Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
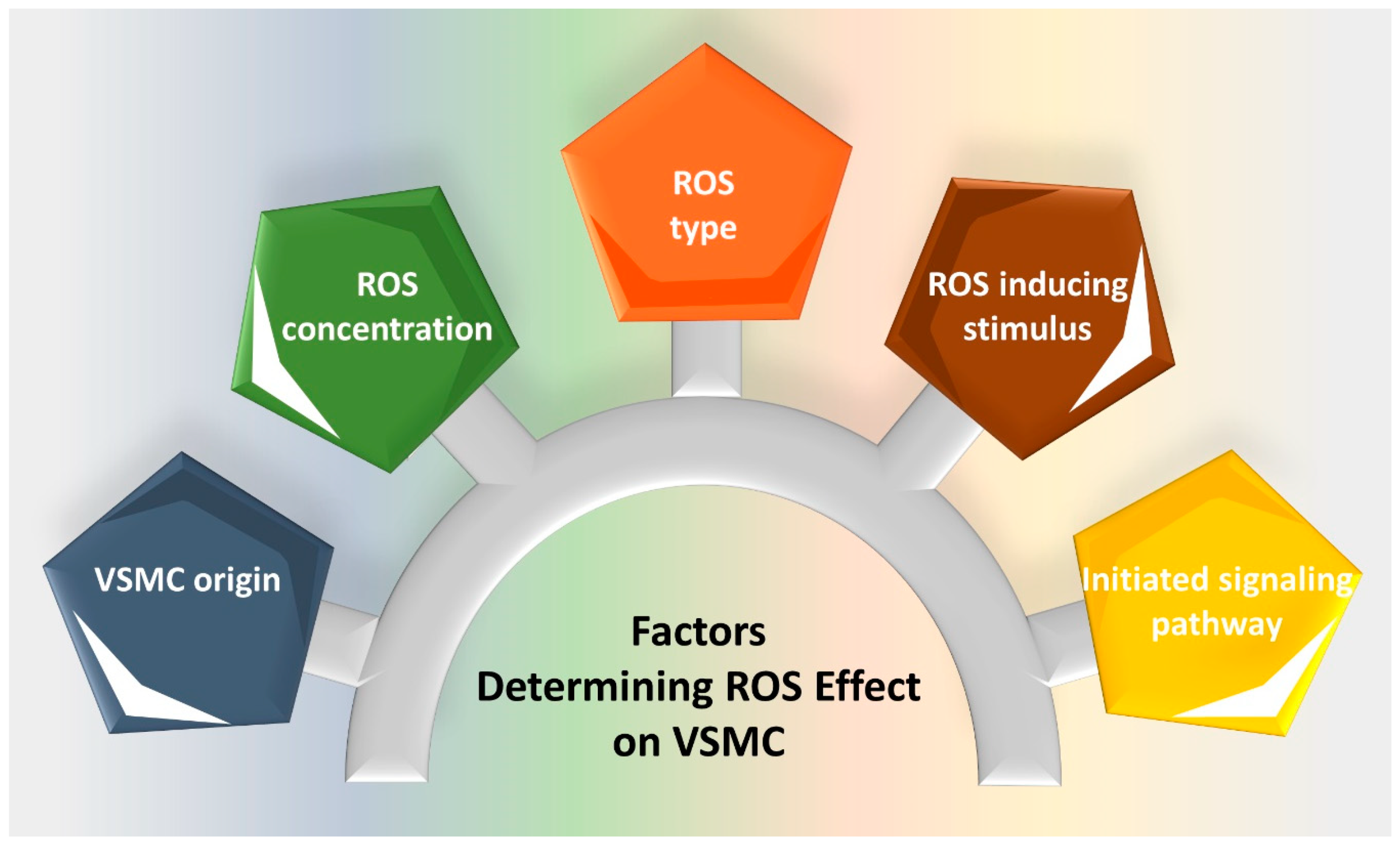
:1. Introduction
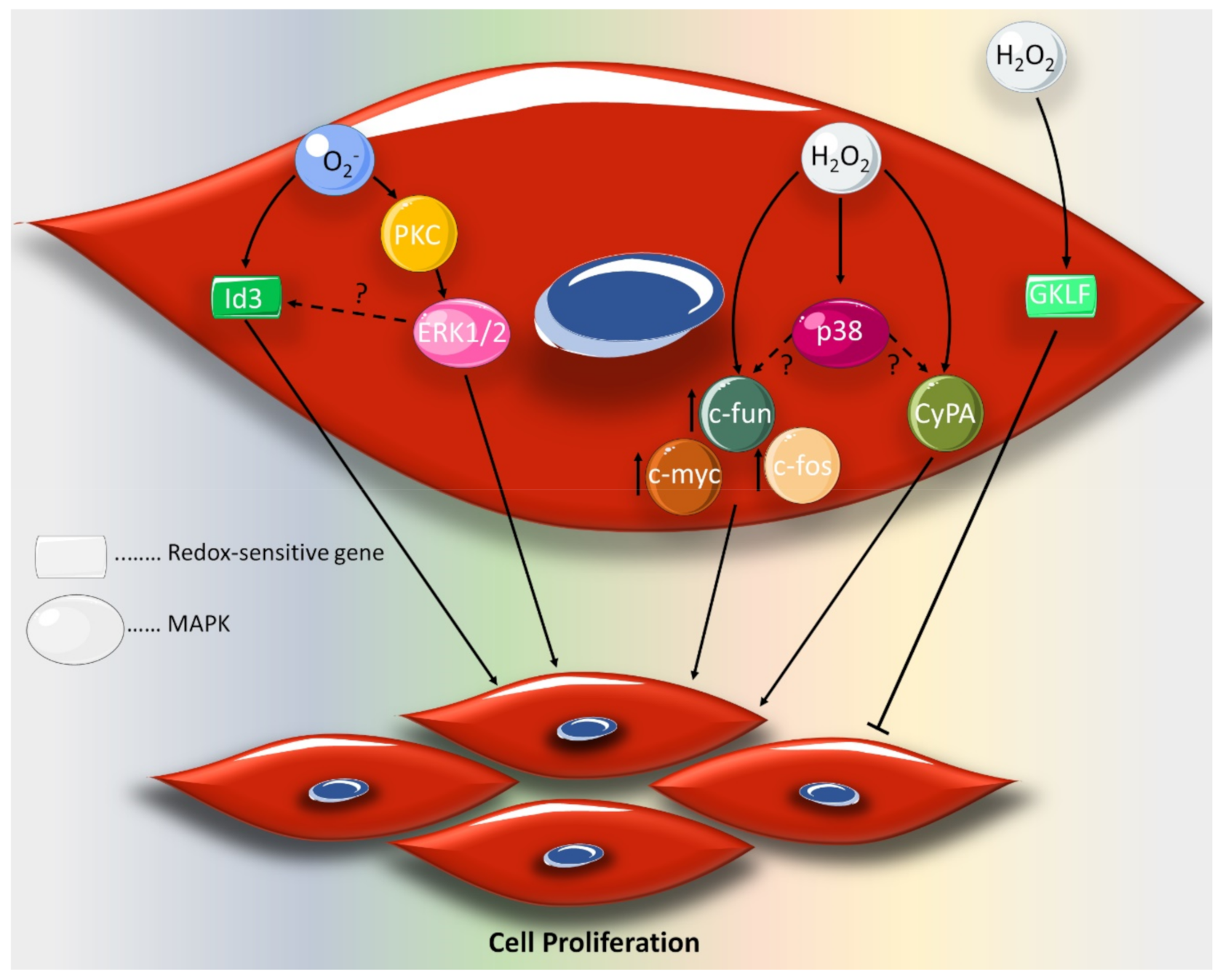
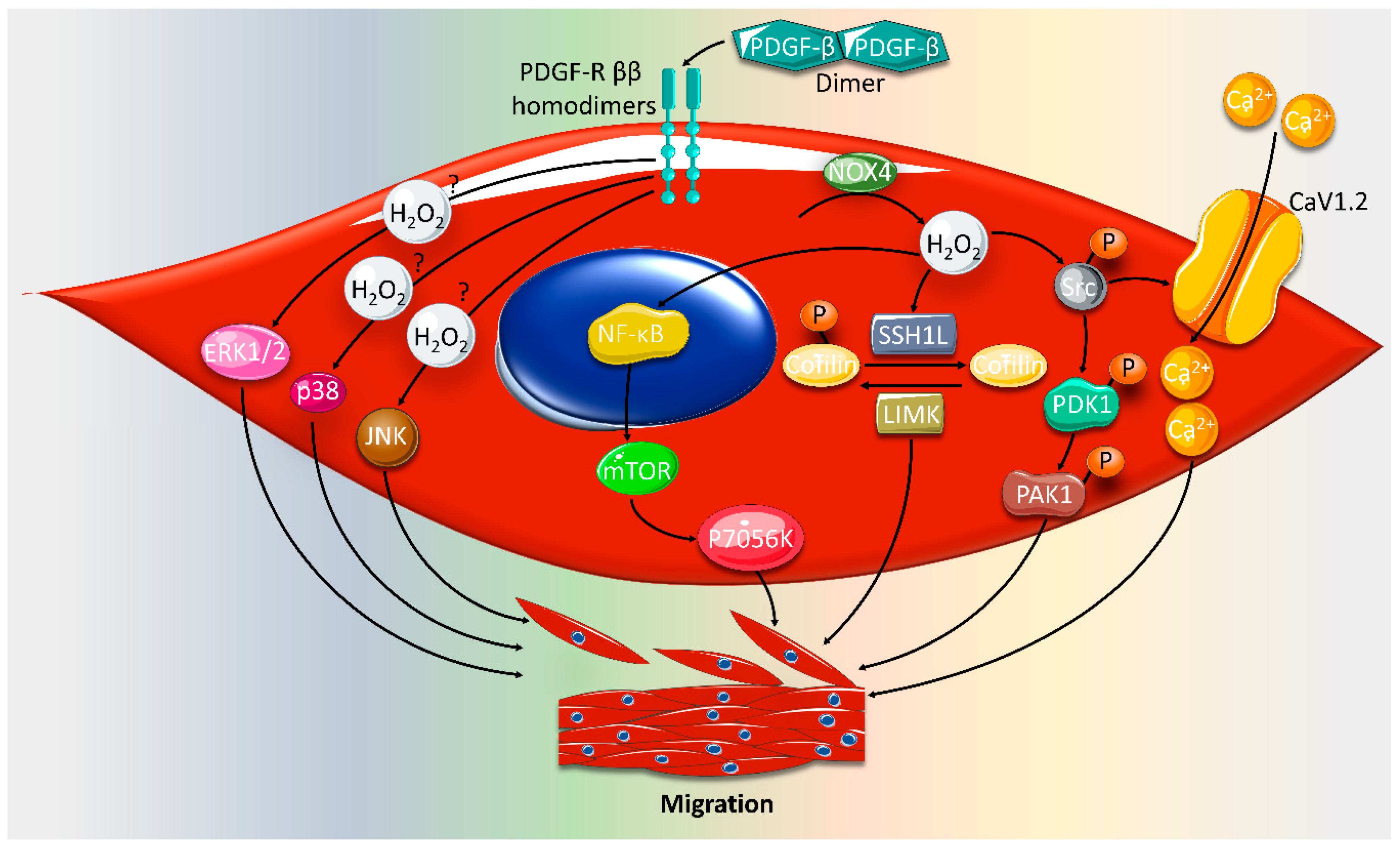
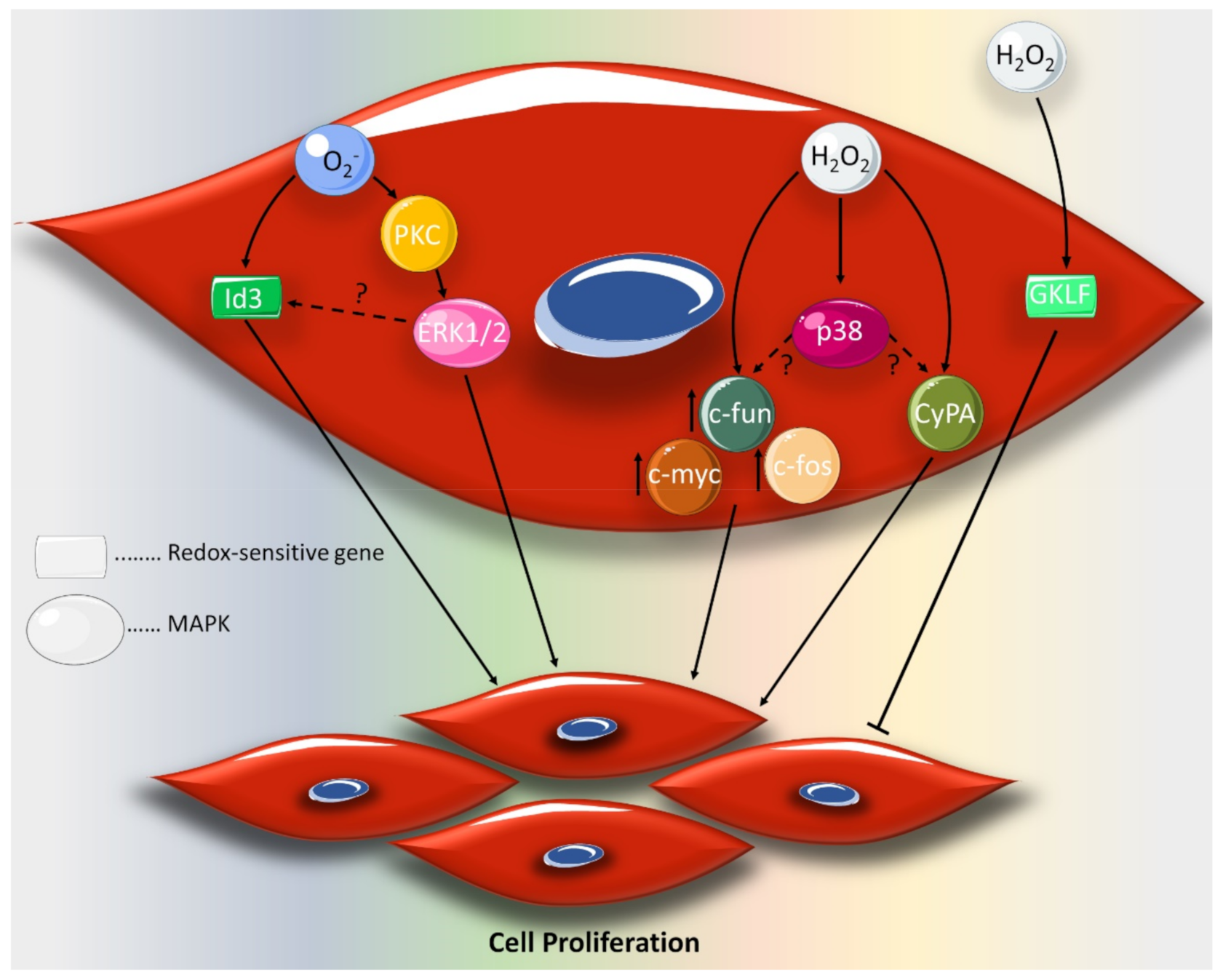
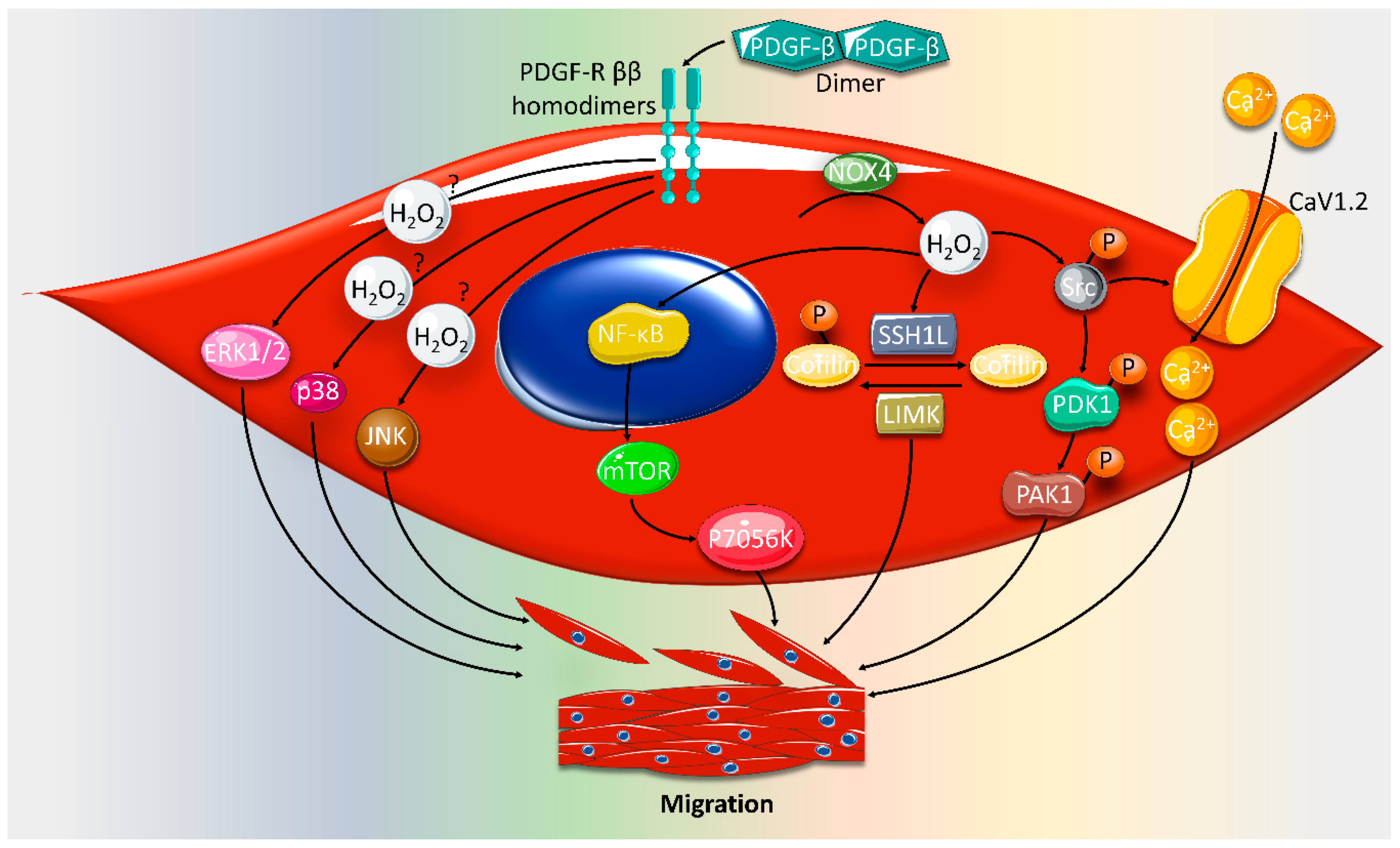
2. Effect of ROS on VSMC Proliferation and Migration
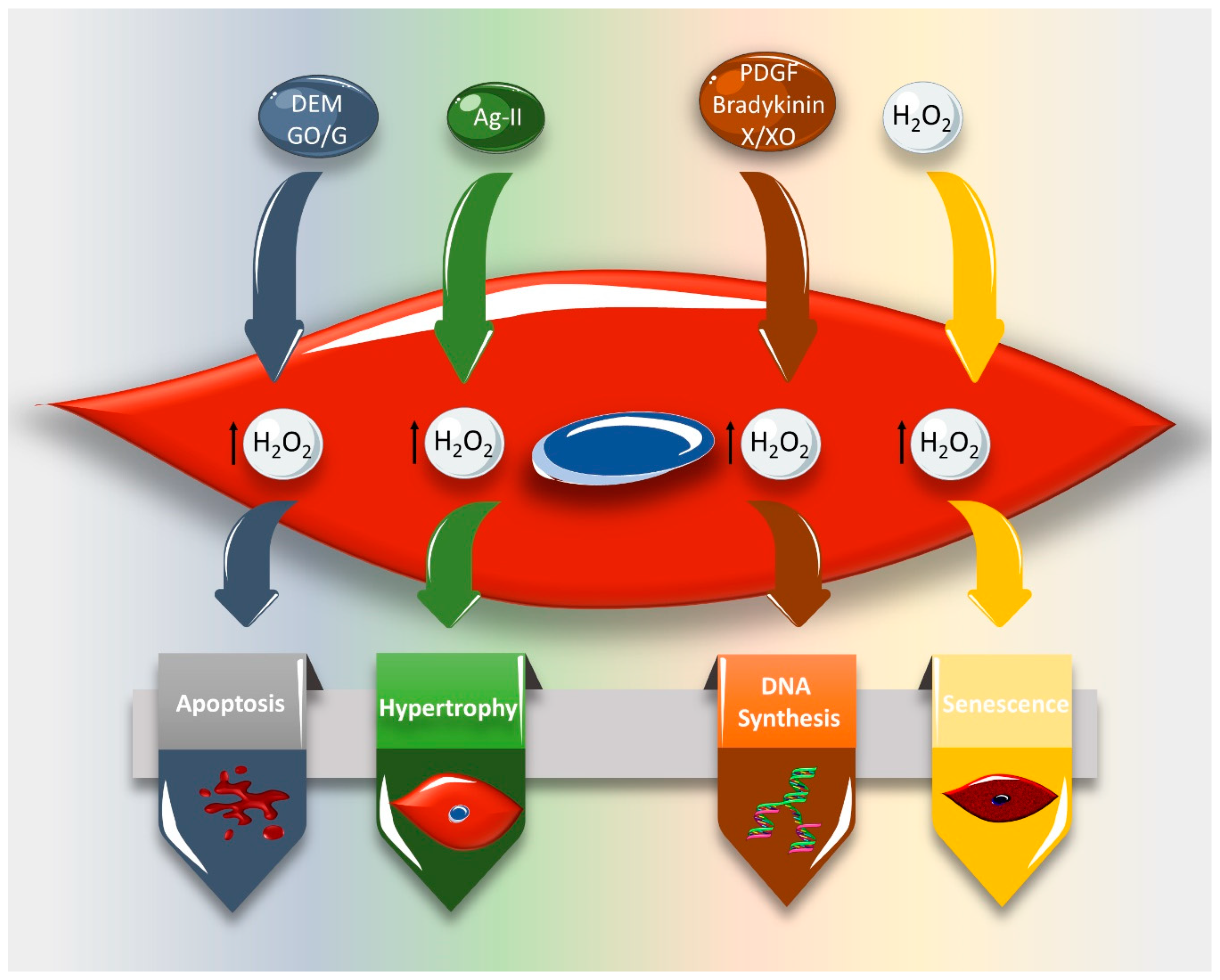
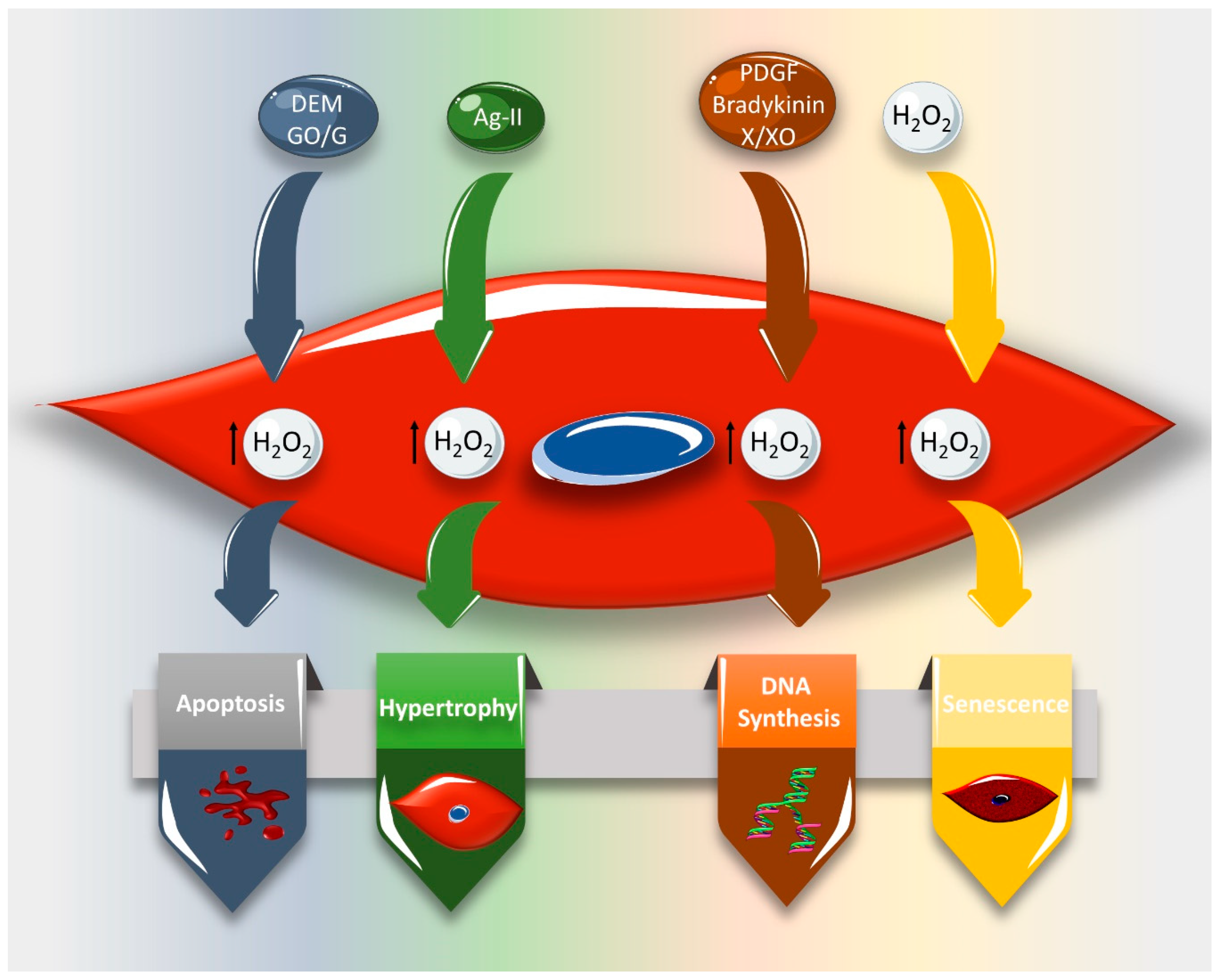
3. Effect of ROS on VSMC Cell Cycle and Cell Fate
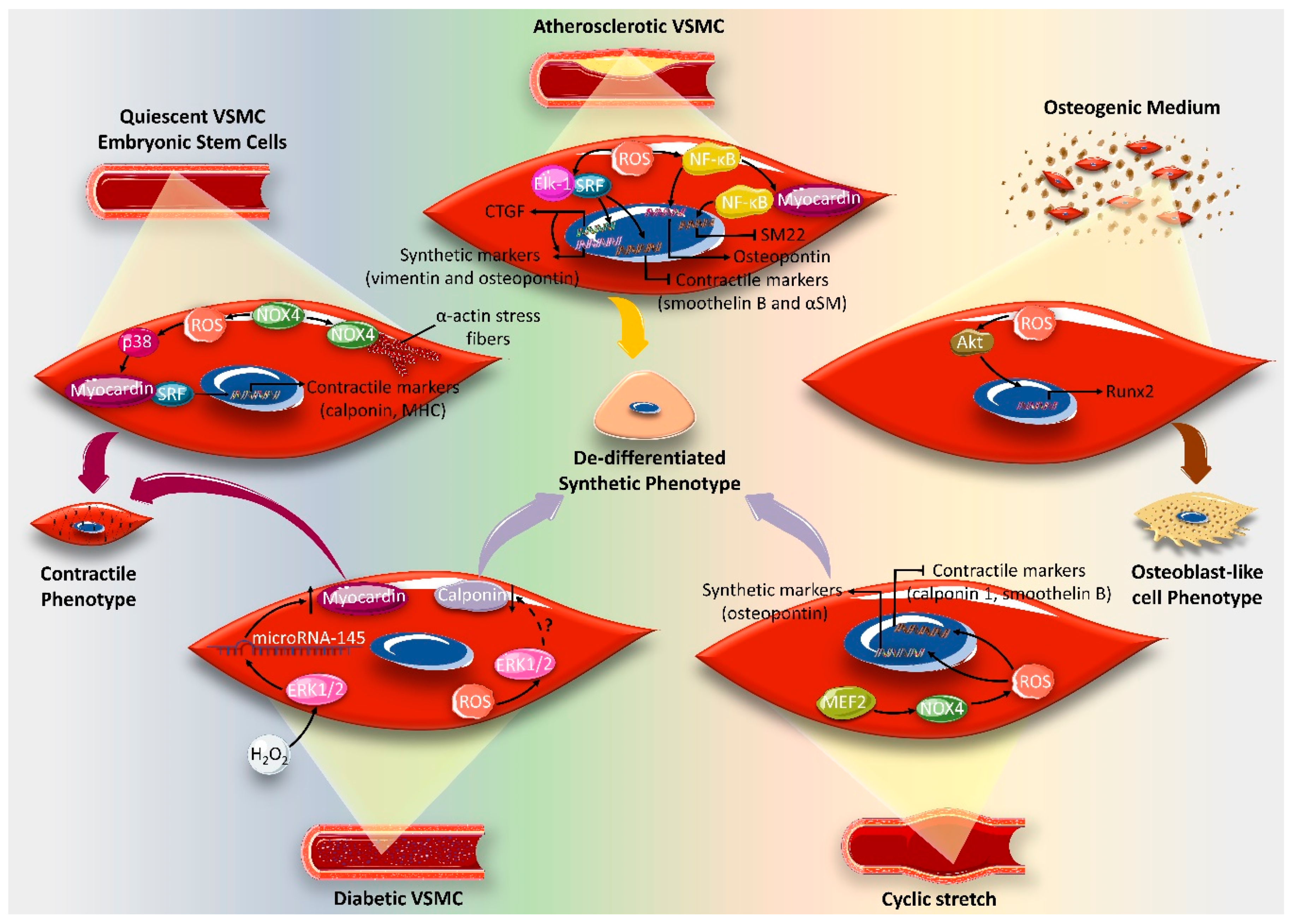
4. Effect of ROS on VSMC Differentiation Markers
5. ROS and VSMC Epigenetics
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ag-II | Angiotensin II |
| bFGF | Basic fibroblast growth factor |
| Cdk | Cyclin-dependent kinase |
| CTGF | Connective tissue growth factor |
| CVD | Cardiovascular disease |
| CyPA | Cyclophilin A |
| ECM | Extracellular matrix |
| EC-SOD | Extracellular superoxide dismutase |
| FGF | Fibroblast growth factor |
| GKLF | Gut-enriched Kruppel-like factor |
| H2O2 | Hydrogen peroxide |
| HAT | Acetyltransferase |
| HDAC | Histone deacetylase |
| HO• | Hydroxyl radical |
| IGF-I | Insulin-like growth factor-I |
| LIMK | LIM kinase |
| MEF2B | Myocyte enhancer factor 2B |
| MCP-1 | Monocyte chemokine protein 1 |
| MMP | Matrix metalloproteinases |
| NAC | N-acetylcysteine |
| NO | Nitric oxide |
| NOX | NADPH Oxidase |
| O2− | Superoxide anion |
| PAK1 | Rac-effector p21-activated protein kinase |
| PDGF | Platelet-derived growth factor |
| PDK-1 | Phosphoinositide-dependent kinase-1 |
| PTDC | Pyrrolidinedithiocarbamate |
| TGF-β | Transforming growth factor |
| ROS | Reactive oxygen species |
| SM | Smooth muscle |
| SOD | Superoxide dismutase |
| SRF | Serum response factor |
| SSHL1 | Slingshot1L |
| TAA | Thoracic aortic aneurysm |
| TET2 | Ten-eleven translocation-2 |
| VEGF | Vascular epidermal growth factor |
| VSMCs | Vascular smooth muscle cells |
References
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhao, H.; Li, H.; Kalyanaraman, B.; Nicolosi, A.C.; Gutterman, D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res. 2003, 93, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 46, 200–206. [Google Scholar] [CrossRef]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Omar, H.; Cherry, P.; Mortelliti, M.; Burke-Wolin, T.; Wolin, M. Inhibition of coronary artery superoxide dismutase attenuates endothelium-dependent and-independent nitrovasodilator relaxation. Circ. Res. 1991, 69, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Vokurkova, M.; Xu, S.; Touyz, R.M. Reactive oxygen species, cell growth, cell cycle progression and vascular remodeling in hypertension. Future Cardiol. 2007, 3, 53–63. [Google Scholar] [CrossRef]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwała, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Żmudka, K. Mechanisms of oxidative stress in human aortic aneurysms—association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozovich, F.; Nicholson, C.; Degen, C.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Stoll, S.; Leimena, C.; Qiu, H. Vascular Smooth Muscle Cell. Vasc. Smooth Muscle Cell. Muscle Cell Tissue–Curr. Status Res. Field 2018, 209–227. [Google Scholar]
- Fardoun, M.; Al-Shehabi, T.; El-Yazbi, A.; Issa, K.; Zouein, F.; Maaliki, D.; Iratni, R.; Eid, A.H. Ziziphus nummularia Inhibits Inflammation-Induced Atherogenic Phenotype of Human Aortic Smooth Muscle Cells. Oxidative Med. Cell. Longev. 2017, 2017, 4134093. [Google Scholar] [CrossRef] [Green Version]
- Rensen, S.; Doevendans, P.; Van Eys, G. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Saleh Al-Shehabi, T.; Iratni, R.; Eid, A.H. Anti-atherosclerotic plants which modulate the phenotype of vascular smooth muscle cells. Phytomedicine Int. J. Phytother. Phytopharm. 2016, 23, 1068–1081. [Google Scholar] [CrossRef]
- Zhu, S.B.; Zhu, J.; Zhou, Z.Z.; Xi, E.P.; Wang, R.P.; Zhang, Y. TGF-beta1 induces human aortic vascular smooth muscle cell phenotype switch through PI3K/AKT/ID2 signaling. Am. J. Transl. Res. 2015, 7, 2764–2774. [Google Scholar]
- Qi, M.; Xin, S. FGF signaling contributes to atherosclerosis by enhancing the inflammatory response in vascular smooth muscle cells. Mol. Med. Rep. 2019, 20, 162–170. [Google Scholar] [CrossRef]
- Ramel, D.; Gayral, S.; Sarthou, M.K.; Auge, N.; Negre-Salvayre, A.; Laffargue, M. Immune and Smooth Muscle Cells Interactions in Atherosclerosis: How to Target a Breaking Bad Dialogue? Front. Pharmacol. 2019, 10, 1276. [Google Scholar] [CrossRef] [Green Version]
- Ljuca, F.; Drevensek, G. Endothelin-1 induced vascular smooth muscle cell proliferation is mediated by cytochrome p-450 arachidonic acid metabolites. Bosn. J. Basic Med Sci. 2010, 10, 223–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Yun, S.J.; Ha, J.M.; Jin, S.Y.; Ha, H.K.; Song, S.H.; Kim, C.D.; Bae, S.S. Prostaglandin D2 stimulates phenotypic changes in vascular smooth muscle cells. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dong, C.Q.; Peng, G.Y.; Huang, H.Y.; Yu, Y.S.; Ji, Z.C.; Shen, Z.Y. MicroRNA-134-5p Regulates Media Degeneration through Inhibiting VSMC Phenotypic Switch and Migration in Thoracic Aortic Dissection. Mol. Ther. Nucleic Acids 2019, 16, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Su, X.; Qin, Q.; Yu, Y.; Jia, M.; Zhang, H.; Li, H.; Pei, L. New insights into phenotypic switching of VSMCs induced by hyperhomocysteinemia: Role of endothelin-1 signaling. Biomed. Pharmacother. 2020, 123, 109758. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.I.; Csanyi, G.; Ranayhossaini, D.J.; Feck, D.M.; Blose, K.J.; Assatourian, L.; Vorp, D.A.; Pagano, P.J. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arter. Thromb Vasc Biol 2015, 35, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Fardoun, M.M.; Issa, K.; Maaliki, D.; Nasser, S.A.; Baydoun, E.; Eid, A.H. Estrogen increases expression of vascular alpha 2C adrenoceptor through the cAMP/Epac/JNK/AP-1 pathway and potentiates cold-induced vasoconstriction. Vasc. Pharmacol. 2020, 131, 106690. [Google Scholar] [CrossRef]
- Wehbe, Z.; Nasser, S.A.; El-Yazbi, A.; Nasreddine, S.; Eid, A.H. Estrogen and Bisphenol A in Hypertension. Curr. Hypertens. Rep. 2020, 22, 23. [Google Scholar] [CrossRef]
- Fardoun, M.; Dehaini, H.; Shaito, A.; Mesmar, J.; El-Yazbi, A.; Badran, A.; Beydoun, E.; Eid, A.H. The hypertensive potential of estrogen: An untold story. Vasc. Pharmacol. 2020, 124, 106600. [Google Scholar] [CrossRef]
- Dehaini, H.; Fardoun, M.; Abou-Saleh, H.; El-Yazbi, A.; Eid, A.A.; Eid, A.H. Estrogen in vascular smooth muscle cells: A friend or a foe? Vasc. Pharmacol. 2018, 111, 15–21. [Google Scholar] [CrossRef]
- Eid, A.H.; Maiti, K.; Mitra, S.; Chotani, M.A.; Flavahan, S.; Bailey, S.R.; Thompson-Torgerson, C.S.; Flavahan, N.A. Estrogen increases smooth muscle expression of alpha2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1955–H1961. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Samaha, A.A.; Baydoun, S.; Iratni, R.; Eid, A.H. Rhus coriaria L. (Sumac) Evokes Endothelium-Dependent Vasorelaxation of Rat Aorta: Involvement of the cAMP and cGMP Pathways. Front. Pharmacol. 2018, 9, 688. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.H. cAMP induces adhesion of microvascular smooth muscle cells to fibronectin via an Epac-mediated but PKA-independent mechanism. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 30, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Chotani, M.A.; Mitra, S.; Eid, A.H.; Han, S.A.; Flavahan, N.A. Distinct cAMP signaling pathways differentially regulate alpha2C-adrenoceptor expression: role in serum induction in human arteriolar smooth muscle cells. Am. J.Physiol. Heart Circ. Physiol. 2005, 288, H69–H76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motawea, H.K.; Jeyaraj, S.C.; Eid, A.H.; Mitra, S.; Unger, N.T.; Ahmed, A.A.; Flavahan, N.A.; Chotani, M.A. Cyclic AMP-Rap1A signaling mediates cell surface translocation of microvascular smooth muscle alpha2C-adrenoceptors through the actin-binding protein filamin-2. Am. J. Physiol. Cell Physiol. 2013, 305, C829–C845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaraj, S.C.; Unger, N.T.; Eid, A.H.; Mitra, S.; Paul El-Dahdah, N.; Quilliam, L.A.; Flavahan, N.A.; Chotani, M.A. Cyclic AMP-Rap1A signaling activates RhoA to induce alpha(2c)-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 303, C499–C511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, A.H.; Chotani, M.A.; Mitra, S.; Miller, T.J.; Flavahan, N.A. Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle alpha2C-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H266–H272. [Google Scholar] [CrossRef] [Green Version]
- Wehbe, N.; Slika, H.; Mesmar, J.; Nasser, S.A.; Pintus, G.; Baydoun, S.; Badran, A.; Kobeissy, F.; Eid, A.H.; Baydoun, E. The Role of Epac in Cancer Progression. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Wehbe, N.; Nasser, S.A.; Al-Dhaheri, Y.; Iratni, R.; Bitto, A.; El-Yazbi, A.F.; Badran, A.; Kobeissy, F.; Baydoun, E.; Eid, A.H. EPAC in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M. Redox control of vascular smooth muscle proliferation. J. Lab. Clin. Med. 1998, 132, 9–15. [Google Scholar] [CrossRef]
- Su, B.; Mitra, S.; Gregg, H.; Flavahan, S.; Chotani, M.; Clark, K.; Goldschmidt-Clermont, P.; Flavahan, N. Redox regulation of vascular smooth muscle cell differentiation. Circ. Res. 2001, 89, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants (Basel) 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- San Martin, A.; Griendling, K.K. Redox control of vascular smooth muscle migration. Antioxid Redox Signal 2010, 12, 625–640. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid Redox Signal 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupte, S.A.; Kaminski, P.M.; George, S.; Kouznestova, L.; Olson, S.C.; Mathew, R.; Hintze, T.H.; Wolin, M.S. Peroxide generation by p47phox-Src activation of Nox2 has a key role in protein kinase C-induced arterial smooth muscle contraction. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1048–H1057. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Chen, X.; Tabet, F.; Yao, G.; He, G.; Quinn, M.T.; Pagano, P.J.; Schiffrin, E.L. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ. Res. 2002, 90, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [Green Version]
- Alexander, R.W. Hypertension and the pathogenesis of atherosclerosis: oxidative stress and the mediation of arterial inflammatory response: a new perspective. Hypertension 1995, 25, 155–161. [Google Scholar] [CrossRef]
- Baas, A.S.; Berk, B.C. Differential activation of mitogen-activated protein kinases by H2O2 and O2− in vascular smooth muscle cells. Circ. Res. 1995, 77, 29–36. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.-X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Alexander, R.; Akers, M.; Griendling, K. p38 MAP kinase is a critical component of the redox-sensitive signaling pathways by angiotensin II: role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998, 273, 15022–15029. [Google Scholar] [CrossRef] [Green Version]
- Patterson, C.; Ruef, J.; Madamanchi, N.R.; Barry-Lane, P.; Hu, Z.; Horaist, C.; Ballinger, C.A.; Brasier, A.R.; Bode, C.; Runge, M.S. Stimulation of a vascular smooth muscle cell NAD(P)H oxidase by thrombin. Evidence that p47(phox) may participate in forming this oxidase in vitro and in vivo. J. Biol. Chem. 1999, 274, 19814–19822. [Google Scholar] [CrossRef] [Green Version]
- Velarde, V.; De La Cerda, P.M.; Duarte, C.; Arancibia, F.; Abbott, E.; Gonzalez, A.; Moreno, F.; Jaffa, A.A. Role of reactive oxygen species in bradykinin-induced proliferation of vascular smooth muscle cells. Biol. Res. 2004, 37, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhang, M.J.; Li, B.H.; Chen, L.; Pi, Y.; Yin, Y.W.; Long, C.Y.; Wang, X.; Sun, M.J.; Chen, X.; et al. PPARgamma Inhibits VSMC Proliferation and Migration via Attenuating Oxidative Stress through Upregulating UCP2. PLoS ONE 2016, 11, e0154720. [Google Scholar] [CrossRef] [Green Version]
- Menshikov, M.; Plekhanova, O.; Cai, H.; Chalupsky, K.; Parfyonova, Y.; Bashtrikov, P.; Tkachuk, V.; Berk, B.C. Urokinase plasminogen activator stimulates vascular smooth muscle cell proliferation via redox-dependent pathways. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Nickenig, G.; Baudler, S.; Müller, C.; Werner, C.; Werner, N.; Welzel, H.; STREHLOW, K.; BöHM, M. Redox-sensitive vascular smooth muscle cell proliferation is mediated by GKLF and Id3 in vitro and in vivo. Faseb J. 2002, 16, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K.; Nigro, P.; Berk, B.C. Oxidative stress and vascular smooth muscle cell growth: a mechanistic linkage by cyclophilin A. Antioxid. Redox Signal. 2010, 12, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.-G.; Melaragno, M.G.; Liao, D.-F.; Yan, C.; Haendeler, J.; Suh, Y.-A.; Lambeth, J.D.; Berk, B.C. Cyclophilin A is a secreted growth factor induced by oxidative stress. Circ. Res. 2000, 87, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.N.; Berk, B.C. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992, 70, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Rao, G.N. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates Ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene 1996, 13, 713. [Google Scholar]
- Brown, M.R.; Miller, F.J., Jr.; Li, W.G.; Ellingson, A.N.; Mozena, J.D.; Chatterjee, P.; Engelhardt, J.F.; Zwacka, R.M.; Oberley, L.W.; Fang, X.; et al. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ. Res. 1999, 85, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, T.; Ishizaka, N.; Rajagopalan, S.; Laursen, J.B.; Capers, Q.t.; Taylor, W.R.; Harrison, D.G.; de Leon, H.; Wilcox, J.N.; Griendling, K.K. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 1997, 80, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arter. Thromb Vasc Biol 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, H.P.; Souza, L.C.; Anastacio, V.M.; Pereira, A.C.; Junqueira, M.L.; Krieger, J.E.; da Luz, P.L.; Augusto, O.; Laurindo, F.R. Vascular oxidant stress early after balloon injury: evidence for increased NAD(P)H oxidoreductase activity. Free Radic. Biol. Med. 2000, 28, 1232–1242. [Google Scholar] [CrossRef]
- Bauersachs, J.; Bouloumie, A.; Fraccarollo, D.; Hu, K.; Busse, R.; Ertl, G. Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation 1999, 100, 292–298. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, L.; Moldovan, N.I.; Sohn, R.H.; Parikh, S.A.; Goldschmidt-Clermont, P.J. Redox changes of cultured endothelial cells and actin dynamics. Circ. Res. 2000, 86, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [Green Version]
- DalleDonne, I.; Milzani, A.; Colombo, R. H2O2-treated actin: assembly and polymer interactions with cross-linking proteins. Biophys. J. 1995, 69, 2710–2719. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, D.B.; Burger, J.M.; Beals, T.F.; Armstrong, B.C.; Hyslop, P.A. Actin polymerization in cellular oxidant injury. Arch. Biochem. Biophys. 1991, 288, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Nishio, E.; Watanabe, Y. The involvement of reactive oxygen species and arachidonic acid in alpha 1-adrenoceptor-induced smooth muscle cell proliferation and migration. Br. J. Pharm. 1997, 121, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen and NF-kappaB in VEGF-induced migration of human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2001, 285, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Helmcke, I.; Palfi, K.; Krause, K.H.; Busse, R.; Brandes, R.P. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arter. Thromb Vasc Biol 2007, 27, 1736–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2004, 36, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Lv, D.D.; Fang, J. Insulin-like growth factor-I induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovasc. Res. 2008, 80, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Haurani, M.J.; Cifuentes, M.E.; Shepard, A.D.; Pagano, P.J. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mRNA and inhibits angiotensin II-induced adventitial myofibroblast migration. Hypertension 2008, 52, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; San Martin, A.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arter. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, D.S.; Taniyama, Y.; Rocic, P.; Seshiah, P.N.; Dechert, M.A.; Gerthoffer, W.T.; Griendling, K.K. Phosphoinositide-dependent kinase 1 and p21-activated protein kinase mediate reactive oxygen species-dependent regulation of platelet-derived growth factor-induced smooth muscle cell migration. Circ. Res. 2004, 94, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Buetow, B.S.; Tappan, K.A.; Crosby, J.R.; Seifert, R.A.; Bowen-Pope, D.F. Chimera analysis supports a predominant role of PDGFRbeta in promoting smooth-muscle cell chemotaxis after arterial injury. Am. J. Pathol. 2003, 163, 979–984. [Google Scholar] [CrossRef]
- Floege, J.; Hudkins, K.L.; Davis, C.L.; Schwartz, S.M.; Alpers, C.E. Expression of PDGF alpha-receptor in renal arteriosclerosis and rejecting renal transplants. J. Am. Soc. Nephrol. Jasn 1998, 9, 211–223. [Google Scholar]
- Rubin, K.; Tingstrom, A.; Hansson, G.K.; Larsson, E.; Ronnstrand, L.; Klareskog, L.; Claesson-Welsh, L.; Heldin, C.H.; Fellstrom, B.; Terracio, L. Induction of B-type receptors for platelet-derived growth factor in vascular inflammation: possible implications for development of vascular proliferative lesions. Lancet 1988, 1, 1353–1356. [Google Scholar] [CrossRef]
- Lu, Q.-B.; Wan, M.-Y.; Wang, P.-Y.; Zhang, C.-X.; Xu, D.-Y.; Liao, X.; Sun, H.-J. Chicoric acid prevents PDGF-BB-induced VSMC dedifferentiation, proliferation and migration by suppressing ROS/NFκB/mTOR/P70S6K signaling cascade. Redox Biol. 2018, 14, 656–668. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Lee, M.Y.; Williams, H.C.; Mizuno, K.; Lassegue, B.; Griendling, K.K. Dual regulation of cofilin activity by LIM kinase and Slingshot-1L phosphatase controls platelet-derived growth factor-induced migration of human aortic smooth muscle cells. Circ Res 2008, 102, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Cordero, J.J.; Magalhaes, M.A.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Kashihara, T.; Nakada, T.; Aoyama, T.; Yamada, M. PDGF-induced migration of synthetic vascular smooth muscle cells through c-Src-activated L-type Ca(2+) channels with full-length CaV1.2 C-terminus. Pflug. Arch. Eur. J. Physiol. 2018, 470, 909–921. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Kim, S.; Izumi, Y.; Izumiya, Y.; Nakao, T.; Miyazaki, H.; Iwao, H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arter. Thromb. Vasc. Biol. 2003, 23, 795–801. [Google Scholar] [CrossRef] [Green Version]
- Uzui, H.; Lee, J.-D.; Shimizu, H.; Tsutani, H.; Ueda, T. The role of protein-tyrosine phosphorylation and gelatinase production in the migration and proliferation of smooth muscle cells. Atherosclerosis 2000, 149, 51–59. [Google Scholar] [CrossRef]
- Mason, D.P.; Kenagy, R.D.; Hasenstab, D.; Bowen-Pope, D.F.; Seifert, R.A.; Coats, S.; Hawkins, S.M.; Clowes, A.W. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ. Res. 1999, 85, 1179–1185. [Google Scholar] [CrossRef]
- Southgate, K.M.; Davies, M.; Booth, R.; Newby, A. Involvement of extracellular-matrix-degrading metalloproteinases in rabbit aortic smooth-muscle cell proliferation. Biochem. J. 1992, 288, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD (P) H oxidase–derived reactive oxygen species. Circ. Res. 2003, 92, e80–e86. [Google Scholar] [CrossRef] [Green Version]
- Branchetti, E.; Poggio, P.; Sainger, R.; Shang, E.; Grau, J.B.; Jackson, B.M.; Lai, E.K.; Parmacek, M.S.; Gorman, R.C.; Gorman, J.H. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc. Res. 2013, 100, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Et Biophys. Acta 2001, 1504, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Li, P.F.; Dietz, R.; von Harsdorf, R. Reactive oxygen species induce apoptosis of vascular smooth muscle cell. Febs Lett. 1997, 404, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.-a. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Bennett, M.R. Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc. Res. 1999, 41, 361–368. [Google Scholar] [CrossRef]
- Li, P.-F.; Dietz, R.; von Harsdorf, R.d. Differential effect of hydrogen peroxide and superoxide anion on apoptosis and proliferation of vascular smooth muscle cells. Circulation 1997, 96, 3602–3609. [Google Scholar] [CrossRef]
- Deshpande, N.N.; Sorescu, D.; Seshiah, P.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Griendling, K.K. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid. Redox Signal. 2002, 4, 845–854. [Google Scholar] [CrossRef]
- Popowich, D.A.; Vavra, A.K.; Walsh, C.P.; Bhikhapurwala, H.A.; Rossi, N.B.; Jiang, Q.; Aalami, O.O.; Kibbe, M.R. Regulation of reactive oxygen species by p53: implications for nitric oxide-mediated apoptosis. Am. J. Physiol.. Heart Circ. Physiol. 2010, 298, H2192–H2200. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Byun, J.K.; Park, M.; Woo Kim, S.; Lee, S.; Kim, J.G.; Lee, I.K.; Choi, Y.K.; Park, K.G. Melatonin inhibits vascular smooth muscle cell proliferation and apoptosis through upregulation of Sestrin2. Exp. Ther. Med. 2020, 19, 3454–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Harrison, D.G. Dual role of reactive oxygen species in vascular growth. Circ Res 1999, 85, 562–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.-C.; Jain, M.; Hsieh, C.-M.; Lee, W.-S.; Yoshizumi, M.; Patterson, C.; Perrella, M.A.; Cooke, C.; Wang, H.; Haber, E. Induction of apoptosis by pyrrolidinedithiocarbamate and N-acetylcysteine in vascular smooth muscle cells. J. Biol. Chem. 1996, 271, 3667–3670. [Google Scholar] [CrossRef] [Green Version]
- Abello, P.A.; Fidler, S.A.; Bulkley, G.B.; Buchman, T.G. Antioxidants modulate induction of programmed endothelial cell death (apoptosis) by endotoxin. Arch. Surg. 1994, 129, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Bristol, L.A.; Hosler, B.; Brown, R.H., Jr.; Kuncl, R.W. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 4155–4159. [Google Scholar] [CrossRef] [Green Version]
- Roederer, M.; Staal, F.J.; Ela, S.W.; Herzenberg, L.A.; Herzenberg, L.A. N-acetylcysteine: potential for AIDS therapy. Pharmacology 1993, 46, 121–129. [Google Scholar] [CrossRef]
- Rao, G.; Lassegue, B.; Griendling, K.; Alexander, R.W. Hydrogen peroxide stimulates transcription of c-jun in vascular smooth muscle cells: role of arachidonic acid. Oncogene 1993, 8, 2759–2764. [Google Scholar]
- Rao, G.N.; Lassegue, B.; Griendling, K.K.; Alexander, R.W.; Berk, B.C. Hydrogen peroxide-induced c-fos expression is mediated by arachidonic acid release: role of protein kinase C. Nucleic Acids Res. 1993, 21, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Fiorani, M.; Cantoni, O.; Tasinato, A.; Boscoboinik, D.; Azzi, A. Hydrogen peroxide-and fetal bovine serum-induced DNA synthesis in vascular smooth muscle cells: positive and negative regulation by protein kinase C isoforms. Biochim. Et Biophys. Acta (Bba)-Mol. Cell Res. 1995, 1269, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Gong, X.; Mo, Y.; Wu, S. Polydatin inhibits the oxidative stress-induced proliferation of vascular smooth muscle cells by activating the eNOS/SIRT1 pathway. Int. J. Mol. Med. 2016, 37, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Przybylska, D.; Janiszewska, D.; Gozdzik, A.; Bielak-Zmijewska, A.; Sunderland, P.; Sikora, E.; Mosieniak, G. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget 2016, 7, 66429–66443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafari, A.M.; Ushio-Fukai, M.; Akers, M.; Yin, Q.; Shah, A.; Harrison, D.G.; Taylor, W.R.; Griendling, K.K. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension 1998, 32, 488–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Baudler, S.; Welzel, H.; Böhm, M.; Nickenig, G. Identification of a novel redox-sensitive gene, Id3, which mediates angiotensin II–induced cell growth. Circulation 2002, 105, 2423–2428. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, O.; Neininger, A.; Schratt, G.; Zinck, R.; Cahill, M.A.; Engel, K.; Kotlyarov, A.; Kraft, R.; Kostka, S.; Gaestel, M. MAPKAP kinase 2 phosphorylates serum response factor in vitro and in vivo. J. Biol. Chem. 1999, 274, 14434–14443. [Google Scholar] [CrossRef] [Green Version]
- Thuerauf, D.J.; Arnold, N.D.; Zechner, D.; Hanford, D.S.; DeMartin, K.M.; McDonough, P.M.; Prywes, R.; Glembotski, C.C. P38 mitogen-activated protein kinase mediates the transcriptional induction of the atrial natriuretic factor gene through a serum response element a potential role for the transcription factor atf6. J. Biol. Chem. 1998, 273, 20636–20643. [Google Scholar] [CrossRef] [Green Version]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Schmidt, H.H.; Lassegue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arter. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Du, K.L.; Chen, M.; Li, J.; Lepore, J.J.; Mericko, P.; Parmacek, M.S. Megakaryoblastic leukemia factor-1 transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J. Biol. Chem. 2004, 279, 17578–17586. [Google Scholar] [CrossRef] [Green Version]
- Sutliff, R.L.; Hilenski, L.L.; Amanso, A.M.; Parastatidis, I.; Dikalova, A.E.; Hansen, L.; Datla, S.R.; Long, J.S.; El-Ali, A.M.; Joseph, G. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2154–2161. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Luo, Z.; Pepe, A.E.; Margariti, A.; Zeng, L.; Xu, Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am. J. Physiol. Cell Physiol. 2009, 296, C711–C723. [Google Scholar] [CrossRef] [Green Version]
- McDonald, O.G.; Wamhoff, B.R.; Hoofnagle, M.H.; Owens, G.K. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J. Clin. Investig. 2006, 116, 36–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, E.S.; Wilson, E.; Ramos, K.S. NF-kappaB and matrix-dependent regulation of osteopontin promoter activity in allylamine-activated vascular smooth muscle cells. Oxid. Med. Cell. Longev. 2012, 2012, 496540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, E.R.; Garvin, M.R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M.; Giachelli, C.M. Osteopontin is synthesized by macrophage, smooth muscle, and endothelial cells in primary and restenotic human coronary atherosclerotic plaques. Arterioscler. Thromb. A J. Vasc. Biol. 1994, 14, 1648–1656. [Google Scholar]
- Tang, R.-H.; Zheng, X.-L.; Callis, T.E.; Stansfield, W.E.; He, J.; Baldwin, A.S.; Wang, D.-Z.; Selzman, C.H. Myocardin inhibits cellular proliferation by inhibiting NF-κB (p65)-dependent cell cycle progression. Proc. Natl. Acad. Sci. USA 2008, 105, 3362–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trion, A.; van der Laarse, A. Vascular smooth muscle cells and calcification in atherosclerosis. Am. Heart J. 2004, 147, 808–814. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [Green Version]
- Chettimada, S.; Ata, H.; Rawat, D.K.; Gulati, S.; Kahn, A.G.; Edwards, J.G.; Gupte, S.A. Contractile protein expression is upregulated by reactive oxygen species in aorta of Goto-Kakizaki rat. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H214–H224. [Google Scholar] [CrossRef] [Green Version]
- Carrillo-Sepulveda, M.A.; Matsumoto, T. Phenotypic modulation of mesenteric vascular smooth muscle cells from type 2 diabetic rats is associated with decreased caveolin-1 expression. Cell. Physiol. Biochem. 2014, 34, 1497–1506. [Google Scholar] [CrossRef]
- Katoh, Y.; Molkentin, J.D.; Dave, V.; Olson, E.N.; Periasamy, M. MEF2B is a component of a smooth muscle-specific complex that binds an A/T-rich element important for smooth muscle myosin heavy chain gene expression. J. Biol. Chem. 1998, 273, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Blank, R.; Owens, G. Platelet-derived growth factor regulates actin isoform expression and growth state in cultured rat aortic smooth muscle cells. J. Cell. Physiol. 1990, 142, 635–642. [Google Scholar] [CrossRef]
- Sung, H.-J.; Eskin, S.G.; Sakurai, Y.; Yee, A.; Kataoka, N.; McIntire, L.V. Oxidative stress produced with cell migration increases synthetic phenotype of vascular smooth muscle cells. Ann. Biomed. Eng. 2005, 33, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Jin, Y.; Tang, W.H.; Qin, L.; Zhang, X.; Tellides, G.; Hwa, J.; Yu, J.; Martin, K.A. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 2013, 128, 2047–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Pons, D.; de Vries, F.R.; van den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Alkemade, F.E.; van Vliet, P.; Henneman, P.; van Dijk, K.W.; Hierck, B.P.; van Munsteren, J.C.; Scheerman, J.A.; Goeman, J.J.; Havekes, L.M.; Gittenberger-de Groot, A.C. Prenatal exposure to apoE deficiency and postnatal hypercholesterolemia are associated with altered cell-specific lysine methyltransferase and histone methylation patterns in the vasculature. Am. J. Pathol. 2010, 176, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Bishop, T.; Ratcliffe, P.J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 2015, 117, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Hypoxia-inducible histone lysine demethylases: impact on the aging process and age-related diseases. Aging Dis. 2016, 7, 180. [Google Scholar]
- Laukkanen, M.O.; Mannermaa, S.; Hiltunen, M.O.; Aittomäki, S.; Airenne, K.; Jänne, J.; Ylä-Herttuala, S. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Doñate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Owens, G.K. Programming smooth muscle plasticity with chromatin dynamics. Circ. Res. 2007, 100, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H. Reactive oxygen species promote vascular smooth muscle cell proliferation. Circ. Res. 2013, 113, 1040–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stampfer, M.J.; Hennekens, C.H.; Manson, J.E.; Colditz, G.A.; Rosner, B.; Willett, W.C. Vitamin E consumption and the risk of coronary disease in women. New Engl. J. Med. 1993, 328, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: the Established Populations for Epidemiologic Studies of the Elderly. Am. J. Clin. Nutr. 1996, 64, 190–196. [Google Scholar] [CrossRef]
- Marques, B.; Trindade, M.; Aquino, J.; Cunha, A.; Gismondi, R.; Neves, M.; Oigman, W. Beneficial effects of acute trans-resveratrol supplementation in treated hypertensive patients with endothelial dysfunction. Clin. Exp. Hypertens. 2018, 40, 218–223. [Google Scholar] [CrossRef]
- Dyck, G.J.; Raj, P.; Zieroth, S.; Dyck, J.R.; Ezekowitz, J.A. The effects of resveratrol in patients with cardiovascular disease and heart failure: a narrative review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, E.R.; Baron, J.A.; Karagas, M.R.; Stukel, T.A.; Nierenberg, D.W.; Stevens, M.M.; Mandel, J.S.; Haile, R.W. Mortality associated with low plasma concentration of beta carotene and the effect of oral supplementation. Jama 1996, 275, 699–703. [Google Scholar] [CrossRef]
- Rapola, J.M.; Virtamo, J.; Ripatti, S.; Huttunen, J.K.; Albanes, D.; Taylor, P.R.; Heinonen, O.P. Randomised trial of α-tocopherol and β-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. Lancet 1997, 349, 1715–1720. [Google Scholar] [CrossRef] [Green Version]
- Eidelman, R.S.; Hollar, D.; Hebert, P.R.; Lamas, G.A.; Hennekens, C.H. Randomized trials of vitamin E in the treatment and prevention of cardiovascular disease. Arch. Intern. Med. 2004, 164, 1552–1556. [Google Scholar] [CrossRef] [Green Version]
- Stephens, N. Randomised controlled trial of vitamin E in patients with coronary disease: Cambrige Heart Antioxidant Study (CHAOS). Lancet 1996, 347, 408–416. [Google Scholar] [CrossRef]
- Stoner, G.D.; Sardo, C.; Apseloff, G.; Mullet, D.; Wargo, W.; Pound, V.; Singh, A.; Sanders, J.; Aziz, R.; Casto, B. Pharmacokinetics of anthocyanins and ellagic acid in healthy volunteers fed freeze-dried black raspberries daily for 7 days. J. Clin. Pharmacol. 2005, 45, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, D. Antioxidants and cardiovascular risk factors. Diseases 2016, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Griffiths, H.; Lunec, J. Ascorbic acid in the 21st century–more than a simple antioxidant. Environ. Toxicol. Pharmacol. 2001, 10, 173–182. [Google Scholar] [CrossRef]
- Duarte, T.L.; Lunec, J. When is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic. Res. 2005, 39, 671–686. [Google Scholar] [CrossRef]
- Goszcz, K.; Deakin, S.J.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Antioxidants in cardiovascular therapy: panacea or false hope? Front. Cardiovasc. Med. 2015, 2, 29. [Google Scholar] [CrossRef]
- Bailey, S.R.; Mitra, S.; Flavahan, S.; Flavahan, N.A. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H243–H250. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badran, A.; Nasser, S.A.; Mesmar, J.; El-Yazbi, A.F.; Bitto, A.; Fardoun, M.M.; Baydoun, E.; Eid, A.H. Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 8764. https://doi.org/10.3390/ijms21228764
Badran A, Nasser SA, Mesmar J, El-Yazbi AF, Bitto A, Fardoun MM, Baydoun E, Eid AH. Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2020; 21(22):8764. https://doi.org/10.3390/ijms21228764
Chicago/Turabian StyleBadran, Adnan, Suzanne A. Nasser, Joelle Mesmar, Ahmed F. El-Yazbi, Alessandra Bitto, Manal M. Fardoun, Elias Baydoun, and Ali H. Eid. 2020. "Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 21, no. 22: 8764. https://doi.org/10.3390/ijms21228764
APA StyleBadran, A., Nasser, S. A., Mesmar, J., El-Yazbi, A. F., Bitto, A., Fardoun, M. M., Baydoun, E., & Eid, A. H. (2020). Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells. International Journal of Molecular Sciences, 21(22), 8764. https://doi.org/10.3390/ijms21228764