The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells


Abstract
1. Introduction
2. Results
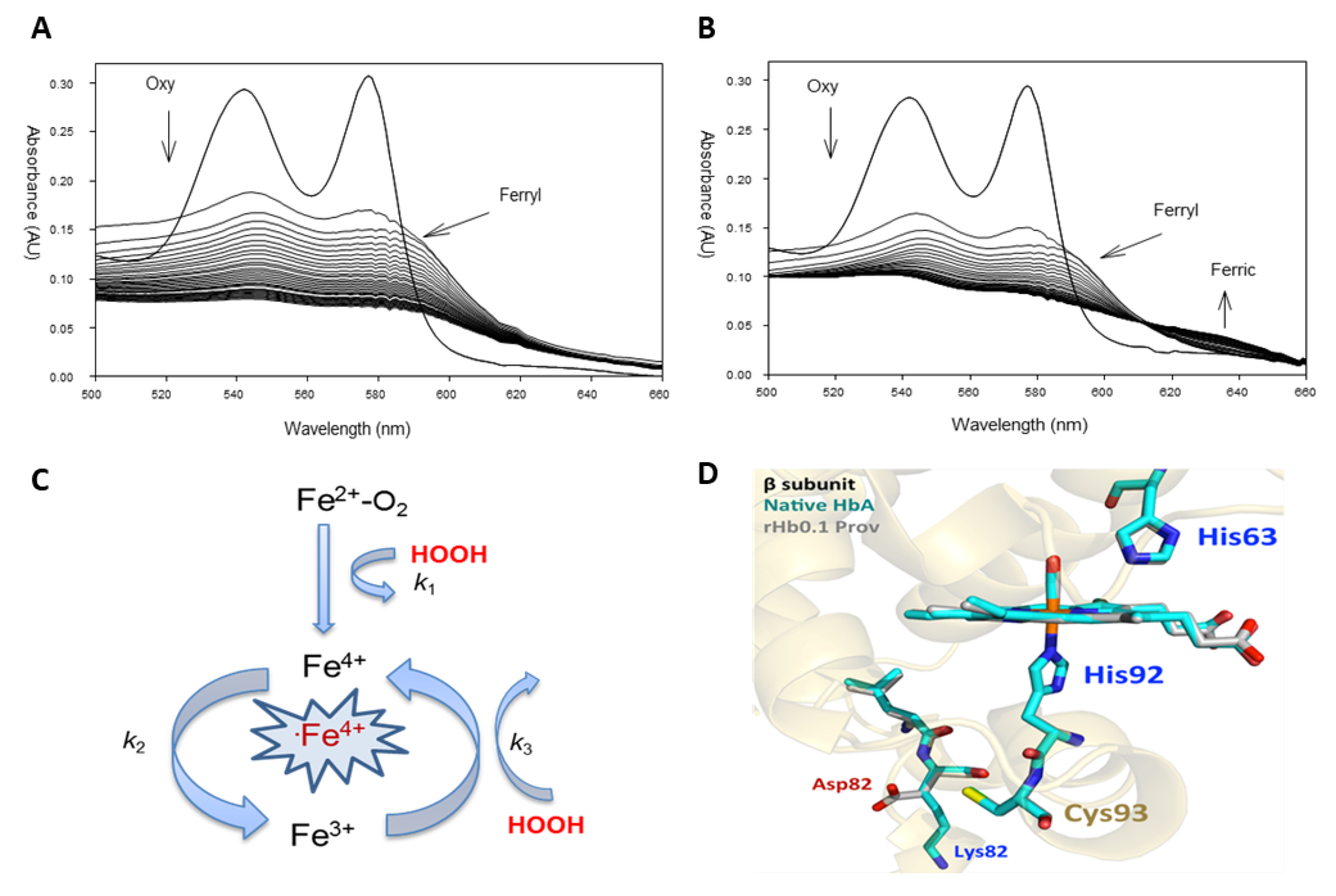
2.1. Pseudoperoxidase Activity of the Providence Mutation
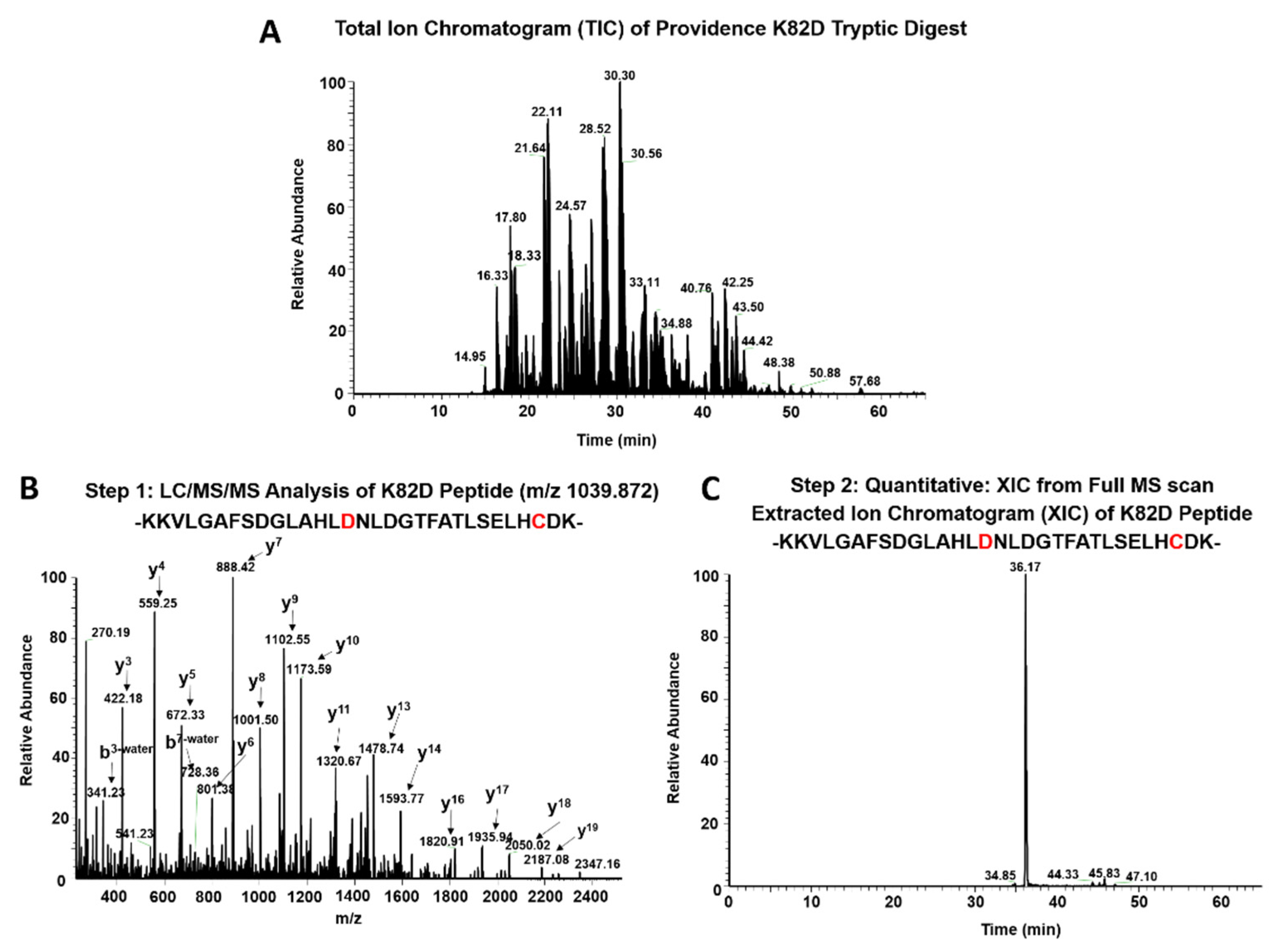
2.2. MS Analysis of βK82D Mutants Treated with Hydrogen Peroxide
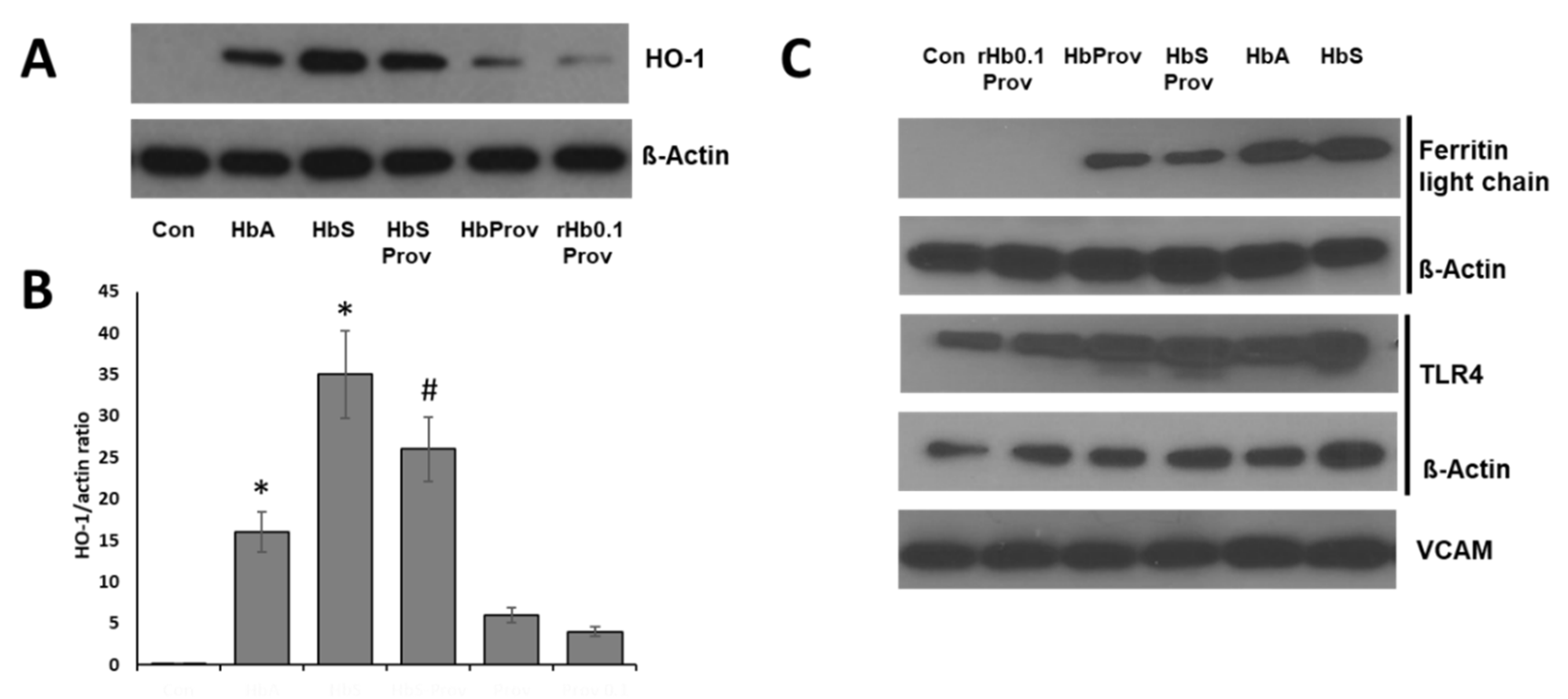
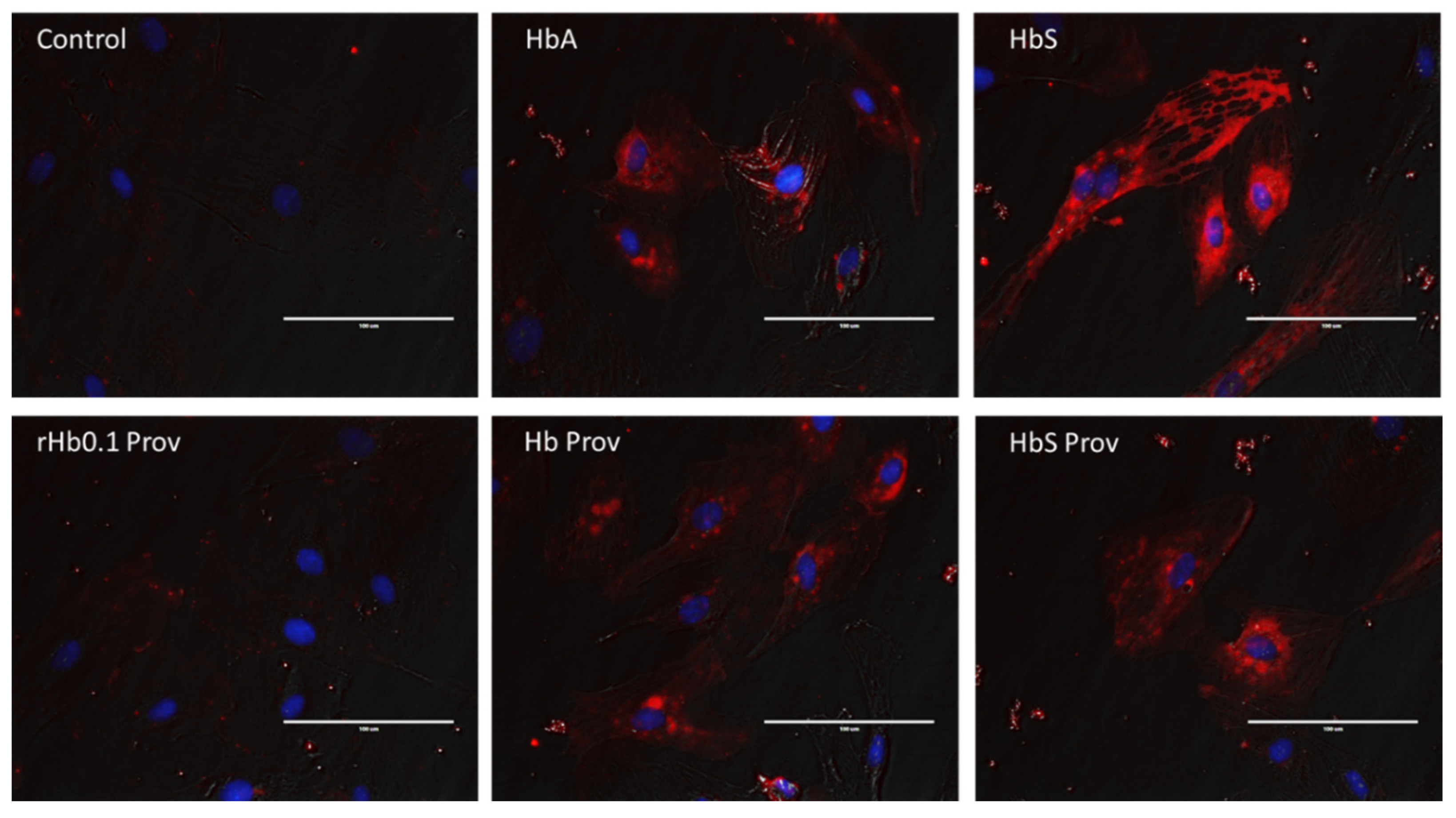
2.3. Endothelial Oxidative Stress Induced by Hemoglobin Mutants
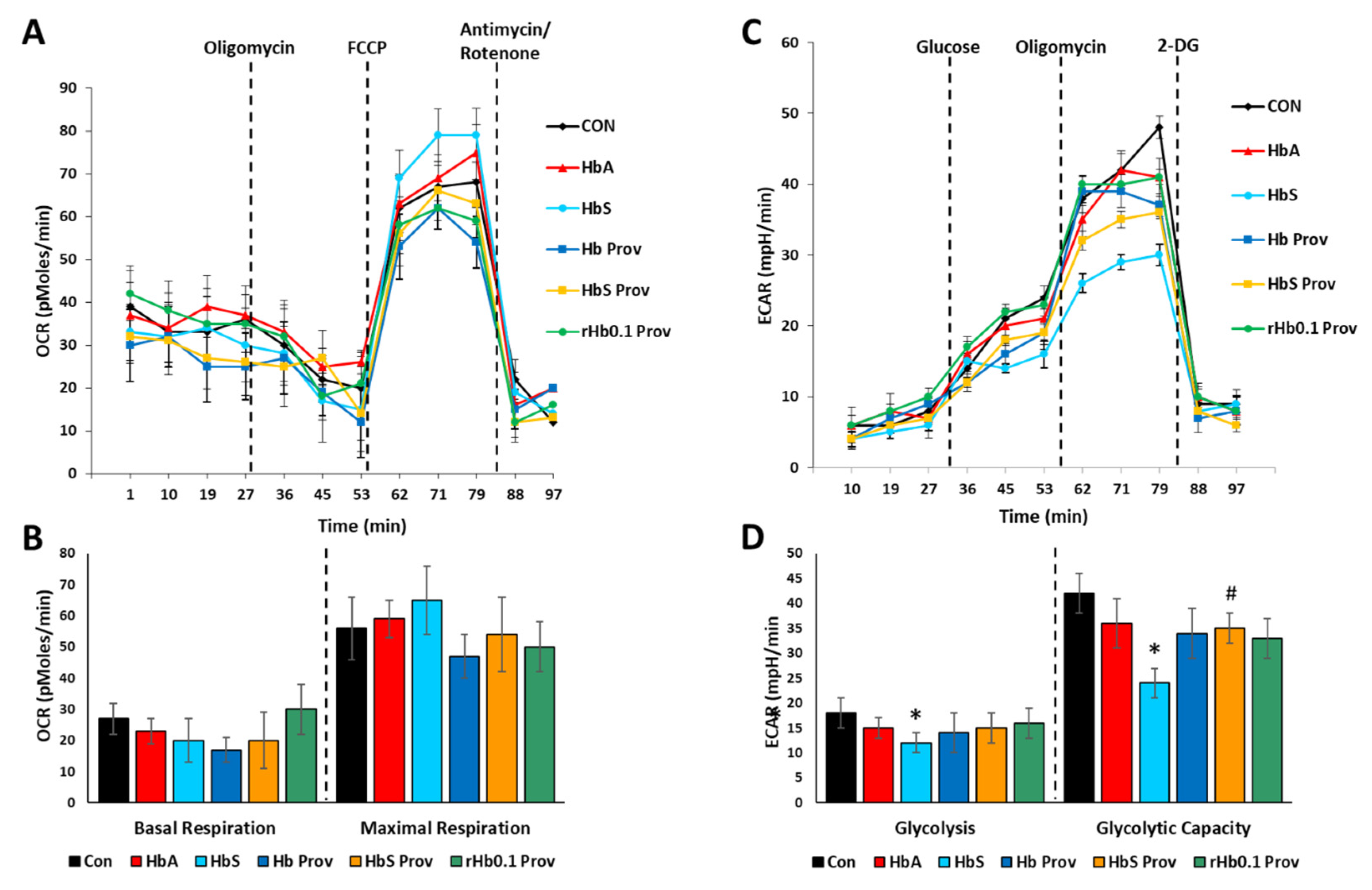
2.4. Impact of Providence Mutation on the Bioenergetic Impairment in HPAECs
3. Discussion
4. Materials and Methods
4.1. Recombinant Hemoglobins
4.2. Oxidation Reactions of Mutant Hemoglobins
4.3. Mass Spectrometry Analysis of Recombinant Hemoglobins
4.4. LC/MS/MS Analysis
4.5. Quantitative MS Analysis
4.6. Endothelial Cell Culture
4.7. Treatment of HPAECs with Hemoglobin Variants
4.8. Gel Electrophoresis and Immunoblotting and Fluorescence Microscopy
4.9. Endothelial Bioenergetic and Glycolytic Flux Measurements
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alayash, A.I. βCysteine 93 in human hemoglobin: A gateway to oxidative stability in health and disease. Lab. Investig. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shikama, K. A controversy on the mechanism of autoxidation of oxymyoglobin and oxyhaemoglobin: Oxidation, dissociation, or displacement? Biochem. J. 1984, 223, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Carrell, R.W. Oxidation of human haemoglobin by copper. Mechanism and suggested role of the thiol group of residue beta-93. Biochem. J. 1977, 165, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, V.; van der Meer, P.F.; de Korte, D.; Seghatchian, J.; Gutiérrez, L. Development of blood transfusion product pathogen reduction treatments: A review of methods, current applications and demands. Transfus. Apher. Sci. 2015, 52, 19–34. [Google Scholar] [CrossRef]
- Buehler, P.W.; Karnaukhova, E.; Gelderman, M.P.; Alayash, A.I. Blood aging, safety, and transfusion: Capturing the “radical” menace. Antioxid. Redox Signal. 2011, 14, 1713–1728. [Google Scholar] [CrossRef] [PubMed]
- Winslow, R.M. Red cell substitutes. Semin. Hematol. 2007, 44, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Estep, T.N. Issues in the development of hemoglobin based oxygen carriers. Semin. Hematol. 2019, 56, 257–261. [Google Scholar] [CrossRef]
- Alayash, A.I. Mechanisms of Toxicity and Modulation of Hemoglobin-Based Oxygen Carriers (HBOCs). Shock 2019, 52, 41–49. [Google Scholar] [CrossRef]
- Alayash, A.I. Oxygen therapeutics: Can we tame haemoglobin? Nat. Rev. Drug Discov. 2004, 3, 152–159. [Google Scholar] [CrossRef]
- Buehler, P.W.; D’Agnillo, F.; Hoffman, V.; Alayash, A.I. Effects of endogenous ascorbate on oxidation, oxygenation, and toxicokinetics of cell-free modified hemoglobin after exchange transfusion in rat and guinea pig. J. Pharmacol. Exp. Ther. 2007, 323, 49–60. [Google Scholar] [CrossRef]
- Fitzgerald, M.C.; Chan, J.Y.; Ross, A.W.; Liew, S.M.; Butt, W.W.; Baguley, D.; Salem, H.H.; Russ, M.K.; Deasy, C.; Martin, K.E.; et al. A synthetic haemoglobin-based oxygen carrier and the reversal of cardiac hypoxia secondary to severe anaemia following trauma. Med. J. Aust. 2011, 194, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Alayash, A.I. Blood substitutes: Why haven’t we been more successful? Trends Biotechnol. 2014, 32, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Buehler, P.W.; Boykins, R.A.; Venable, R.M.; Alayash, A.I. Structural basis of peroxide-mediated changes in human hemoglobin: A novel oxidative pathway. J. Biol. Chem. 2007, 282, 4894–4907. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.; Hicks, W.; Jia, Y.; Baek, J.H.; Miller, J.L.; Alayash, A.I. Isolated Hb Providence beta82Asn and beta82Asp fractions are more stable than native HbA(0) under oxidative stress conditions. Biochemistry 2011, 50, 9752–9766. [Google Scholar] [CrossRef]
- Strader, M.B.; Bangle, R.; Parker Siburt, C.J.; Varnado, C.L.; Soman, J.; Benitez Cardenas, A.S.; Samuel, P.S.; Singleton, E.W.; Crumbliss, A.L.; Olson, J.S.; et al. Engineering Oxidative Stability in Human Hemoglobin based on the Hb Providence (betaK82D) mutation and Genetic Crosslinking. Biochem. J. 2017. [Google Scholar] [CrossRef]
- Meng, F.; Kassa, T.; Strader, M.B.; Soman, J.; Olson, J.S.; Alayash, A.I. Substitutions in the β subunits of sickle-cell hemoglobin improve oxidative stability and increase the delay time of sickle-cell fiber formation. J. Biol. Chem. 2019, 294, 4145–4159. [Google Scholar] [CrossRef]
- Strader, M.B.; Alayash, A.I. Exploring Oxidative Reactions in Hemoglobin Variants Using Mass Spectrometry: Lessons for Engineering Oxidatively Stable Oxygen Therapeutics. Antioxid. Redox Signal. 2017, 26, 777–793. [Google Scholar] [CrossRef]
- Kassa, T.; Jana, S.; Strader, M.B.; Meng, F.; Jia, Y.; Wilson, M.T.; Alayash, A.I. Sickle Cell Hemoglobin in the Ferryl State Promotes betaCys-93 Oxidation and Mitochondrial Dysfunction in Epithelial Lung Cells (E10). J. Biol. Chem. 2015, 290, 27939–27958. [Google Scholar] [CrossRef]
- Jana, S.; Strader, M.B.; Meng, F.; Hicks, W.; Kassa, T.; Tarandovskiy, I.; De Paoli, S.; Simak, J.; Heaven, M.R.; Belcher, J.D.; et al. Hemoglobin oxidation-dependent reactions promote interactions with band 3 and oxidative changes in sickle cell-derived microparticles. JCI Insight 2018, 3, e120451. [Google Scholar] [CrossRef]
- Strader, M.B.; Jana, S.; Meng, F.; Heaven, M.R.; Shet, A.S.; Thein, S.L.; Alayash, A.I. Post-translational modification as a response to cellular stress induced by hemoglobin oxidation in sickle cell disease. Sci. Rep. 2020, 10, 14218. [Google Scholar] [CrossRef]
- Jana, S.; Meng, F.; Hirsch, R.E.; Friedman, J.M.; Alayash, A.I. Oxidized Mutant Human Hemoglobins S and E Induce Oxidative Stress and Bioenergetic Dysfunction in Human Pulmonary Endothelial Cells. Front. Physiol. 2017, 8, 1082. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Soares, M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 2014, 20, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Garrod, A. An address on the place of biochemistry in medicine. Br. Med. J. 1928, 1, 1099–1101. [Google Scholar] [CrossRef][Green Version]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin variants: Biochemical properties and clinical correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858. [Google Scholar] [CrossRef]
- Kassa, T.; Brad Strader, M.; Nakagawa, A.; Zapol, W.M.; Alayash, A.I. Targeting betaCys93 in hemoglobin S with an antisickling agent possessing dual allosteric and antioxidant effects. Metallomics 2017, 9, 1260–1270. [Google Scholar] [CrossRef]
- Pimenova, T.; Pereira, C.P.; Gehrig, P.; Buehler, P.W.; Schaer, D.J.; Zenobi, R. Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J. Proteome Res. 2010, 9, 4061–4070. [Google Scholar] [CrossRef]
- Kassa, T.; Wood, F.; Strader, M.B.; Alayash, A.I. Antisickling Drugs Targeting βCys93 Reduce Iron Oxidation and Oxidative Changes in Sickle Cell Hemoglobin. Front. Physiol. 2019, 10, 931. [Google Scholar] [CrossRef]
- Bonaventura, J.; Bonaventura, C.; Sullivan, B.; Ferruzzi, G.; McCurdy, P.R.; Fox, J.; Moo-Penn, W.F. Hemoglobin providence. Functional consequences of two alterations of the 2,3-diphosphoglycerate binding site at position beta 82. J. Biol. Chem. 1976, 251, 7563–7571. [Google Scholar] [PubMed]
- Charache, S.; Fox, J.; McCurdy, P.; Kazazian, H., Jr.; Winslow, R.; Hathaway, P.; van Beneden, R.; Jessop, M. Postsynthetic deamidation of hemoglobin Providence (beta 82 Lys replaced by Asn, Asp) and its effect on oxygen transport. J. Clin. Investig. 1977, 59, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.E.; Blair, N.E.; Huehns, E.R.; Clegg, J.B. Hb A-like sickle haemoglobin: Hb S-providence. Br. J. Haematol. 1988, 70, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, R.F.; Lima, E.S. Oxidative stress in sickle cell disease. Rev. Bras. Hematol. Hemoter. 2013, 35, 16–17. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Barst, R.J.; Castro, O.L.; Gordeuk, V.R.; Hillery, C.A.; Kato, G.J.; Kim-Shapiro, D.B.; Machado, R.; Morris, C.R.; Steinberg, M.H.; et al. Pulmonary hypertension and NO in sickle cell. Blood 2010, 116, 852–854. [Google Scholar] [CrossRef]
- Morris, C.R.; Vichinsky, E.P. Pulmonary hypertension in thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 205–213. [Google Scholar] [CrossRef]
- Davidson, S.M.; Duchen, M.R. Endothelial mitochondria: Contributing to vascular function and disease. Circ. Res. 2007, 100, 1128–1141. [Google Scholar] [CrossRef]
- Groschner, L.N.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Endothelial mitochondria--less respiration, more integration. Pflug. Arch. 2012, 464, 63–76. [Google Scholar] [CrossRef]
- Kaczara, P.; Motterlini, R.; Kus, K.; Zakrzewska, A.; Abramov, A.Y.; Chlopicki, S. Carbon monoxide shifts energetic metabolism from glycolysis to oxidative phosphorylation in endothelial cells. FEBS Lett. 2016, 590, 3469–3480. [Google Scholar] [CrossRef]
- Kaczara, P.; Motterlini, R.; Rosen, G.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef]
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015, 290, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, B.; Gallo, D.; Csizmadia, E.; Harris, C.; Belcher, J.; Vercellotti, G.M.; Penacho, N.; Seth, P.; Sukhatme, V.; Ahmed, A.; et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 2013, 73, 7009–7021. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Gori, T.; Münzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Kassa, T.; Jana, S.; Wood, F.; Zhang, X.; Jia, Y.; D’Agnillo, F.; Alayash, A.I. Comprehensive Biochemical and Biophysical Characterization of Hemoglobin-Based Oxygen Carrier Therapeutics: All HBOCs Are Not Created Equally. Bioconjugate Chem. 2018, 29, 1560–1575. [Google Scholar] [CrossRef]
- Looker, D.; Mathews, A.J.; Neway, J.O.; Stetler, G.L. Expression of recombinant human hemoglobin in Escherichia coli. Methods Enzymol. 1994, 231, 364–374. [Google Scholar]
- Meng, F.; Alayash, A.I. Determination of extinction coefficients of human hemoglobin in various redox states. Anal. Biochem. 2017, 521, 11–19. [Google Scholar] [CrossRef]
- Strader, M.B.; Hicks, W.A.; Kassa, T.; Singleton, E.; Soman, J.; Olson, J.S.; Weiss, M.J.; Mollan, T.L.; Wilson, M.T.; Alayash, A.I. Post-translational transformation of methionine to aspartate is catalyzed by heme iron and driven by peroxide: A novel subunit-specific mechanism in hemoglobin. J. Biol. Chem. 2014, 289, 22342–22357. [Google Scholar] [CrossRef]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Conditions HPAECs | Cys93 Oxidation | Cys93 Oxidation | Cys93 Oxidation |
|---|---|---|---|
| 2.5:1 | 5:1 | 10:1 | |
| (H2O2:heme) | (H2O2:heme) | (H2O2:heme) | |
| HbA [18] | 21 ± 4.2% | 31 ± 2.8% | 58 ± 3.8% |
| HbS (βE6V) [18] | 32.9 ± 2.7% | 64 ± 5.0% | 67% ± 6.8 |
| Hb Prov (βK82D) | 6.8 ± 3.1% | 11.6 ± 0.8% | 9.5 ± 4.3% |
| HbS Prov (βE6V/βK82D) [16] | 11.9 ± 2.0% | N/A | 22 ± 1.3% |
| Crosslinked Providence (rHb0.1 Prov) [15] | Below detection | Below detection | 2.7 ± 0.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jana, S.; Strader, M.B.; Alayash, A.I. The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 9453. https://doi.org/10.3390/ijms21249453
Jana S, Strader MB, Alayash AI. The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells. International Journal of Molecular Sciences. 2020; 21(24):9453. https://doi.org/10.3390/ijms21249453
Chicago/Turabian StyleJana, Sirsendu, Michael Brad Strader, and Abdu I. Alayash. 2020. "The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells" International Journal of Molecular Sciences 21, no. 24: 9453. https://doi.org/10.3390/ijms21249453
APA StyleJana, S., Strader, M. B., & Alayash, A. I. (2020). The Providence Mutation (βK82D) in Human Hemoglobin Substantially Reduces βCysteine 93 Oxidation and Oxidative Stress in Endothelial Cells. International Journal of Molecular Sciences, 21(24), 9453. https://doi.org/10.3390/ijms21249453