Multi-Omics Integration Highlights the Role of Ubiquitination in CCl4-Induced Liver Fibrosis


 , , , , , , ,
, , , , , , ,  ,
, 
 , , , ,
, , , ,  and add
Show full author list
and add
Show full author list
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis: An Interplay between Injury, DNA Damage Response (DDR), and Regeneration
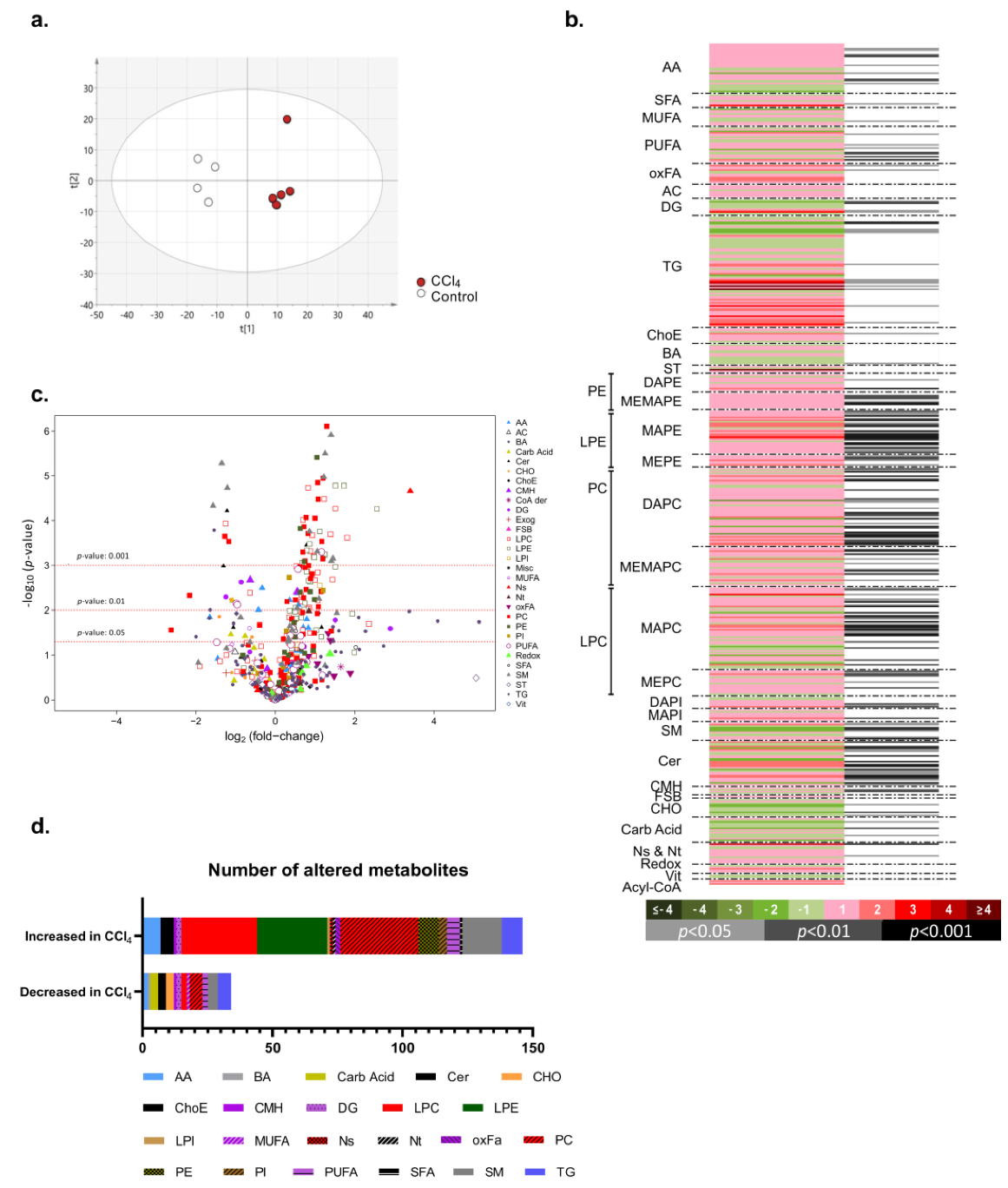
2.2. Metabolic Reprogramming in Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
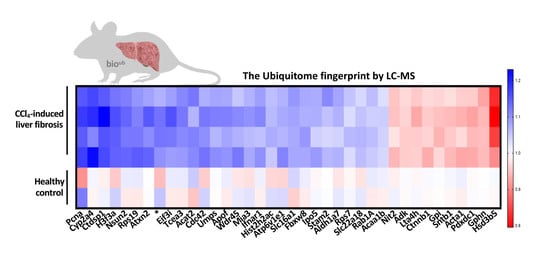
2.3. Ubiquitination Is Aberrant in Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
2.4. Characterization of the Ubiquitinated Proteome in Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
2.5. PCNA Ubiquitination in DNA Damage Repair (DDR) during Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
3. Discussion
4. Materials and Methods
4.1. Mouse Models
4.2. Carbon Tetrachloride (CCl4)-Induced Liver Fibrosis
4.3. Histology Analysis
4.4. Metabolomics Analysis by Liquid Chromatography-Mass Spectrometry (LC-MS) Analysis
4.5. Transcriptomics Analysis
4.6. Real-Time Polymerase-Chain Reactions (RT-PCR)
4.7. Pulldown Assay
4.8. Proteomics Analysis by LC–MS/MS Analysis
4.9. Western Blotting
4.10. HepG2 Cell Line Treatments
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Caballería, L.; Pera, G.; Arteaga, I.; Rodríguez, L.; Alumà, A.; Morillas, R.M.; De La Ossa, N.; Díaz, A.; Expósito, C.; Miranda, D.; et al. High Prevalence of Liver Fibrosis Among European Adults With Unknown Liver Disease: A Population-Based Study. Clin. Gastroenterol. Hepatol. 2018, 16, 1138–1145.e5. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachiondo-Ortega, S.; Mercado-Gómez, M.; Serrano-Macia, M.; Lopitz-Otsoa, F.; Salas-Villalobos, T.B.; Varela-Rey, M.; Delgado, T.C.; Martínez-Chantar, M.L. Ubiquitin-Like Post-Translational Modifications (Ubl-PTMs): Small Peptides with Huge Impact in Liver Fibrosis. Cells 2019, 8, 1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, M.; Yamauchi, M.; Toda, G.; Takada, K.; Hirakawa, T.; Ohkawa, K. Serum Ubiquitin Levels in Patients With Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 1999, 23, 76S–80S. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Shen, X.Z.; Zhou, C.H.; Wang, J.Y. Abnormal expression of Smurf2 during the process of rat liver fibrosis. Chin. J. Dig. Dis. 2006, 7, 237–245. [Google Scholar] [CrossRef]
- Zhang, T.; Kho, D.H.; Wang, Y.; Harazono, Y.; Nakajima, K.; Xie, Y.; Raz, A. Gp78, an E3 Ubiquitin Ligase Acts as a Gatekeeper Suppressing Nonalcoholic Steatohepatitis (NASH) and Liver Cancer. PLoS ONE 2015, 10, e0118448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.L.; Murphy, L.B.; Leslie, J.; Kendrick, S.; French, J.; Fox, C.R.; Sheerin, N.S.; Fisher, A.J.; Robinson, J.H.; Tiniakos, D.G.; et al. Ubiquitin C-terminal hydrolase 1: A novel functional marker for liver myofibroblasts and a therapeutic target in chronic liver disease. J. Hepatol. 2015, 63, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, D.; Fujii, R.; Yagishita, N.; Matsumoto, N.; Aratani, S.; Izumi, T.; Azakami, K.; Nakazawa, M.; Fujita, H.; Sato, T.; et al. E3 Ubiquitin Ligase Synoviolin Is Involved in Liver Fibrogenesis. PLoS ONE 2010, 5, e13590. [Google Scholar] [CrossRef]
- He, H.; Dai, J.; Feng, J.; He, Q.; Chen, X.; Dai, W.; Xu, A.; Huang, H. FBXO31 modulates activation of hepatic stellate cells and liver fibrogenesis by promoting ubiquitination of Smad7. J. Cell. Biochem. 2019, 121, 3711–3719. [Google Scholar] [CrossRef]
- Li, B.; Cong, M.; Zhu, Y.; Xiong, Y.; Jin, W.; Wan, Y.; Zhou, Y.; Ao, Y.; Wang, H. Indole-3-Carbinol Induces Apoptosis of Hepatic Stellate Cells through K63 De-Ubiquitination of RIP1 in Rats. Cell. Physiol. Biochem. 2017, 41, 1481–1490. [Google Scholar] [CrossRef]
- Lopitz-Otsoa, F.; Rodriguez-Suarez, E.; Aillet, F.; Casado-Vela, J.; Lang, V.; Matthiesen, R.; Elortza, F.; Rodriguez, M.S. Integrative analysis of the ubiquitin proteome isolated using Tandem Ubiquitin Binding Entities (TUBEs). J. Proteom. 2012, 75, 2998–3014. [Google Scholar] [CrossRef] [PubMed]
- Barbier-Torres, L.; Beraza, N.; Fernández-Tussy, P.; Lopitz-Otsoa, F.; Fernández-Ramos, D.; Zubiete-Franco, I.; Varela-Rey, M.; Delgado, T.C.; Gutiérrez, V.; Anguita, J.; et al. Histone deacetylase 4 promotes cholestatic liver injury in the absence of prohibitin-1. Hepatology 2015, 62, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martínez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin Profiling in Liver Using a Transgenic Mouse with Biotinylated Ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Kumagai, K.; Tabu, K.; Sasaki, F.; Takami, Y.; Morinaga, Y.; Mawatari, S.; Hashimoto, S.; Tanoue, S.; Kanmura, S.; Tamai, T.; et al. Glycoprotein Nonmetastatic Melanoma B (Gpnmb)-Positive Macrophages Contribute to the Balance between Fibrosis and Fibrolysis during the Repair of Acute Liver Injury in Mice. PLoS ONE 2015, 10, e0143413. [Google Scholar] [CrossRef] [Green Version]
- Zubiete-Franco, I.; Fernández-Tussy, P.; Barbier-Torres, L.; Simon, J.; Fernández-Ramos, D.; Lopitz-Otsoa, F.; Juan, V.G.; De Davalillo, S.L.; Duce, A.M.; Iruzubieta, P.; et al. Deregulated neddylation in liver fibrosis. Hepatology 2017, 65, 694–709. [Google Scholar] [CrossRef] [Green Version]
- Qu, A.; Shah, Y.M.; Matsubara, T.; Yang, Q.; Gonzalez, F.J. PPARα-Dependent Activation of Cell Cycle Control and DNA Repair Genes in Hepatic Nonparenchymal Cells. Toxicol. Sci. 2010, 118, 404–410. [Google Scholar] [CrossRef]
- Snel, B. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Chen, J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol. 2012, 19, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.-L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nat. Cell Biol. 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Olson, M.J. DNA strand breaks induced by hydrogen peroxide in isolated rat hepatocytes. J. Toxicol. Environ. Health Part A 1988, 23, 407–423. [Google Scholar] [CrossRef]
- Chang, H.; Meng, H.-Y.; Liu, S.-M.; Wang, Y.; Yang, X.-X.; Lu, F.; Wang, H.-Y. Identification of key metabolic changes during liver fibrosis progression in rats using a urine and serum metabolomics approach. Sci. Rep. 2017, 7, 11433. [Google Scholar] [CrossRef] [Green Version]
- Shea, B.S. Sphingolipid Regulation of Tissue Fibrosis. Open Rheumatol. J. 2012, 6, 123–129. [Google Scholar] [CrossRef]
- Tsugane, K.; Tamiya-Koizumi, K.; Nagino, M.; Nimura, Y.; Yoshida, S. A possible role of nuclear ceramide and sphingosine in hepatocyte apoptosis in rat liver. J. Hepatol. 1999, 31, 8–17. [Google Scholar] [CrossRef]
- Kumar, A.; Oskouian, B.; Fyrst, H.; Zhang, M.; Paris, F.; Saba, J.D. S1P lyase regulates DNA damage responses through a novel sphingolipid feedback mechanism. Cell Death Dis. 2011, 2, e119. [Google Scholar] [CrossRef] [Green Version]
- Carroll, B.; Donaldson, J.C.; Obeid, L.M. Sphingolipids in the DNA damage response. Adv. Biol. Regul. 2014, 58, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nat. Cell Biol. 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Von Stechow, L.; Typas, D.; Puigvert, J.C.; Oort, L.; Siddappa, R.; Pines, A.; Vrieling, H.; Van De Water, B.; Mullenders, L.H.F.; Danen, E.H.J. The E3 Ubiquitin Ligase ARIH1 Protects against Genotoxic Stress by Initiating a 4EHP-Mediated mRNA Translation Arrest. Mol. Cell. Biol. 2015, 35, 1254–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, Y.; Nakashima, R.; Matsuda, K.; Okamaoto, K.; Hirano, R.; Kawasaki, H.; Miuma, S.; Miyaaki, H.; Malhi, H.; Abiru, S.; et al. Detection of DNA damage response in nonalcoholic fatty liver disease via p53-binding protein 1 nuclear expression. Mod. Pathol. 2019, 32, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Borude, P.; Bhushan, B.; Apte, U. DNA Damage Response Regulates Initiation of Liver Regeneration Following Acetaminophen Overdose. Gene Expr. 2018, 18, 115–123. [Google Scholar] [CrossRef]
- Weake, V.M.; Workman, J.L. Histone Ubiquitination: Triggering Gene Activity. Mol. Cell 2008, 29, 653–663. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, X.; Rosenfeld, M.G. Histone H2A ubiquitination in transcriptional regulation and DNA damage repair. Int. J. Biochem. Cell Biol. 2009, 41, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Liu, J.; Cheruiyot, A.; Lee, J.-H.; Ordog, T.; Lou, Z.; You, Z.; Zhang, Z. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016, 30, 946–959. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, B.; Rezaeian, A.H.; Xu, X.; Chou, P.-C.; Jin, G.; Han, F.; Pan, B.-S.; Wang, C.-Y.; Long, J.; et al. H3 ubiquitination by NEDD4 regulates H3 acetylation and tumorigenesis. Nat. Commun. 2017, 8, 14799. [Google Scholar] [CrossRef] [Green Version]
- Simon, J.; Nuñez-García, M.; Fernández-Tussy, P.; Barbier-Torres, L.; Fernández-Ramos, D.; Gómez-Santos, B.; Buqué, X.; Lopitz-Otsoa, F.; Goikoetxea-Usandizaga, N.; Serrano-Macia, M.; et al. Targeting Hepatic Glutaminase 1 Ameliorates Non-alcoholic Steatohepatitis by Restoring Very-Low-Density Lipoprotein Triglyceride Assembly. Cell Metab. 2020, 31, 605–622.e10. [Google Scholar] [CrossRef]
- Barr, J.; Caballería, J.; Martínez-Arranz, I.; Domínguez-Díez, A.; Alonso, C.; Muntané, J.; Pérez-Cormenzana, M.; García-Monzón, C.; Mayo, R.; Martín-Duce, A.; et al. Obesity-Dependent Metabolic Signatures Associated with Nonalcoholic Fatty Liver Disease Progression. J. Proteome Res. 2012, 11, 2521–2532. [Google Scholar] [CrossRef]
- Martínez-Arranz, I.; Mayo, R.; Perez-Cormenzana, M.; Mincholé, I.; Salazar, L.; Alonso, C.; Mato, J.M. Enhancing metabolomics research through data mining. J. Proteom. 2015, 127, 275–288. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercado-Gómez, M.; Lopitz-Otsoa, F.; Azkargorta, M.; Serrano-Maciá, M.; Lachiondo-Ortega, S.; Goikoetxea-Usandizaga, N.; Rodríguez-Agudo, R.; Fernández-Ramos, D.; Bizkarguenaga, M.; Juan, V.G.-d.; et al. Multi-Omics Integration Highlights the Role of Ubiquitination in CCl4-Induced Liver Fibrosis. Int. J. Mol. Sci. 2020, 21, 9043. https://doi.org/10.3390/ijms21239043
Mercado-Gómez M, Lopitz-Otsoa F, Azkargorta M, Serrano-Maciá M, Lachiondo-Ortega S, Goikoetxea-Usandizaga N, Rodríguez-Agudo R, Fernández-Ramos D, Bizkarguenaga M, Juan VG-d, et al. Multi-Omics Integration Highlights the Role of Ubiquitination in CCl4-Induced Liver Fibrosis. International Journal of Molecular Sciences. 2020; 21(23):9043. https://doi.org/10.3390/ijms21239043
Chicago/Turabian StyleMercado-Gómez, Maria, Fernando Lopitz-Otsoa, Mikel Azkargorta, Marina Serrano-Maciá, Sofia Lachiondo-Ortega, Naroa Goikoetxea-Usandizaga, Rubén Rodríguez-Agudo, David Fernández-Ramos, Maider Bizkarguenaga, Virginia Gutiérrez-de Juan, and et al. 2020. "Multi-Omics Integration Highlights the Role of Ubiquitination in CCl4-Induced Liver Fibrosis" International Journal of Molecular Sciences 21, no. 23: 9043. https://doi.org/10.3390/ijms21239043
APA StyleMercado-Gómez, M., Lopitz-Otsoa, F., Azkargorta, M., Serrano-Maciá, M., Lachiondo-Ortega, S., Goikoetxea-Usandizaga, N., Rodríguez-Agudo, R., Fernández-Ramos, D., Bizkarguenaga, M., Juan, V. G.-d., Lectez, B., Aloria, K., Arizmendi, J. M., Simon, J., Alonso, C., Lozano, J. J., Avila, M. A., Banales, J. M., Marin, J. J. G., ... Delgado, T. C. (2020). Multi-Omics Integration Highlights the Role of Ubiquitination in CCl4-Induced Liver Fibrosis. International Journal of Molecular Sciences, 21(23), 9043. https://doi.org/10.3390/ijms21239043