The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-yl)(phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies

 , , ,
, , ,
Abstract
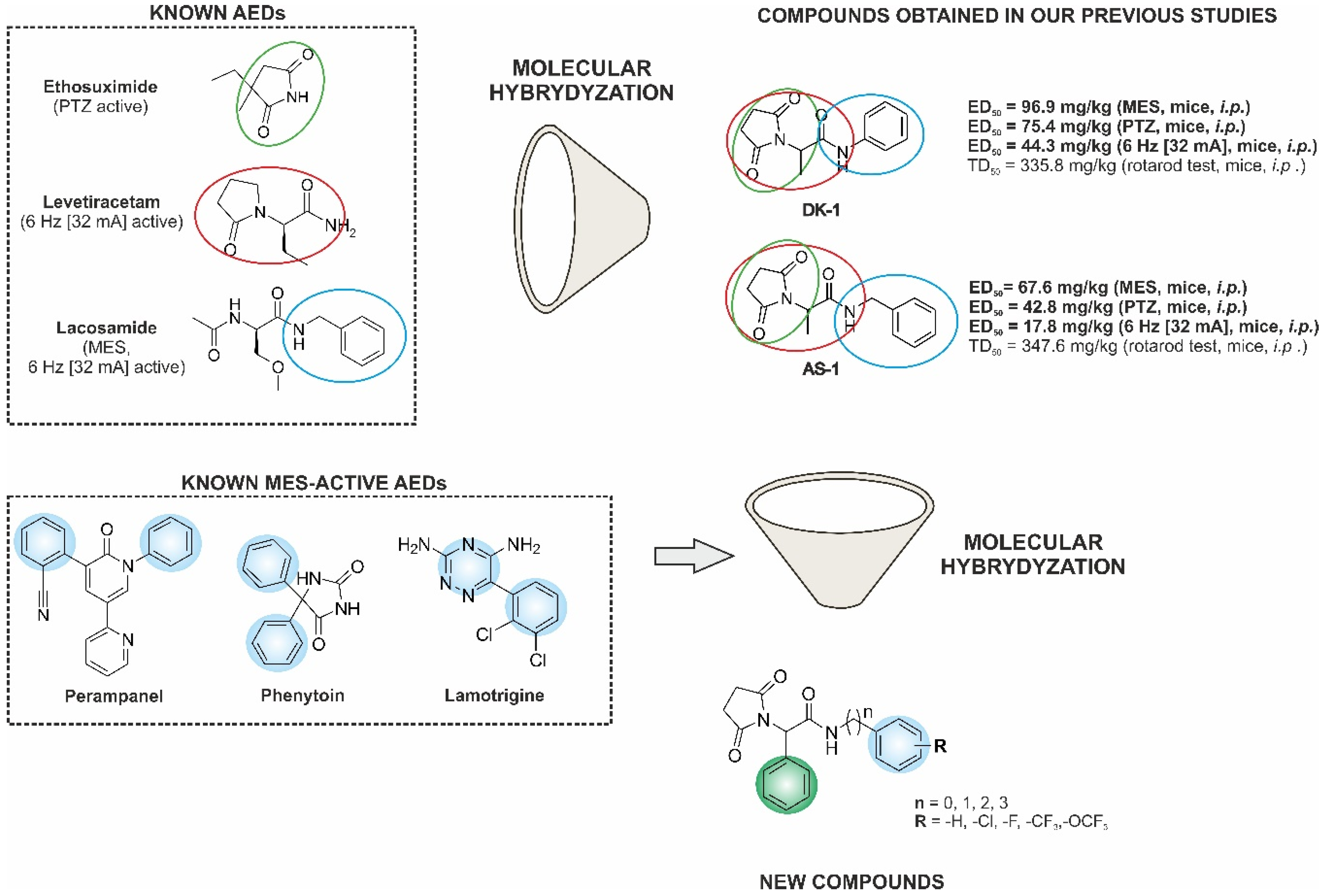
:1. Introduction
2. Results and Discussion
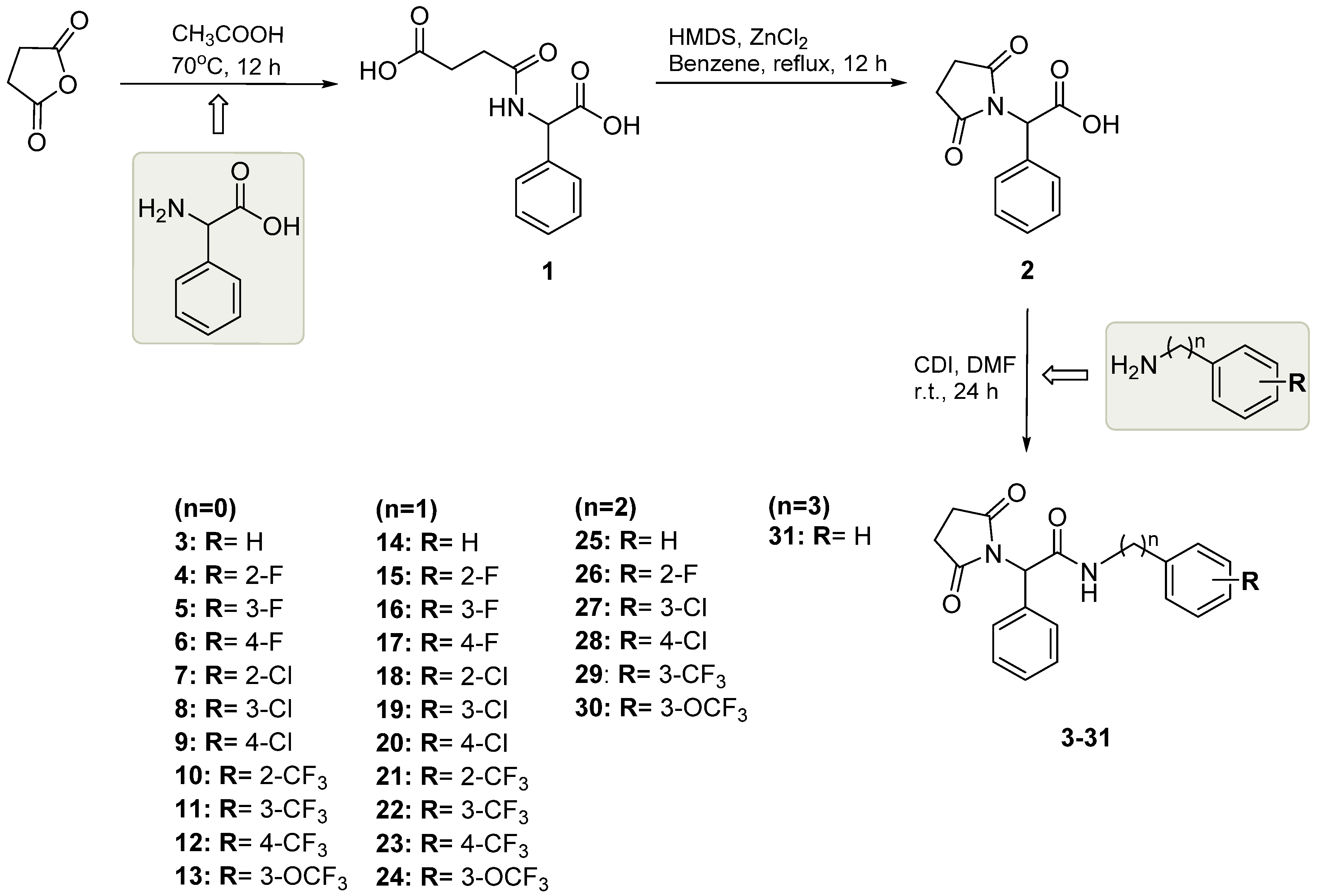
2.1. Chemistry
2.2. Anticonvulsant Activity
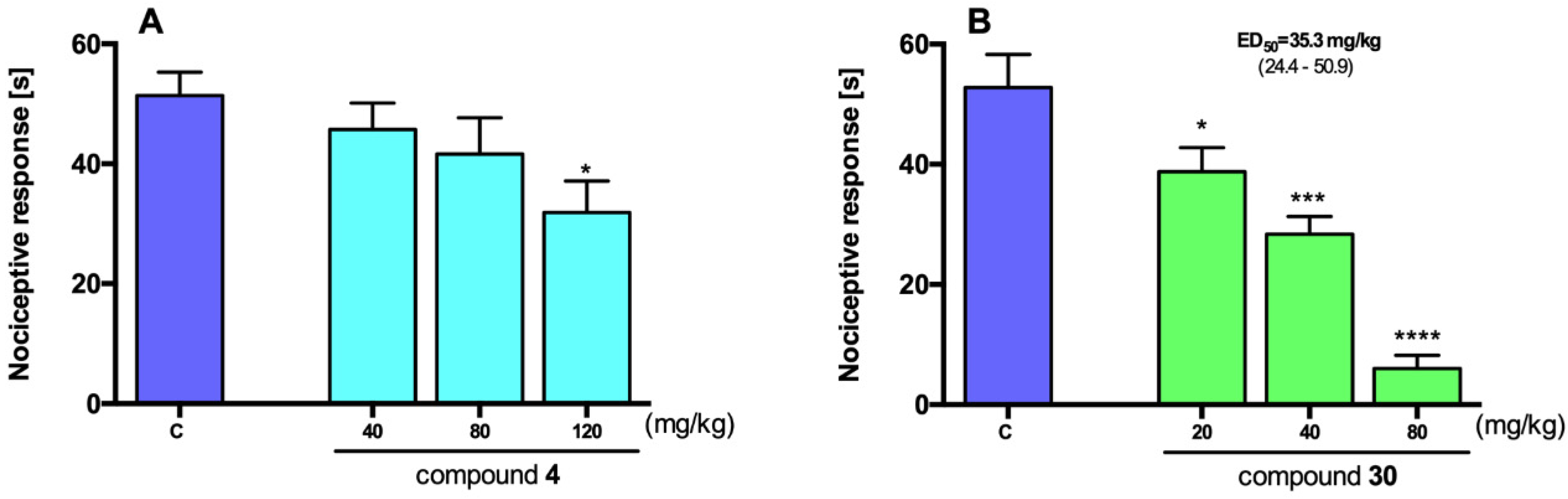
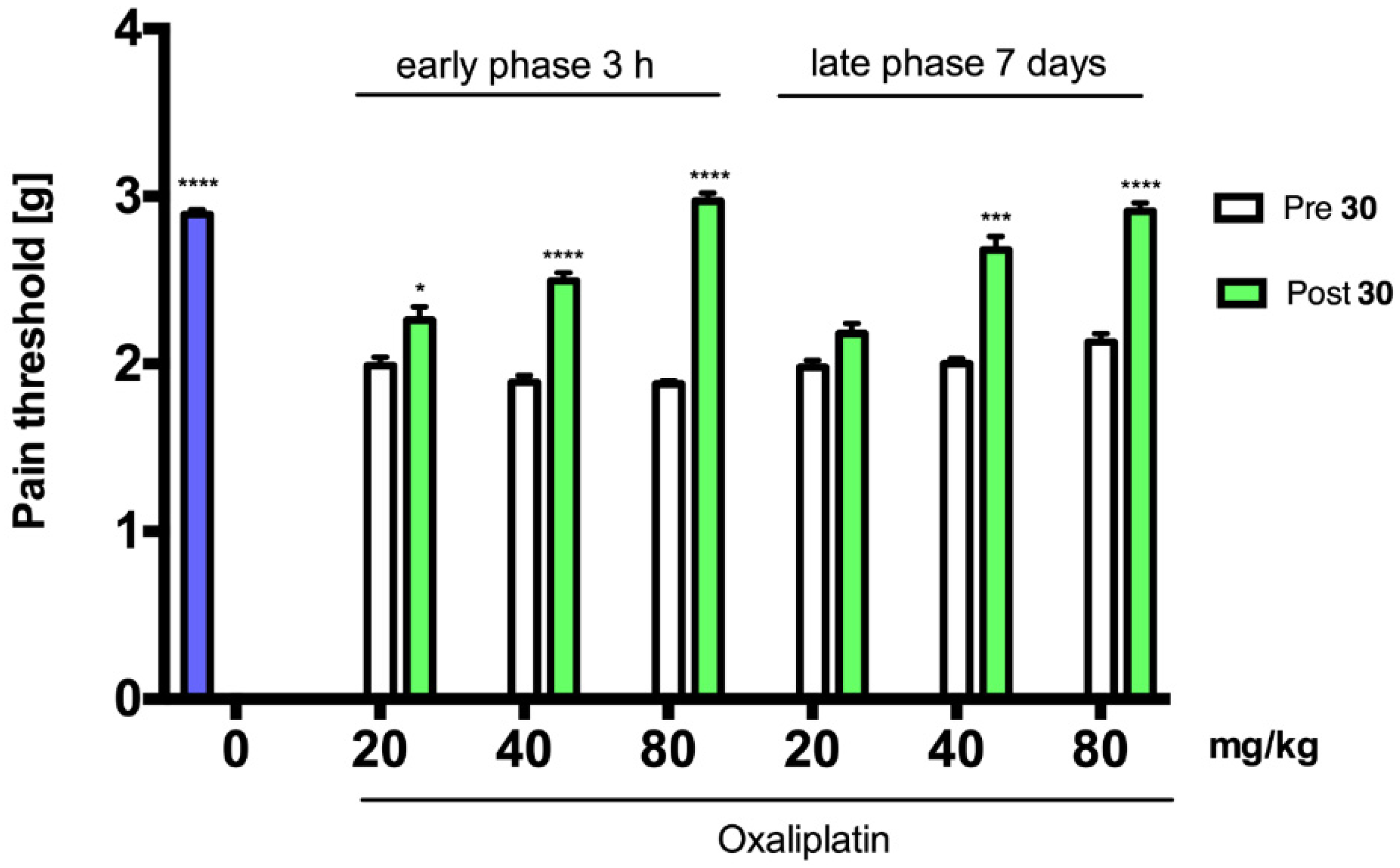
2.3. Antinociceptive Activity
2.4. In Vitro Radioligand Binding Studies and Functional Assays
2.5. In Vitro ADME-Tox Assays
2.5.1. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.5.2. Metabolic Stability
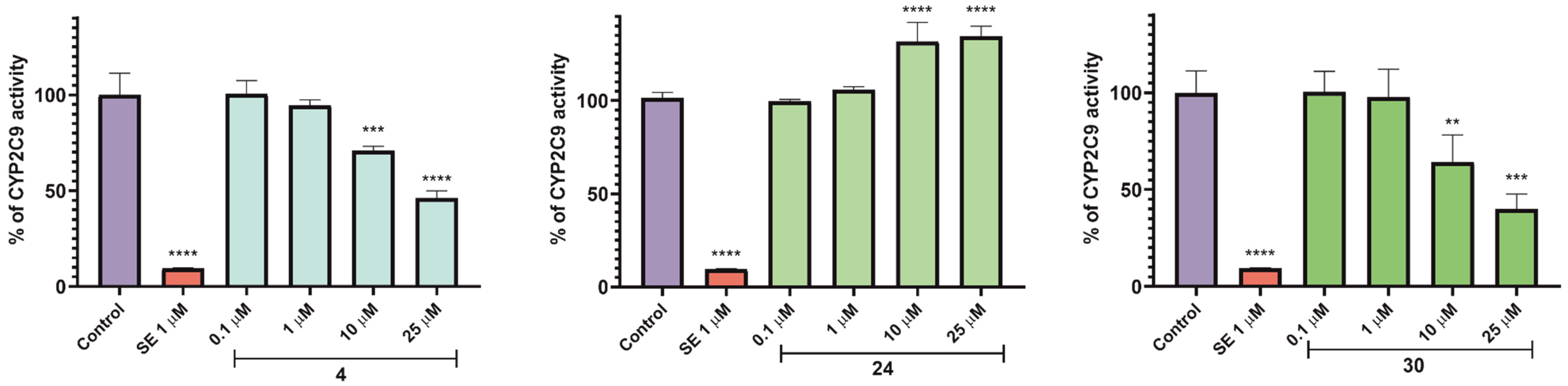
2.5.3. Influence on Function of Cytochrome P450 Isoforms—CYP3A4, CYP2D6, and CYP2C9
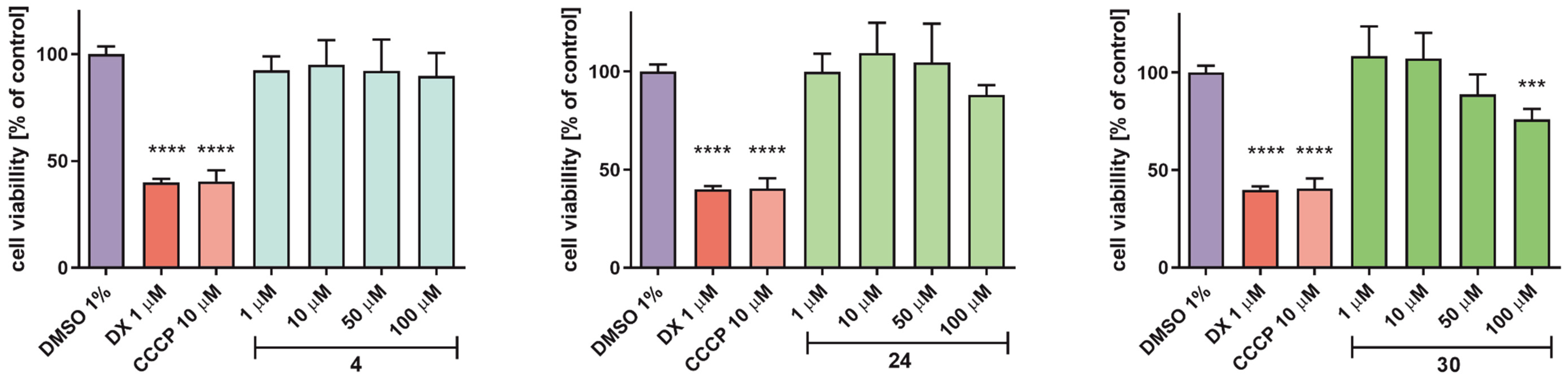
2.5.4. Determination of Hepatotoxicity Risk
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthetic Procedure for Succinamic Acid (1)
4-((Carboxy(phenyl)methyl)amino)-4-oxobutanoic Acid (1)
3.1.2. Synthetic Procedure for Monocarboxylic Acid (2)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenylacetic Acid (2)
3.1.3. Synthetic Procedure for Target Compounds (3–31)
2-(2,5-Dioxopyrrolidin-1-yl)-N,2-diphenylacetamide (3)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(2-fluorophenyl)-2-phenylacetamide (4)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(3-fluorophenyl)-2-phenylacetamide (5)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(4-fluorophenyl)-2-phenylacetamide (6)
N-(2-Chlorophenyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (7)
N-(3-Chlorophenyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (8)
N-(4-Chlorophenyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (9)
(2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(2-(trifluoromethyl)phenyl)acetamide) (10)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethyl)phenyl)acetamide (11)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(4-(trifluoromethyl)phenyl)acetamide (12)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethoxy)phenyl)acetamide (13)
N-Benzyl-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (14)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(2-fluorobenzyl)-2-phenylacetamide (15)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(3-fluorobenzyl)-2-phenylacetamide (16)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(4-fluorobenzyl)-2-phenylacetamide (17)
N-(2-Chlorobenzyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (18)
N-(3-Chlorobenzyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (19)
N-(4-Chlorobenzyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (20)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(2-(trifluoromethyl)benzyl)acetamide (21)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethyl)benzyl)acetamide (22)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(4-(trifluoromethyl)benzyl)acetamide (23)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethoxy)benzyl)acetamide (24)
2-(2,5-Dioxopyrrolidin-1-yl)-N-phenethyl-2-phenylacetamide (25)
2-(2,5-Dioxopyrrolidin-1-yl)-N-(2-fluorophenethyl)-2-phenylacetamide (26)
N-(3-Chlorophenethyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (27)
N-(4-Chlorophenethyl)-2-(2,5-dioxopyrrolidin-1-yl)-2-phenylacetamide (28)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethyl)phenethyl)acetamide (29)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-(trifluoromethoxy)phenethyl)acetamide (30)
2-(2,5-Dioxopyrrolidin-1-yl)-2-phenyl-N-(3-phenylpropyl)acetamide (31)
3.2. Anticonvulsant Activity
3.3. Antinociceptive Activity
3.4. In Vivo Data Analysis
3.4.1. Anticonvulsant Activity and Neurotoxicity Studies
3.4.2. Antinociceptive Activity Studies
3.5. In Vitro ADME-Tox Studies
3.5.1. Permeability
3.5.2. Metabolic Stability
3.5.3. Influence on Recombinant Human CYP3A4, CYP2D6, and CYP2C9 Cytochrome P450 Isoforms
3.5.4. Hepatotoxicity Assessment
3.6. In Silico Studies
3.7. In Vitro Binding and Functional Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADME-Tox | Absorption, distribution, metabolism, excretion, toxicity |
| AEDs | Antiepileptic drugs |
| CCCP | 3-Chlorophenylhydrazone |
| CDI | Carbonyldiimidazole |
| CFN | Caffeine |
| CNS | Central nervous system |
| DCM | Dichloromethane |
| DMF | Dimethylformamide |
| DX | Doxorubicin |
| HLMs | Human liver microsomes |
| HMDS | Hexamethyldisilazane |
| 6 Hz | 6 Hz seizure test |
| KE | Ketoconazole |
| LCS | Lacosamide |
| LiAlH4 | Lithium aluminum hydride |
| LTG | Lamotrigine |
| Lacosamide | Acetonitrile |
| MES | Maximal electroshock seizure test |
| MeOH | Methanol |
| NFX | Norfloxacin |
| OXPT | Oxaliplatin |
| PAMPA | Parallel artificial membrane permeability assay |
| PI | Protective index (TD50/ED50) |
| QD | Quinidine |
| SE | Sulfaphenazole |
| THF | Tetrahydrofuran |
| TPE | Time of peak effect |
| VPA | Valproic acid |
References
- Golyala, A.; Kwan, P. Drug development for refractory epilepsy: The past 25 years and beyond. Seizure 2017, 44, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Media Centre. Epilepsy. Fact Sheet No. 999. May 2015. Available online: http://www.who.int/mediacentre/factsheets/fs999/en/ (accessed on 19 November 2020).
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [PubMed]
- Salpekar, J.A.; Mula, M. Common psychiatric comorbidities in epilepsy: How big of a problem is it? Epilepsy Behav. 2019, 98, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Klitgaard, H.; Twyman, R.E.; Schmidt, D. New avenues for anti-epileptic drug discovery and development. Nat. Rev. Drug Discov. 2013, 12, 757–776. [Google Scholar] [CrossRef]
- Petrelli, A.; Valabrega, G. Multitarget drugs: The present and the future of cancer therapy. Expert Opin. Pharmacother. 2009, 10, 589–600. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Kupershmidt, L.; Amit, T.; Weinreb, O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, S132–S136. [Google Scholar] [CrossRef]
- Simone Tranches Dias, K.; Viegas, C. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Abram, M.; Zagaja, M.; Mogilski, S.; Andres-Mach, M.; Latacz, G.; Baś, S.; Łuszczki, J.J.; Kieć-Kononowicz, K.; Kamiński, K. Multifunctional Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-yl)-3-methoxypropanamides with Anticonvulsant and Antinociceptive Properties. J. Med. Chem. 2017, 60, 8565–8579. [Google Scholar] [CrossRef]
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Kamiński, K.; Rapacz, A.; Łuszczki, J.J.; Latacz, G.; Obniska, J.; Kieć-Kononowicz, K.; Filipek, B. Design, synthesis and biological evaluation of new hybrid anticonvulsants derived from N-benzyl-2-(2,5-dioxopyrrolidin-1-yl)propanamide and 2-(2,5-dioxopyrrolidin-1-yl)butanamide derivatives. Bioorg. Med. Chem. 2015, 23, 2548–2561. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Rapacz, A.; Filipek, B.; Obniska, J. Design, synthesis and anticonvulsant activity of new hybrid compounds derived from N-phenyl-2-(2,5-dioxopyrrolidin-1-yl)-propanamides and -butanamides. Bioorg. Med. Chem. 2016, 24, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Zagaja, M.; Łuszczki, J.J.; Rapacz, A.; Andres-Mach, M.; Latacz, G.; Kieć-Kononowicz, K. Design, Synthesis, and Anticonvulsant Activity of New Hybrid Compounds Derived from 2-(2,5-Dioxopyrrolidin-1-yl)propanamides and 2-(2,5-Dioxopyrrolidin-1-yl)butanamides. J. Med. Chem. 2015, 58, 5274–5286. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Lubelska, A.; Kamiński, R.M.; Kamiński, K. Multitargeted Compounds Derived from (2,5-Dioxopyrrolidin-1-yl)(phenyl)-Acetamides as Candidates for Effective Anticonvulsant and Antinociceptive Agents. ACS Chem. Neurosci. 2020, 11, 1996–2008. [Google Scholar] [CrossRef]
- Mäkinen, J.; Rainesalo, S.; Raitanen, J.; Peltola, J. The effect of newer antiepileptic drugs in combination therapy. Epilepsy Res. 2017, 132, 15–20. [Google Scholar] [CrossRef]
- Castel-Branco, M.M.; Alves, G.L.; Figueiredo, I.V.; Falcão, A.; Caramona, M.M. The maximal electroshock seizure (MES) model in the preclinical assessment of potential new antiepileptic drugs. Methods Find. Exp. Clin. Pharmacol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.Y.; Kondo, S.; Toru, T.; Ueno, Y. Lewis Acid and Hexamethyldisilazane-Promoted Efficient Synthesis of N-Alkyl- and N-Arylimide Derivatives. J. Org. Chem. 1997, 62, 2652–2654. [Google Scholar] [CrossRef]
- Galanopoulou, A.S.; Kokaia, M.; Loeb, J.A.; Nehlig, A.; Pitkänen, A.; Rogawski, M.A.; Staley, K.J.; Whittemore, V.H.; Dudek, F.E. Epilepsy therapy development: Technical and methodologic issues in studies with animal models. Epilepsia 2013, 54, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; Steve White, H. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, J.T.; Wilcoxon, F.A. Simplified Method of Evaluating Dose-Effect Experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar] [PubMed]
- Salinas-Abarca, A.B.; Avila-Rojas, S.H.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Granados-Soto, V. Formalin injection produces long-lasting hypersensitivity with characteristics of neuropathic pain. Eur. J. Pharmacol. 2017, 797, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Muley, M.M.; Krustev, E.; McDougall, J.J. Preclinical Assessment of Inflammatory Pain. CNS Neurosci. Ther. 2016, 22, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, T.M.; Tram, K.V.; Wilcox, G.L.; Birnbaum, A.K. Comparison of Antiepileptic Drugs Tiagabine, Lamotrigine, and Gabapentin in Mouse Models of Acute, Prolonged, and Chronic Nociception. J. Pharmacol. Exp. Ther. 2002, 302, 1168–1175. [Google Scholar] [CrossRef] [Green Version]
- Yashpal, K.; Fisher, K.; Chabot, J.-G.; Coderre, T.J. Differential effects of NMDA and group I mGluR antagonists on both nociception and spinal cord protein kinase C translocation in the formalin test and a model of neuropathic pain in rats. Pain 2001, 94, 17–29. [Google Scholar] [CrossRef]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef]
- Roca-Lapirot, O.; Radwani, H.; Aby, F.; Nagy, F.; Landry, M.; Fossat, P. Calcium signalling through L-type calcium channels: Role in pathophysiology of spinal nociceptive transmission. Br. J. Pharmacol. 2018, 175, 2362–2374. [Google Scholar] [CrossRef]
- Radwani, H.; Lopez-Gonzalez, M.J.; Cattaert, D.; Roca-Lapirot, O.; Dobremez, E.; Bouali-Benazzouz, R.; Eiríksdóttir, E.; Langel, Ü.; Favereaux, A.; Errami, M.; et al. Cav1.2 and Cav1.3 L-type calcium channels independently control short- and long-term sensitization to pain. J. Physiol. 2016, 594, 6607–6626. [Google Scholar] [CrossRef]
- Yaksh, T.L. Calcium Channels as Therapeutic Targets in Neuropathic Pain. J. Pain 2006, 7, S13–S30. [Google Scholar] [CrossRef]
- Perucca, E.; Meador, K.J. Adverse effects of antiepileptic drugs. Acta Neurol. Scand. 2005, 112, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, K.R.; Broekhuizen, K.; Groeneveld, G.J. Clinical trial simulations of the interaction between cannabidiol and clobazam and effect on drop-seizure frequency. Br. J. Clin. Pharmacol. 2020, 86, 380–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handoko, K.B.; Souverein, P.C.; Staa, T.P.V.; Meyboom, R.H.B.; Leufkens, H.G.M.; Egberts, T.C.G.; Bemt, P.M.L.A.V. Risk of Aplastic Anemia in Patients Using Antiepileptic Drugs. Epilepsia 2006, 47, 1232–1236. [Google Scholar] [CrossRef] [Green Version]
- Phillips, K.A.; Veenstra, D.L.; Oren, E.; Lee, J.K.; Sadee, W. Potential Role of Pharmacogenomics in Reducing Adverse Drug Reactions: A Systematic Review. JAMA 2001, 286, 2270–2279. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, K.; Socała, K.; Zagaja, M.; Andres-Mach, M.; Abram, M.; Jakubiec, M.; Pieróg, M.; Nieoczym, D.; Rapacz, A.; Gawel, K.; et al. N-Benzyl-(2,5-dioxopyrrolidin-1-yl)propanamide (AS-1) with Hybrid Structure as a Candidate for a Broad-Spectrum Antiepileptic Drug. Neurotherapeutics 2020, 17, 309–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiński, K.; Mogilski, S.; Abram, M.; Rapacz, A.; Latacz, G.; Szulczyk, B.; Walczak, M.; Kuś, K.; Matyjaszczyk, K.; Kamiński, R.M. KA-104, a new multitargeted anticonvulsant with potent antinociceptive activity in preclinical models. Epilepsia 2020, 61, 2119–2128. [Google Scholar] [CrossRef]
- Kehne, J.H.; Klein, B.D.; Raeissi, S.; Sharma, S. The National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem. Res. 2017, 42, 1894–1903. [Google Scholar] [CrossRef] [Green Version]
- Beirith, A.; Santos, A.R.S.; Rodrigues, A.L.S.; Creczynski-Pasa, T.B.; Calixto, J.B. Spinal and supraspinal antinociceptive action of dipyrone in formalin, capsaicin and glutamate tests. Study of the mechanism of action. Eur. J. Pharmacol. 1998, 345, 233–245. [Google Scholar] [CrossRef]
- Mogilski, S.; Kubacka, M.; Redzicka, A.; Kazek, G.; Dudek, M.; Malinka, W.; Filipek, B. Antinociceptive, anti-inflammatory and smooth muscle relaxant activities of the pyrrolo[3,4-d]pyridazinone derivatives: Possible mechanisms of action. Pharmacol. Biochem. Behav. 2015, 133, 99–110. [Google Scholar] [CrossRef]
- Sałat, K.; Cios, A.; Wyska, E.; Sałat, R.; Mogilski, S.; Filipek, B.; Więckowski, K.; Malawska, B. Antiallodynic and antihyperalgesic activity of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one compared to pregabalin in chemotherapy-induced neuropathic pain in mice. Pharmacol. Biochem. Behav. 2014, 122, 173–181. [Google Scholar] [CrossRef]
- Cali, J.J.; Ma, D.; Sobol, M.; Simpson, D.J.; Frackman, S.; Good, T.D.; Daily, W.J.; Liu, D. Luminogenic cytochrome P450 assays. Expert Opin. Drug Metab. Toxicol. 2006, 2, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P.; et al. KA-11, a Novel Pyrrolidine-2,5-dione Derived Broad-Spectrum Anticonvulsant: Its Antiepileptogenic, Antinociceptive Properties and in Vitro Characterization. ACS Chem. Neurosci. 2019, 10, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Carosati, E.; De Boeck, B.; Ethirajulu, K.; Mackie, C.; Howe, T.; Vianello, R. MetaSite: Understanding Metabolism in Human Cytochromes from the Perspective of the Chemist. J. Med. Chem. 2005, 48, 6970–6979. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TPE (h) a | ED50 MES (mg/kg) b | ED50 6 Hz (32 mA) (mg/kg) c | TD50 (rotarod) (mg/kg) d | PI (TD50/ED50) e |
|---|---|---|---|---|---|
| 3 | 0.5 | >130 | 92.5 (72.3–118.3) | >300 | >3.2 (6 Hz) |
| 4 | 0.5 | 73.2 (58.3–92.8) | – | >300 | >4.1 (MES) |
| 6 | 0.5 | 117.7 (92.6–149.6) | 57.6 (33.9–97.9) | >300 | >2.5 (MES) >5.2 (6 Hz) |
| 8 | 0.5 | 73.5 (59.3–90.4) | – | >300 | >4.1 (MES) |
| 9 | 0.5 | 105.8 (100.3–111.6) | – | >300 | >2.8 (MES) |
| 14 | 0.5 | >130 | 62.9 (45.7–86.7) | >300 | >4.7 (6 Hz) |
| 19 | 0.5 | 90.7 (82.6–99.5) | 89.9 (74.4–108.5) | >300 | >3.3 (MES) >3.3 (6 Hz) |
| 21 | 0.5 | 49.6 (44.3–55.7) | – | >300 | >6.0 (6 Hz) |
| 22 | 0.5 | >130 | 57.6 (33.9–97.9) | >300 | >5.2 (6 Hz) |
| 24 | 0.5 | >130 | 43.8 (33.9–53.8) | >300 | >6.8 (6 Hz) |
| 25 | 0.5 | 87.9 (82.0–94.3) | 88.9 (83.3–94.9) | 210.5 (195.0–227.1) | 2.4 (MES) 2.4 (6 Hz) |
| 27 | 0.5 | – | 36.3 (24.9– 53.1) | >300 | 8.3 (6 Hz) |
| 28 | 0.5 | 117.4 (108.4–127.3) | 88.1 (81.8–95.0) | 272.5 (253.2–293.3) | 2.3 (MES) 2.9 (6 Hz) |
| 29 | 0.5 | 55.4 (41.5–65.4) | 49.3 (38.0–62.1) | 178.8 (164.1–188.5) | 3.2 (MES) 3.6 (6 Hz) |
| 30 | 0.5 | 45.6 (35.8–58.2) | 39.5 (28.3–55.2) | 162.4 (144.0–183.1) | 3.5 (MES) 4.1 (6 Hz) |
| 31 | 0.5 | – | 103.8 (86.1–125.2) | >300 | 2.9 (6 Hz) |
| LCSf | 0.5 | 9.2 (8.5–10.0) | 5.3 (3.5–7.8) | 46.2 (44.5–48.0) | 5.0 (MES) 8.8 (6 Hz) |
| LTGf | 1.0 | 3.1 (1.9–4.7) | 6.4 (4.1–9.9) | 48.3 (37.4–62.6) | 15.6 (MES) 7.5 (6 Hz) |
| VPAf | 0.5 | 252.7 (220.1–290.2) | 130.6 (117.6–145.2) | 430.7 (407.9–454.9) | 1.7 (MES) 3.3 (6 Hz) |
| % Inhibition of Control Specific Binding a | |||||
|---|---|---|---|---|---|
| Assay | Na+ Channel (Site 2) (Antagonist Radioligand) *,# | Cav1.2 Channel (Antagonist Radioligand) * | Cav2.2 Channel (Antagonist Radioligand) * | GABA-A Receptor (α1,β2,γ2) (Agonist Radioligand) * | Potassium Channel (hERG) ** |
| Concentration (µM) | 10 | 10 | 10 | 10 | 100 |
| Compound | |||||
| 3 | 10.8 | 9.0 | 3.6 | 5.2 | 0.8 |
| 4 | 23.5 | 5.0 | 1.1 | 2.8 | 2.3 |
| 21 | 18.8 | 26.6 | 0.2 | 1.2 | 6.3 |
| 24 | 22.3 | 36.5 | 5.5 | −0.8 | 4.4 |
| 25 | 14.8 | 19.0 | 2.5 | 3.6 | 4.8 |
| 27 | 20.1 | 38.3 | 3.8 | 0.7 | 2.7 |
| 29 | 18.3 | 44.2 | −0.6 | 2.1 | 3.4 |
| 30 | 15.8 | 47.6 | 4.0 | 0.3 | 1.7 |
| Molecular Target | % Inhibition of Control Agonist Response a |
|---|---|
| Assay | Cav1.2 (h) Calcium Ion Channel Cell-Based Antagonist Calcium Flux Assay (Human Recombinant (HEK-293 Cells)) |
| Compound | |
| 21 | 27.0 |
| 24 | 38.8 |
| 27 | 40.0 |
| 29 | 45.0 |
| 30 | 49.0 |
| Compound | Pe (10−6 cm/s) ± SD a |
|---|---|
| CFN b | 12.22 ± 0.9 |
| NFX c | 0.056 ± 0.0 |
| 4 | 11.25 ± 0.7 |
| 24 | 10.87 ± 0.5 |
| 30 | 13.20 ± 1.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abram, M.; Jakubiec, M.; Rapacz, A.; Mogilski, S.; Latacz, G.; Kamiński, R.M.; Kamiński, K. The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-yl)(phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies. Int. J. Mol. Sci. 2020, 21, 8780. https://doi.org/10.3390/ijms21228780
Abram M, Jakubiec M, Rapacz A, Mogilski S, Latacz G, Kamiński RM, Kamiński K. The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-yl)(phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies. International Journal of Molecular Sciences. 2020; 21(22):8780. https://doi.org/10.3390/ijms21228780
Chicago/Turabian StyleAbram, Michał, Marcin Jakubiec, Anna Rapacz, Szczepan Mogilski, Gniewomir Latacz, Rafał M. Kamiński, and Krzysztof Kamiński. 2020. "The Search for New Anticonvulsants in a Group of (2,5-Dioxopyrrolidin-1-yl)(phenyl)Acetamides with Hybrid Structure—Synthesis and In Vivo/In Vitro Studies" International Journal of Molecular Sciences 21, no. 22: 8780. https://doi.org/10.3390/ijms21228780