after Re-Watering Different in Roots and Leaves
2.6. Changes of SiP5CS Expression Increase Proline Content and Drought Tolerance
3. Discussion
4. Materials and Methods
4.1. Plants and Experimental Design
4.2. Measurement of Relative Water Content
4.3. Assessment of Antioxidant Enzymesactivities, P5CS Activity and Proline and Malondialdehyde Content
4.4. RNA Extraction, cDNA Library Construction and Sequencing
4.5. RNA-seq Data Analysis and Functional Annotation
4.6. RT-qPCR Validation of DEGs
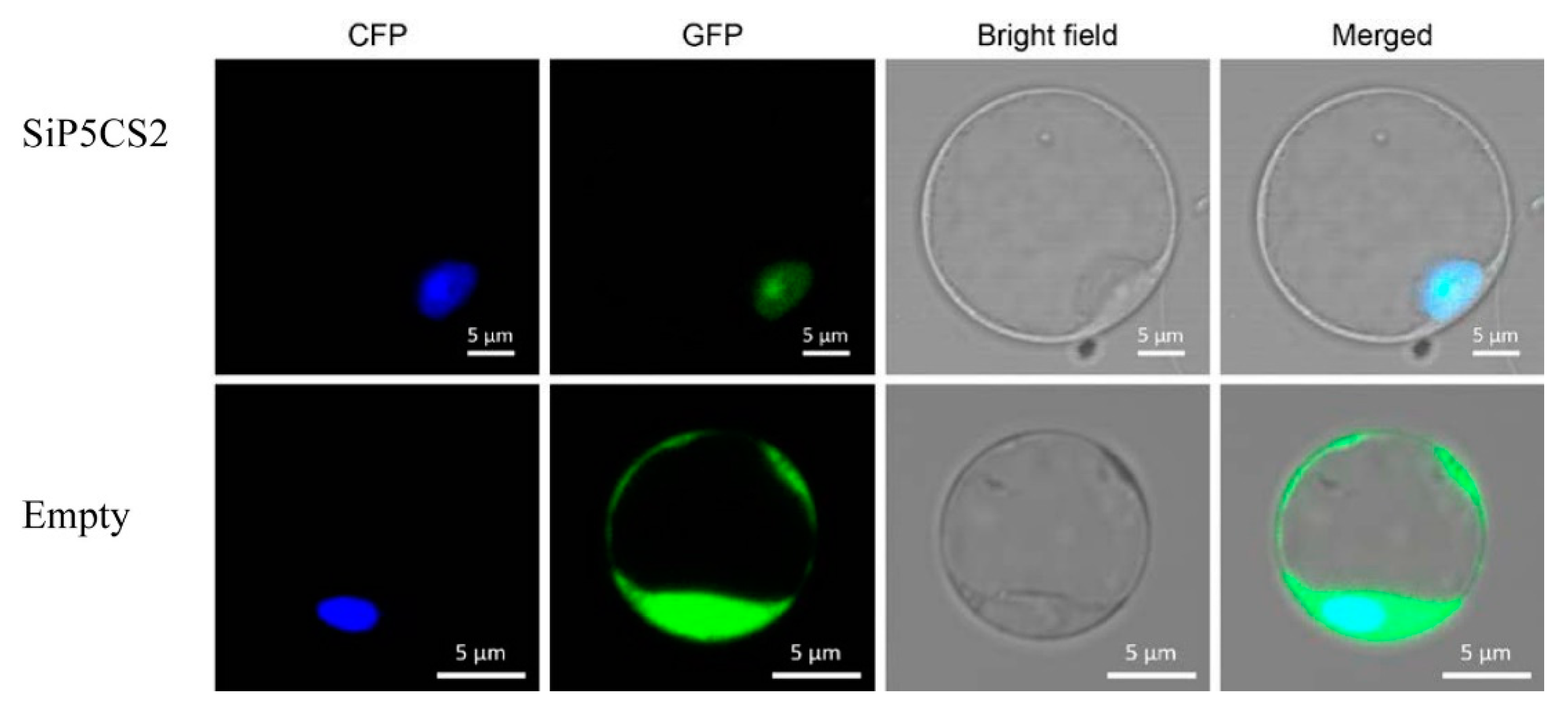
4.7. Subcellular Localization of SiP5CS2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability Statement
Abbreviations
| ABA | Abscisic acid |
| AP2/ERF | APETALA2/Ethylene responsive factor |
| bHLH | Basic helix-loop-helix |
| bZIP | Basic region leucine zipper |
| CAT | Catalase |
| CK | Control |
| D | Drought |
| DEGs | Differentially expressed genes |
| DREB | Dehydration responsive element binding protein |
| ERF | Ethylene responsive factor |
| FDR | False discovery rate |
| GFP | Green fluorescent protein |
| GO | Gene ontology |
| HD-Zip | Homeodomain-leucine zipper |
| KEGG | the Kyoto Encyclopedia of Genes and Genomes |
| LWC | Leaf water content |
| MYB | v-myb avian myeloblastosis viral oncogene homolog |
| NAC | NAM/ATAF1/2/CUC1/2 |
| POD | Peroxidase |
| R | Rehydration |
| RPKM | Reads per kb per million reads |
| SOD | Superoxide dismutase |
| TF | Transcription factor |
| WUE | Water use efficiency |
References
- Gong, Z.; Xiong, L.; Shi, H.; Yang, S.; Zhu, J.K. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [PubMed]
- Delauney, A.J.; Verma, D.P.S. Proline biosynthesis and osmoregulation in plants. Plant J. 1993, 4, 215–223. [Google Scholar] [CrossRef]
- Tang, S.; Li, L.; Wang, Y.; Chen, Q.; Zhang, W.; Jia, G.; Zhi, H.; Zhao, B.; Diao, X. Genotype-specific physiological and transcriptomic responses to drought stress in Setaria italica (an emerging model for panicoideae grasses). Sci. Rep. 2017, 7, 10009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Liu, X.; Quan, Z.; Cheng, S.; Xu, X.; Pan, S.; Xie, M.; Zeng, P.; Yue, Z.; Wang, W.; et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 2012, 30, 549–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennetzen, J.L.; Schmutz, J.; Wang, H.; Percifield, R.; Hawkins, J.; Pontaroli, A.C.; Estep, M.; Feng, L.; Vaughn, J.N.; Grimwood, J.; et al. Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 2012, 30, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Brutnell, T.P. Setaria viridis and Setaria italica, model genetic systems for the panicoid grasses. J. Exp. Bot. 2011, 62, 3031–3037. [Google Scholar] [CrossRef] [Green Version]
- Pant, S.R.; Irigoyen, S.; Doust, A.N.; Scholthof, K.B.; Mandadi, K.K. Setaria: A food crop and translational research model for c4 grasses. Front. Plant Sci. 2016, 7, 1885. [Google Scholar] [CrossRef]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Verbruggen, N.; Hermans, C. Proline accumulation in plants: A review. Amino Acids 2008, 35, 753–759. [Google Scholar] [CrossRef]
- Igarashi, Y.; Yoshiba, Y.; Sanada, Y.; Yamaguchi-Shinozaki, K.; Wada, K.; Shinozaki, K. Characterization of the gene for Δ1-pyrroline-5carboxylate synthetase and correlation between the expression of the gene and salt tolerance in Oryza sativa L. Plant Mol. Biol. 1997, 33, 857–865. [Google Scholar] [CrossRef]
- Strizhov, N.; Ábrahám, E.; Ökrész, L.; Blickling, S.; Zilberstein, A.; Schell, J.; Koncz, C.; Szabados, L. Differential expression of two P5CS genes controlling proline accumulation during salt stress requires ABA and is regulated by ABA1, ABI1 and AXR2 in Arabidopsis. Plant J. 1997, 12, 557–569. [Google Scholar] [PubMed]
- Armengaud, P.; Thiery, L.; Buhot, N.; Grenier-de March, G.; Savouré, A. Transcriptional regulation of proline biosynthesis in Medicago truncatula reveals developmental and environmental specific features. Physiol. Plant 2004, 120, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Jung, K.H.; Lee, C.-H.; An, G. Stress-inducible OsP5CS2 gene is essential for salt and cold tolerance in rice. Plant Sci. 2004, 167, 417–426. [Google Scholar] [CrossRef]
- Yoshiba, Y.; Kiyosue, T.; Katagiri, T.; Ueda, H.; Mizoguchi, T.; Yamaguchi-Shinozaki, K.; Wada, K.; Harada, Y.; Shinozaki, K. Correlation between the induction of a gene for Δ1-pyrroline-5-carboxylate synthetase and the accumulation of proline in Arabidopsis thaliana under osmotic stress. Plant J. 1995, 7, 751–760. [Google Scholar] [CrossRef]
- Székely, G.; Ábrahám, E.; Cséplö, Á.; Rigó, G.; Zsigmond, L.; Csiszár, J.; Ayaydin, F.; Strizhov, N.; Jã¡sik, J.; Schmelzer, E.; et al. Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 2007, 53, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, R.; Falasca, G.; Sabatini, S.; Altamura, M.M.; Costantino, P.; Trovato, M. The proline biosynthetic genes P5CS1 and P5CS2 play overlapping roles in Arabidopsis flower transition but not in embryo development. Physiol. Plant 2009, 137, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Funck, D.; Winter, G.; Baumgarten, L.; Forlani, G. Requirement of proline synthesis during Arabidopsis reproductive development. BMC Plant Biol. 2012, 12, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.B.; Nam, Y.W. A novel Δ1-pyrroline-5-carboxylate synthetase gene of Medicago truncatula plays a predominant role in stress-induced proline accumulation during symbiotic nitrogen fixation. J. Plant Physiol. 2013, 170, 291–302. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Tang, S.; Liu, J.; Zhang, H.; Zhi, H.; Jia, G.; Diao, X. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. BMC Genet. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.Q.; Gao, X.L.; Gao, J.F.; Li, J.; Yang, P.; Feng, B.L. Transcriptome profiling using RNA-seq to provide insights into foxtail millet seedling tolerance to short-term water deficit stress induced by PEG-6000. J. Integr. Agric. 2019, 18, 2457–2471. [Google Scholar] [CrossRef]
- Zhang, D.-F.; Zeng, T.-R.; Liu, X.-Y.; Gao, C.-X.; Li, Y.-X.; Li, C.-H.; Song, Y.-C.; Shi, Y.-S.; Wang, T.; Li, Y. Transcriptomic profiling of sorghum leaves and roots responsive to drought stress at the seedling stage. J. Integr. Agric. 2019, 18, 1980–1995. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lata, C.; Mishra, A.K.; Muthamilarasan, M.; Bonthala, V.S.; Khan, Y.; Prasad, M. Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS ONE 2014, 9, e113092. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Si, W.; Ji, W.; Qin, Q.; Zhao, M.L.; Jiang, H.Y. Genome-wide investigation and expression profiling of HD-Zip transcription factors in foxtail millet (Setaria italica L.). BioMed Res. Int. 2018, 2018, 8457614. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Wang, X.; Hu, L.; Qi, X.; Ge, L.; Xu, W.Y. MYB-like transcription factor SiMYB42 from foxtail millet (Setaria italica L.) enhances Arabidopsis tolerance to low-nitrogen stress. Hereditas 2018, 40, 327–338. [Google Scholar] [PubMed]
- Zhang, L.; Shu, H.; Zhang, A.Y.; Liu, B.L.; Xing, G.F.; Xue, J.A. Foxtail millet WRKY genes and drought stress. J. Agric. Sci. 2017, 155, 777–790. [Google Scholar] [CrossRef]
- Lata, C.; Bhutty, S.; Bahadur, R.P.; Majee, M.; Prasad, M. Association of an SNP in a novel DREB2-like gene SiDREB2 with stress tolerance in foxtail millet [Setaria italica (L.)]. J. Exp. Bot. 2011, 62, 3387–3401. [Google Scholar] [CrossRef] [Green Version]
- Puranik, S.; Bahadur, R.P.; Srivastava, P.S.; Prasad, M. Molecular cloning and characterization of a membrane associated NAC family gene, SiNAC from foxtail millet [Setaria italica (L.) P. Beauv]. Mol. Biotechnol. 2011, 49, 138–150. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Xu, J.; Duan, S.; Wang, Q.; Li, G.; Jin, L. Transcriptome profiling reveals effects of drought stress on gene expression in diploid potato genotype P3-198. Int. J. Mol. Sci. 2019, 20, 852. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Xie, S.; Liu, Y.; Yi, F.; Yu, J. Genome-wide annotation of genes and noncoding RNAs of foxtail millet in response to simulated drought stress by deep sequencing. Plant Mol. Biol. 2013, 83, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yue, J.; Wu, X.; Xu, C.; Yu, J. An ABA-responsive DRE-binding protein gene from Setaria italica, SiARDP, the target gene of SiAREB, plays a critical role under drought stress. J. Exp. Bot. 2014, 65, 5415–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthamilarasan, M.; Bonthala, V.S.; Mishra, A.K.; Khandelwal, R.; Khan, Y.; Roy, R.; Prasad, M. C2H2 type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct. Integr. Genom. 2014, 14, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Shweta, S.; Muthamilarasan, M.; Rani, R.; Prasad, M. Study on aquaporins of Setaria italica suggests the involvement of SiPIP3;1 and SiSIP1;1 in abiotic stress response. Funct. Integr. Genom. 2019, 19, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Chen, E.Y.; Zhang, Y.T.; Yang, Y.B.; Kong, Q.H.; Zhang, H.W.; Wang, H.L.; Wang, R.F.; Guan, Y.N.; Ruan, Y. Effects of drought and rehydration on proline accumulation and SiP5CR gene expression from foxtail millet (Setaria italica (L.) beauv). Mol. Plant Breed. 2018, 16, 7460–7465. [Google Scholar]
- Qin, L.; Yang, Y.B.; Guan, Y.A.; Zhang, H.W.; Wang, H.L.; Liu, B. Identification of drought tolerance at germination period of foxtail millet cultivars developed from different ecological. J. Plant Genet. Resour. 2013, 14, 148–153. [Google Scholar]
- Rodríguez, M.; Canales, E.; Orlando, B.-H. Molecular aspects of abiotic stress in plants. Biotecnol. Appl. 2005, 22, 1–10. [Google Scholar]
- Ashraf, M. Inducing drought tolerance in plants: Recent advances. Biotechnol. Adv. 2010, 28, 169–183. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K.; Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 2003, 6, 410–417. [Google Scholar] [CrossRef]
- Neumann, P.M. Coping mechanisms for crop plants in droughtprone environments. Ann. Bot. 2008, 101, 901–907. [Google Scholar] [CrossRef]
- Shi, W.; Cheng, J.; Wen, X.; Wang, J.; Shi, G.; Yao, J.; Hou, L.; Sun, Q.; Xiang, P.; Yuan, X.; et al. Transcriptomic studies reveal a key metabolic pathway contributing to a well-maintained photosynthetic system under drought stress in foxtail millet (Setaria italica L.). PeerJ 2018, 6, e4752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Liu, H.; Yang, C.; Wang, J.; Yan, G.J.; Si, P. Genome-wide identification of MYB genes and expression analysis under different biotic and abiotic stresses in Helianthus annuus L. Ind. Crops Prod. 2020, 143, 111924. [Google Scholar] [CrossRef]
- Qin, X.; Zeevaart, J.A.D. The 9-cis-epoxycarotenoid cleavage reaction is the key regulatory step of abscisic acid biosynthesis in water-stressed bean. Proc. Natl. Acad. Sci. USA 1999, 96, 15354–15361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwakarma, K.; Upadhyay, N.; Kumar, N.; Yadav, G.; Singh, J.; Mishra, R.K.; Kumar, V.; Verma, R.; Upadhyay, R.G.; Pandey, M.; et al. Abscisic acid signaling and abiotic stress tolerance in plants: A review on currentknowledge and future prospects. Front. Plant Sci. 2017, 8, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1172408. [Google Scholar] [CrossRef] [PubMed]
- Klay, I.; Gouia, S.; Liu, M.; Mila, I.; Khoudi, H.; Bernadac, A.; Bouzayen, M.; Pirrello, J. Ethylene Response Factors (ERF) are deferentially regulated by deferent abiotic stress types in tomato plants. Plant Sci. 2018, 274, 137–145. [Google Scholar] [CrossRef]
- Wang, P.; Wang, H.; Wang, Y.; Ren, F.; Liu, W. Analysis of bHLH genes from foxtail millet (Setaria italica) and their potential relevance to drought stress. PLoS ONE 2018, 13, e0207344. [Google Scholar] [CrossRef]
- Muthamilarasan, M.; Khandelwal, R.; Yadav, C.B.; Bonthala, V.S.; Khan, Y.; Prasad, M. Identification and molecular characterization of MYB Transcription Factor Superfamily in C4 model plant foxtail millet (Setaria italica L.). PLoS ONE 2014, 3, e109920. [Google Scholar] [CrossRef] [Green Version]
- Nott, A.; Jung, H.S.; Koussevitzky, S.; Chory, J. Plastid-to-nucleus retrograde signaling. Annu. Rev. Plant Biol. 2006, 57, 739–759. [Google Scholar] [CrossRef]
- Pruneda-Paz, J.L.; Kay, S.A. An expanding universe of circadian networks in higher plants. Trends Plant Sci. 2010, 15, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Staneloni, R.J.; Rodriguez-Batiller, M.J.; Casal, J.J. Abscisic acid, high-light, and oxidative stress down-regulate a photosynthetic gene via a promoter motif not involved in phytochrome-mediated transcriptional regulation. Mol. Plant 2008, 1, 75–83. [Google Scholar] [CrossRef]
- Liu, X.-F.; Sun, W.-M.; Li, Z.-Q.; Bai, R.-X.; Li, J.-X.; Shi, Z.-H.; Geng, H.; Zheng, Y.; Zhang, J.; Zhang, G.-F. Over-expression of ScMnSOD, a SOD gene derived from jojoba, improve drought tolerance in arabidopsis. J. Integr. Agric. 2013, 12, 1722–1730. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, J.; Li, X.; Xu, J.; Wang, L. Identification and characterization of a PutCu/Zn-SOD gene from Puccinellia tenuiflora (Turcz.) Scribn. et. Merr. Plant Growth Regul. 2016, 79, 55–64. [Google Scholar] [CrossRef]
- Han, X.M.; Chen, Q.X.; Yang, Q.; Zeng, Q.Y.; Lan, T.; Liu, Y.J. Genome-wide analysis of superoxide dismutase genes in Larix kaempferi. Gene 2019, 686, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 2002, 7, 405–410. [Google Scholar] [CrossRef]
- Verma, D.; Lakhanpal, N.; Singh, K. Genome-wide identification and characterization of abiotic-stress responsive SOD (superoxide dismutase) gene family in Brassica juncea and B. rapa. BMC Genom. 2019, 20, 227. [Google Scholar] [CrossRef] [Green Version]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Chen, J.B.; Wang, S.M.; Jing, R.L.; Mao, X.G. Cloning the pvp5cs gene from common bean (phaseolus vulgaris) and its expression patterns under abiotic stresses. J. Plant Physiol. 2009, 166, 12–19. [Google Scholar] [CrossRef]
- Hmidi, D.; Abdelly, C.; Athar, H.; Ashraf, M.; Messedi, D. Effect of salinity on osmotic adjustment, proline accumulation and possible role of ornithine-δ-aminotransferase in proline biosynthesis in Cakile maritima. Physiol. Mol. Biol. Plants 2018, 24, 1017–1033. [Google Scholar] [CrossRef]
- Giannopolitis, C.N.; Ries, S.K. Superoxide dismutases. I. Occur. High. Plants 1977, 59, 309–314. [Google Scholar]
- Sigrid, K.; Ina, Z.; Heinz, C. Scavenging of hydrogen peroxide in the endosperm of ricinus communis by ascorbate peroxidase. Plant Cell Physiol. 1990, 31, 1005–1013. [Google Scholar]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar] [PubMed]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Stewart, R.R.C.; Bewley, J.D. Lipid peroxidation associated with accelerated aging of soybean axes. Plant Physiol. 1980, 65, 245–248. [Google Scholar] [CrossRef] [Green Version]
- Spoljarević, M.; Agić, D.; Lisjak, M.; Gumze, A.; Wilson, I.D.; Hancock, J.T.; Teklić, T. The relationship of proline content and metabolism on the productivity of maize plants. Plant Signal. Behav. 2011, 6, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Xue, W.; Xing, Y.; Weng, X.; Zhao, Y.; Tang, W.; Wang, L. Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat. Genet. 2008, 40, 761–776. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leaves | Roots | |||||
|---|---|---|---|---|---|---|
| Sample Name | LCK | LD | LR | RCK | RD | RR |
| Total reads | 60224561 | 57522977 | 66828528 | 43141689 | 44366653 | 48433784 |
| Total mapped | 58424526 (97.00%) | 55680409 (96.81%) | 64842416 (97.02%) | 40677294 (94.28%) | 42250499 (95.22%) | 45618663 (94.26%) |
| Uniquely mapped | 57107274 (94.82%) | 53658224 (93.28%) | 63175538 (94.54%) | 39536790 (91.64%) | 42587443 (93.09%) | 44445300 (91.84%) |
| Multiple mapped | 1317252 (2.19%) | 2022185 (3.53%) | 1666878 (2.49%) | 1140504 (2.64%) | 944568 (2.14%) | 1173363 (2.42%) |
| Reads mapped to ‘+’ | 28529557 (47.37%) | 26802873 (46.59%) | 31558786 (47.23%) | 19754833 (45.79%) | 20635877 (46.51%) | 22204397 (45.88%) |
| Reads mapped to ‘−’ | 28577717 (47.45%) | 26855350 (46.68%) | 31616752 (47.31%) | 19781957 (45.85%) | 20670055 (46.58%) | 22240903 (45.96%) |
| Non-spliced reads | 34533551 (57.33%) | 32164993 (55.92%) | 38000455 (56.85%) | 26139840 (60.56%) | 27001368 (60.84%) | 29114060 (60.25%) |
| Spliced reads | 22573723 (37.49%) | 21493230 (37.36%) | 25175083 (37.69%) | 13396950 (31.08%) | 14304564 (32.24%) | 15331240 (31.58%) |
| Comparison | DEGs | Upregulated | Downregulated |
|---|---|---|---|
| LCK-LD | 4202 | 1652 | 2550 |
| LCK-LR | 3266 | 2164 | 1102 |
| LD-LR | 5049 | 3287 | 1762 |
| RCK-RD | 6374 | 2751 | 3623 |
| RCK-RR | 4152 | 2050 | 2102 |
| RD-RR | 4020 | 2391 | 1629 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, L.; Chen, E.; Li, F.; Yu, X.; Liu, Z.; Yang, Y.; Wang, R.; Zhang, H.; Wang, H.; Liu, B.; et al. Genome-Wide Gene Expression Profiles Analysis Reveal Novel Insights into Drought Stress in Foxtail Millet (Setaria italica L.). Int. J. Mol. Sci. 2020, 21, 8520. https://doi.org/10.3390/ijms21228520
Qin L, Chen E, Li F, Yu X, Liu Z, Yang Y, Wang R, Zhang H, Wang H, Liu B, et al. Genome-Wide Gene Expression Profiles Analysis Reveal Novel Insights into Drought Stress in Foxtail Millet (Setaria italica L.). International Journal of Molecular Sciences. 2020; 21(22):8520. https://doi.org/10.3390/ijms21228520
Chicago/Turabian StyleQin, Ling, Erying Chen, Feifei Li, Xiao Yu, Zhenyu Liu, Yanbing Yang, Runfeng Wang, Huawen Zhang, Hailian Wang, Bin Liu, and et al. 2020. "Genome-Wide Gene Expression Profiles Analysis Reveal Novel Insights into Drought Stress in Foxtail Millet (Setaria italica L.)" International Journal of Molecular Sciences 21, no. 22: 8520. https://doi.org/10.3390/ijms21228520
APA StyleQin, L., Chen, E., Li, F., Yu, X., Liu, Z., Yang, Y., Wang, R., Zhang, H., Wang, H., Liu, B., Guan, Y., & Ruan, Y. (2020). Genome-Wide Gene Expression Profiles Analysis Reveal Novel Insights into Drought Stress in Foxtail Millet (Setaria italica L.). International Journal of Molecular Sciences, 21(22), 8520. https://doi.org/10.3390/ijms21228520