Acid Ceramidase Depletion Impairs Neuronal Survival and Induces Morphological Defects in Neurites Associated with Altered Gene Transcription and Sphingolipid Content


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
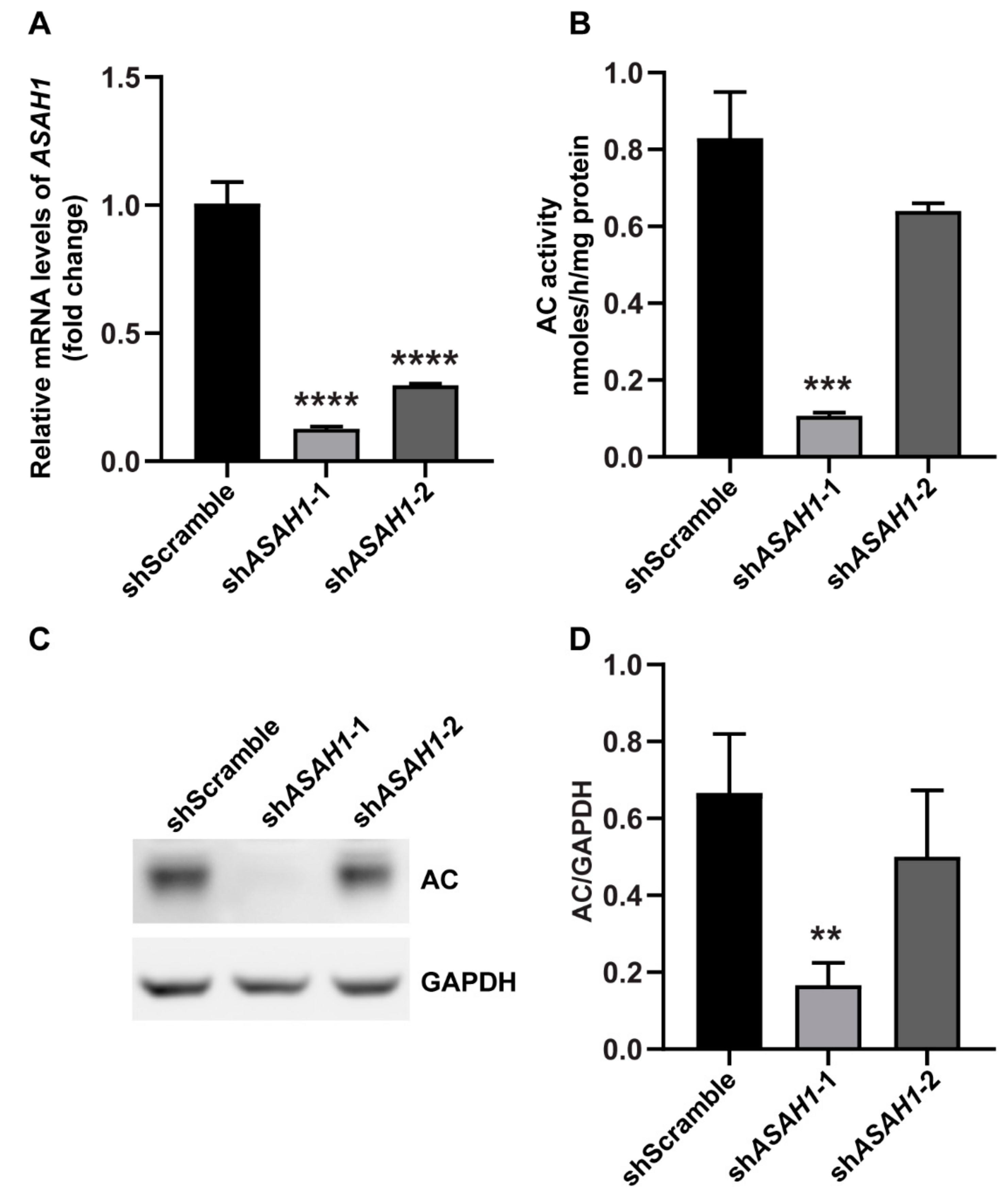
2.1. Establishment of Stable ASAH1 Knockdown Cell Lines
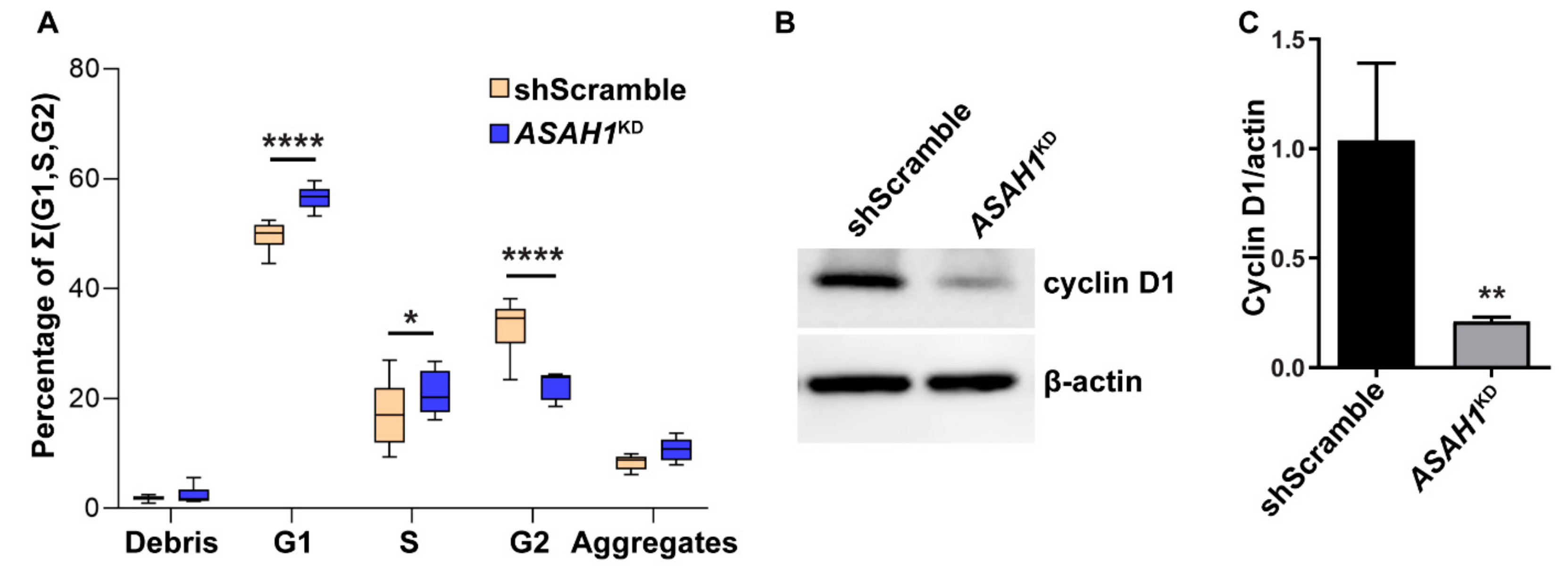
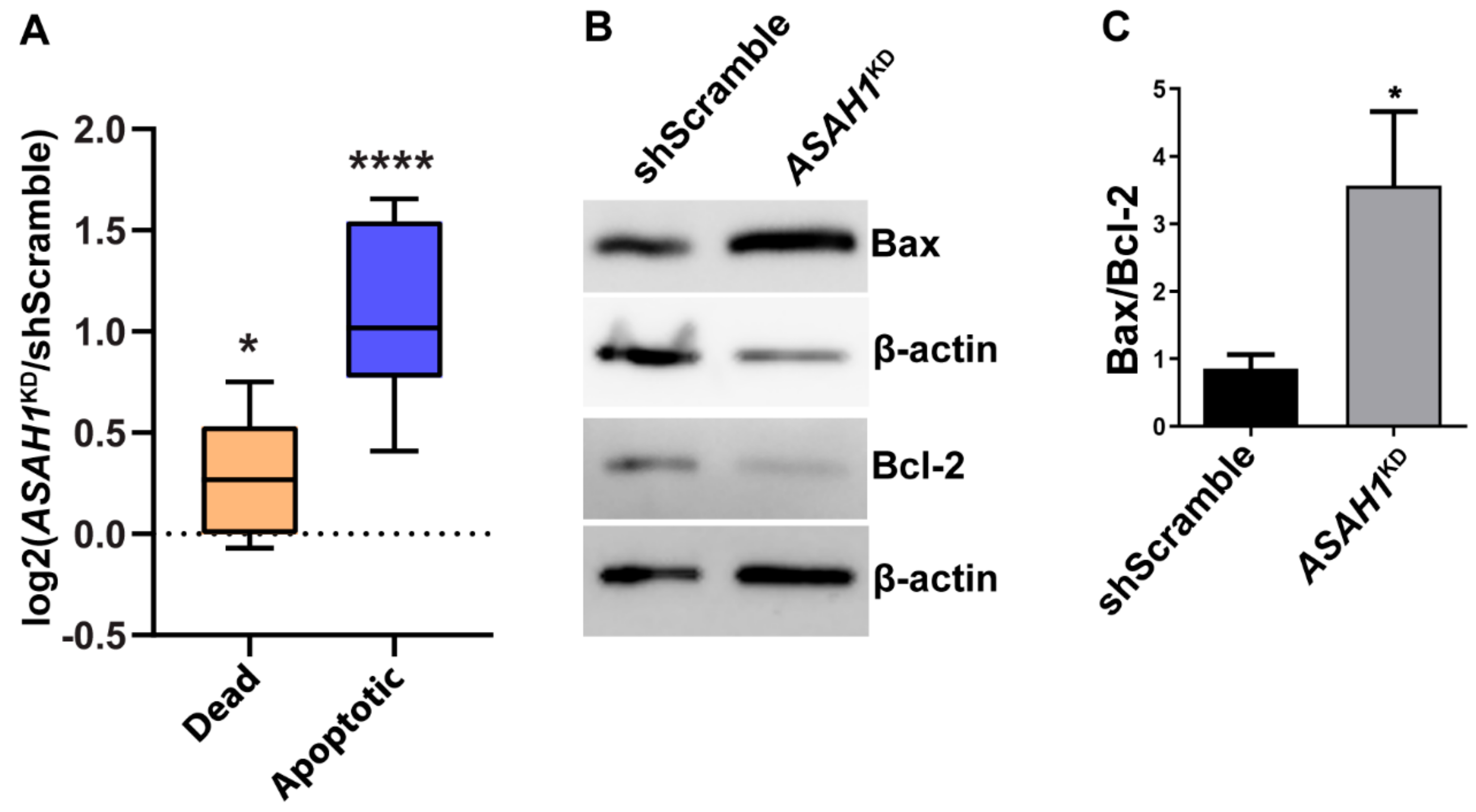
2.2. AC Reduction Induces Cell Cycle Arrest at G1/S Phase and Apoptosis
2.3. AC Depletion Leads to a Diffuse Distribution Pattern of Lysosomes
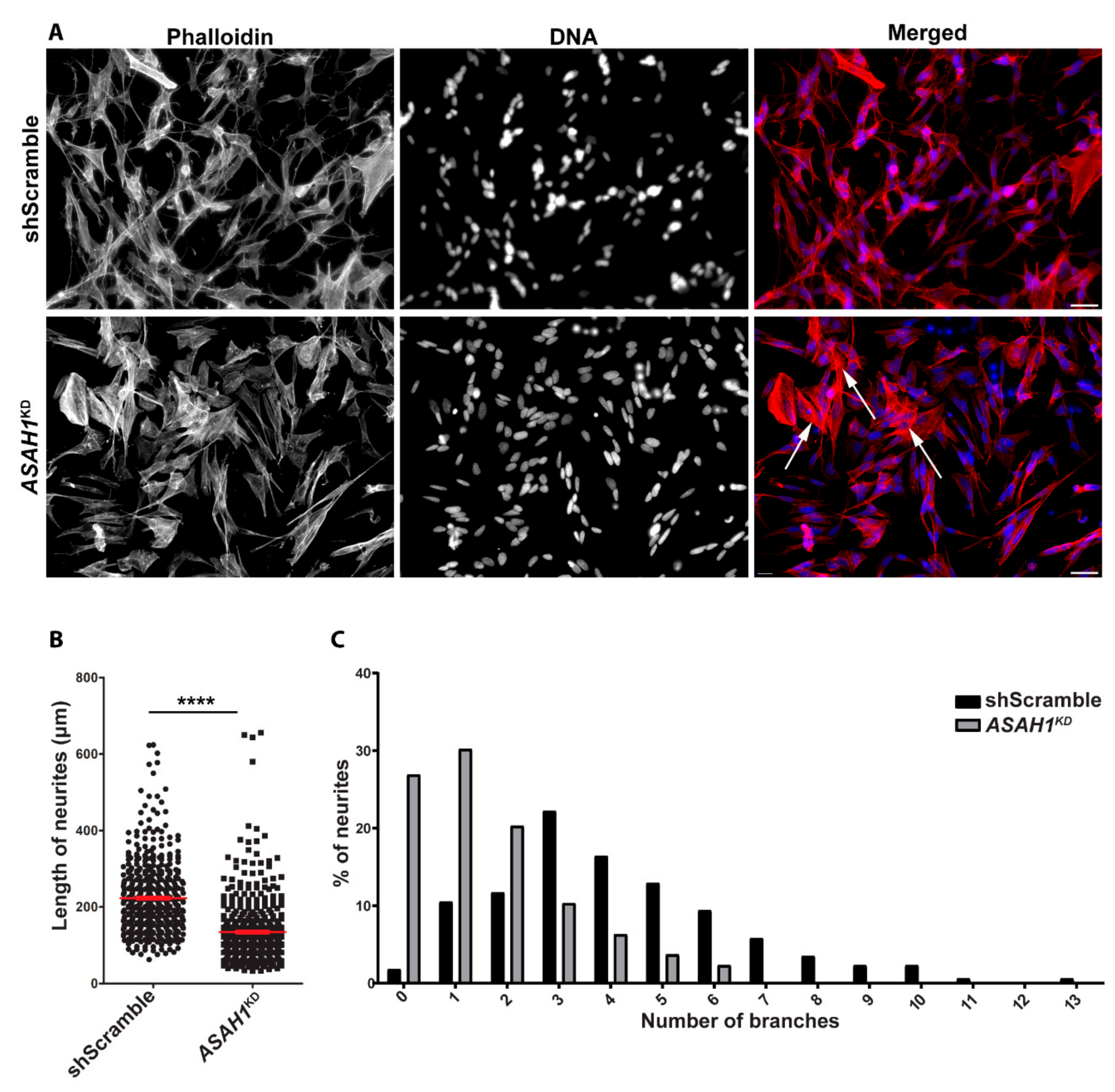
2.4. ASAH1KD Cells Display Shorter Neurites and Fewer Branches Per Neurite
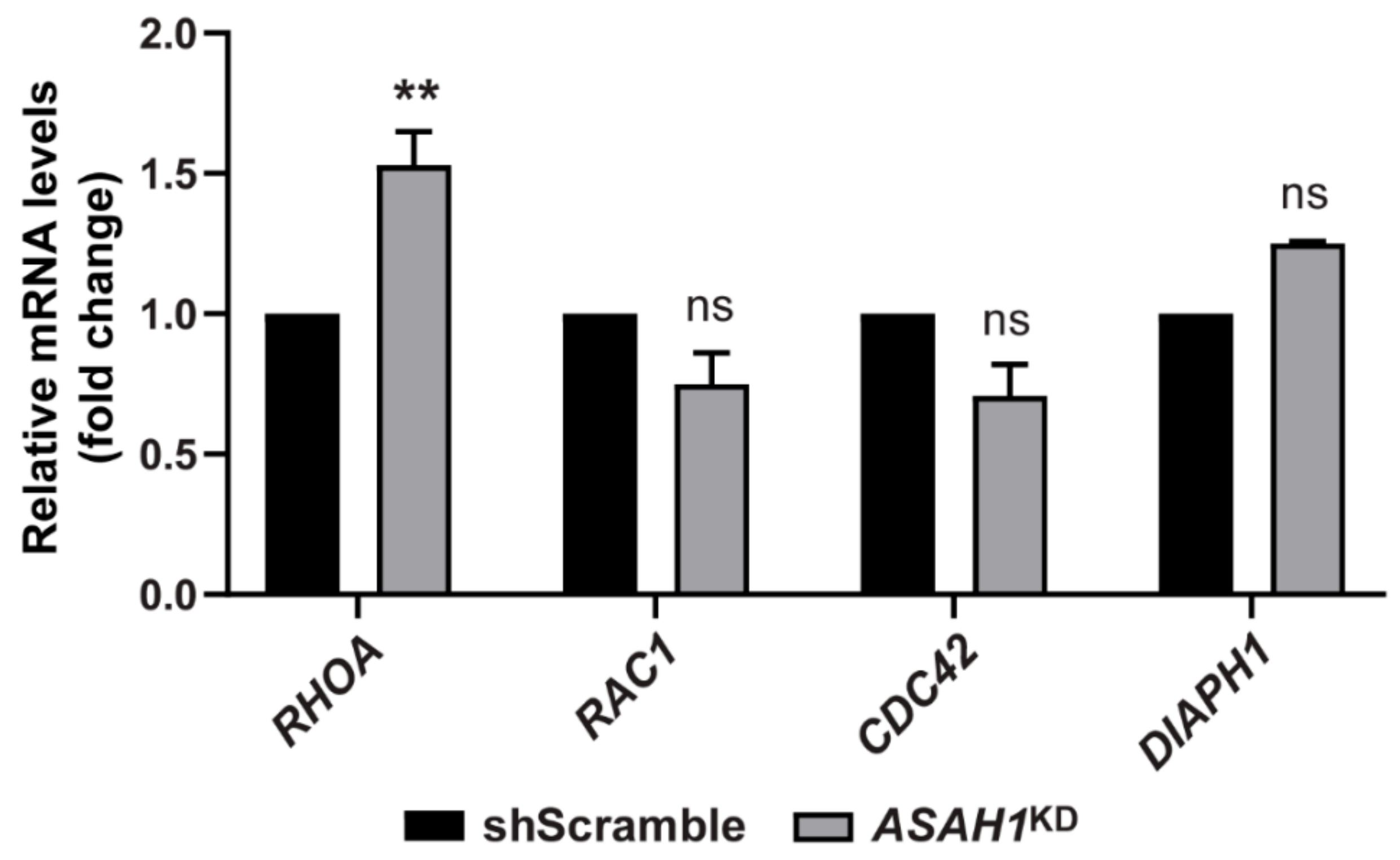
2.5. AC Depletion Causes Significant Up-Regulation of the RHOA Transcript
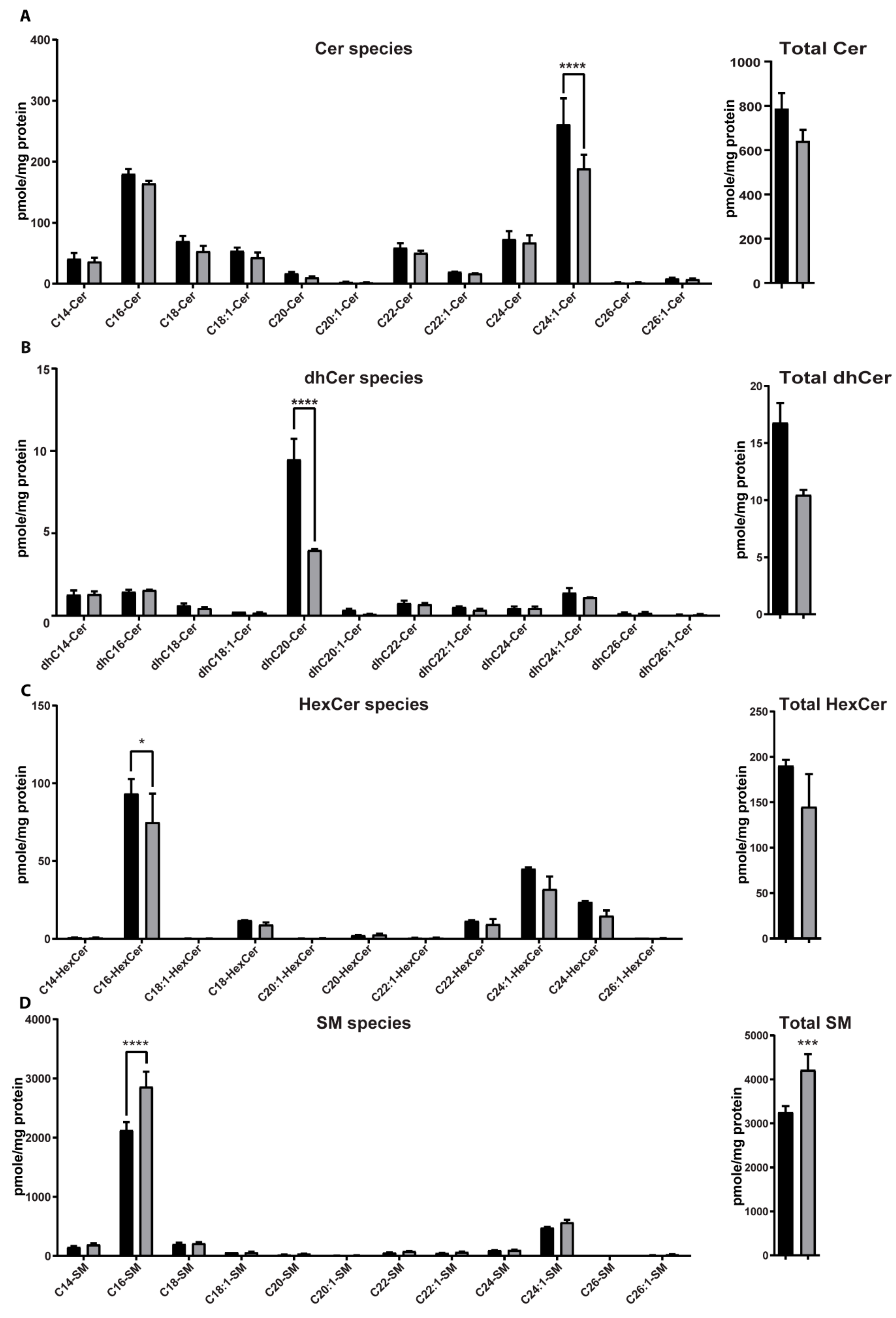
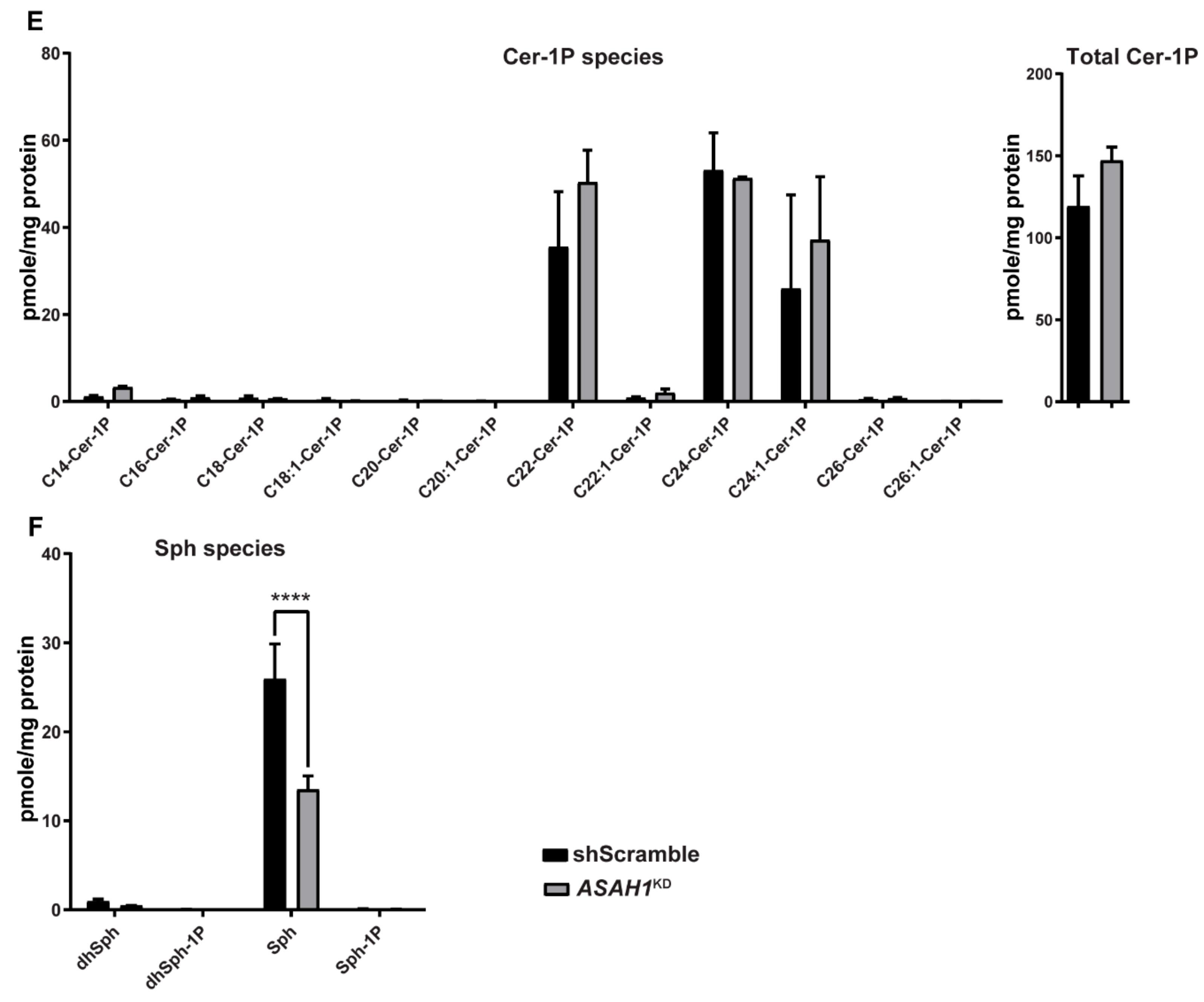
2.6. AC Depletion Leads to Changes in Sphingolipid Content
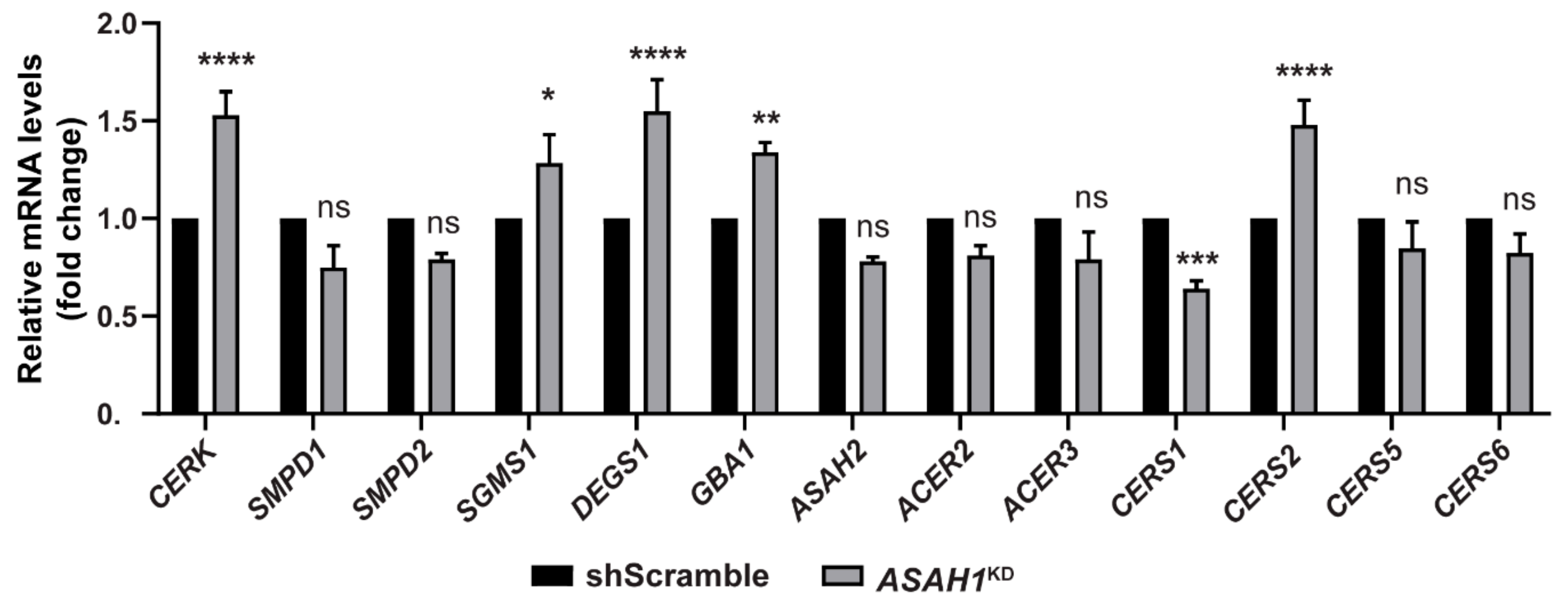
2.7. AC Depletion Causes Alteration in Genes that Control Cer Homeostasis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Differentiation
4.2. Generation of Stable ASAH1 Knockdown SH-SY5Y Cell Lines
4.3. Cell Viability Calculation
4.4. Neurite Outgrowth
4.5. Reverse-Transcription Quantitative PCR (RT-qPCR)
4.6. Enzyme Activity
4.7. Flow Cytometry: Apoptosis and Cell Cycle Assays
4.8. Immunofluorescence Staining and Microscopy
4.9. Western Blot Analysis
4.10. Quantification of Sphingolipids by HPLC/MS-MS
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AC | acid ceramidase |
| ACER2 | plasma membrane ceramidase |
| ACER3 | golgi ceramidase |
| ASAH1 | lysosomal ceramidase |
| ASAH2 | endoplasmic reticulum and Golgi ceramidase |
| Cer | ceramide |
| Cer-1P | ceramide-1-phosphate |
| CERK | ceramide kinase |
| CERS1,2,5,6 | ceramide synthases |
| DEGS1 | dihydroceramide desaturase |
| dhCer | dihydroceramide |
| dhSph | dihydrosphingosine |
| dhSph-1P | dihydrosphingosine-1-phospate |
| FD | Farber disease |
| GBA1 | glucosylceramidase beta1 |
| GluCer | glucosylceramide |
| HexCer | hexosylceramide |
| SGMS1 | sphingomyelin synthase 1 |
| SM | sphingomyelin |
| SMA | spinal muscular atrophy |
| SMA-PME | spinal muscular atrophy with progressive myoclonic epilepsy |
| SMPD1 | lysosomal sphingomyelin phosphodiesterase 1 |
| SMPD2 | neutral sphingomyelin phosphodiesterase 2 |
| Sph | sphingosine |
| Sph-1P | sphingosine-1-phospate |
References
- Sugita, M.; Dulaney, J.T.; Moser, H.W. Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science 1972, 178, 1100–1102. [Google Scholar] [CrossRef]
- Park, J.H.; Schuchman, E.H. Acid ceramidase and human disease. Biochim. Biophys. Acta 2006, 1758, 2133–2138. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tawk, M.; Tiziano, F.D.; Veillet, J.; Bayes, M.; Nolent, F.; Garcia, V.; Servidei, S.; Bertini, E.; Castro-Giner, F.; et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am. J. Hum. Genet. 2012, 91, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Filosto, M.; Aureli, M.; Castellotti, B.; Rinaldi, F.; Schiumarini, D.; Valsecchi, M.; Lualdi, S.; Mazzotti, R.; Pensato, V.; Rota, S.; et al. ASAH1 variant causing a mild SMA phenotype with no myoclonic epilepsy: A clinical, biochemical and molecular study. Eur. J. Hum. Genet. EJHG 2016, 24, 1578–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.M.; Park, J.H.; Simonaro, C.M.; He, X.; Gordon, R.E.; Friedman, A.H.; Ehleiter, D.; Paris, F.; Manova, K.; Hepbildikler, S.; et al. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics 2002, 79, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Tanimukai, H.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Elevation of the level and activity of acid ceramidase in Alzheimer’s disease brain. Eur. J. Neurosci. 2004, 20, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Dworski, S.; Jones, E.E.; Kamani, M.A.; Micsenyi, M.C.; Sawada, T.; Le Faouder, P.; Bertrand-Michel, J.; Dupuy, A.; Dunn, C.K.; et al. Acid Ceramidase Deficiency in Mice Results in a Broad Range of Central Nervous System Abnormalities. Am. J. Pathol. 2017, 187, 864–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef]
- Jana, A.; Hogan, E.L.; Pahan, K. Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci. 2009, 278, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, A.; Futerman, A.H. Distinct roles for ceramide and glucosylceramide at different stages of neuronal growth. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 2929–2938. [Google Scholar] [CrossRef]
- Harel, R.; Futerman, A.H. Inhibition of sphingolipid synthesis affects axonal outgrowth in cultured hippocampal neurons. J. Biol. Chem. 1993, 268, 14476–14481. [Google Scholar] [PubMed]
- Schwarz, A.; Rapaport, E.; Hirschberg, K.; Futerman, A.H. A regulatory role for sphingolipids in neuronal growth. Inhibition of sphingolipid synthesis and degradation have opposite effects on axonal branching. J. Biol. Chem. 1995, 270, 10990–10998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuya, S.; Ono, K.; Hirabayashi, Y. Sphingolipid biosynthesis is necessary for dendrite growth and survival of cerebellar Purkinje cells in culture. J. Neurochem. 1995, 65, 1551–1561. [Google Scholar] [CrossRef]
- Brann, A.B.; Scott, R.; Neuberger, Y.; Abulafia, D.; Boldin, S.; Fainzilber, M.; Futerman, A.H. Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 8199–8206. [Google Scholar] [CrossRef] [Green Version]
- Furuya, S.; Mitoma, J.; Makino, A.; Hirabayashi, Y. Ceramide and its interconvertible metabolite sphingosine function as indispensable lipid factors involved in survival and dendritic differentiation of cerebellar Purkinje cells. J. Neurochem. 1998, 71, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Bodennec, J.; Pelled, D.; Riebeling, C.; Trajkovic, S.; Futerman, A.H. Phosphatidylcholine synthesis is elevated in neuronal models of Gaucher disease due to direct activation of CTP:phosphocholine cytidylyltransferase by glucosylceramide. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1814–1816. [Google Scholar] [CrossRef]
- Pelled, D.; Riebeling, C.; van Echten-Deckert, G.; Sandhoff, K.; Futerman, A.H. Reduced rates of axonal and dendritic growth in embryonic hippocampal neurones cultured from a mouse model of Sandhoff disease. Neuropathol. Appl. Neurobiol. 2003, 29, 341–349. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, M.F.; Ashworth, A.; Hall, A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 1995, 269, 1270–1272. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Ikeda, K.; Himeno, M. A role for small GTPase RhoA in regulating intracellular membrane traffic of lysosomes in invasive rat hepatoma cells. Histochem. J. 2002, 34, 189–213. [Google Scholar] [CrossRef]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Uehata, M.; Himeno, M. Small guanosine triphosphatase Rho/Rho-associated kinase as a novel regulator of intracellular redistribution of lysosomes in invasive tumor cells. Cell Tissue Res. 2000, 301, 341–351. [Google Scholar] [CrossRef]
- Martin, E.; Schule, R.; Smets, K.; Rastetter, A.; Boukhris, A.; Loureiro, J.L.; Gonzalez, M.A.; Mundwiller, E.; Deconinck, T.; Wessner, M.; et al. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 92, 238–244. [Google Scholar] [CrossRef] [Green Version]
- Asensio-Juan, E.; Gallego, C.; Martinez-Balbas, M.A. The histone demethylase PHF8 is essential for cytoskeleton dynamics. Nucleic Acids Res. 2012, 40, 9429–9440. [Google Scholar] [CrossRef]
- Jacquier, A.; Buhler, E.; Schafer, M.K.; Bohl, D.; Blanchard, S.; Beclin, C.; Haase, G. Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann. Neurol. 2006, 60, 105–117. [Google Scholar] [CrossRef]
- Bowerman, M.; Shafey, D.; Kothary, R. Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J. Mol. Neurosci. MN 2007, 32, 120–131. [Google Scholar] [CrossRef]
- Nolle, A.; Zeug, A.; van Bergeijk, J.; Tonges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.S.; Wynne, J.; Treisman, R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 1995, 81, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A.; Loucks, F.A. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Front. Biosci. A J. Virtual Libr. 2008, 13, 657–676. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; D’Mello, S.R. p21-Activated kinase-1 is necessary for depolarization-mediated neuronal survival. J. Neurosci. Res. 2005, 79, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Loucks, F.A.; Le, S.S.; Zimmermann, A.K.; Ryan, K.R.; Barth, H.; Aktories, K.; Linseman, D.A. Rho family GTPase inhibition reveals opposing effects of mitogen-activated protein kinase kinase/extracellular signal-regulated kinase and Janus kinase/signal transducer and activator of transcription signaling cascades on neuronal survival. J. Neurochem. 2006, 97, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, P.; Ridley, A.J. Rho GTPases and cell cycle control. Growth Factors 2006, 24, 159–164. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Petit, D.; Bertoglio, J. Cell cycle regulation of Rho signaling pathways. Cell Cycle 2012, 11, 3003–3010. [Google Scholar] [CrossRef] [Green Version]
- Sands, M.S. Farber disease: Understanding a fatal childhood disorder and dissecting ceramide biology. EMBO Mol. Med. 2013, 5, 799–801. [Google Scholar] [CrossRef]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Bonifacino, J.S.; Neefjes, J. Moving and positioning the endolysosomal system. Curr. Opin. Cell Biol. 2017, 47, 1–8. [Google Scholar] [CrossRef]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.; Berlin, I.; Wijdeven, R.H.; Janssen, L.; Janssen, G.M.; Garstka, M.A.; Janssen, H.; Mensink, M.; van Veelen, P.A.; Spaapen, R.M.; et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 2016, 166, 152–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bright, N.A.; Davis, L.J.; Luzio, J.P. Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr. Biol. CB 2016, 26, 2233–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Tokuda, K.; Himeno, M. Overexpression of ROCK in human breast cancer cells: Evidence that ROCK activity mediates intracellular membrane traffic of lysosomes. Pathol. Oncol. Res. POR 2003, 9, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jaattela, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fennelly, C.; Amaravadi, R.K. Lysosomal Biology in Cancer. Methods Mol. Biol. 2017, 1594, 293–308. [Google Scholar] [PubMed] [Green Version]
- Li, G.; Huang, D.; Hong, J.; Bhat, O.M.; Yuan, X.; Li, P.L. Control of lysosomal TRPML1 channel activity and exosome release by acid ceramidase in mouse podocytes. Am. J. Physiol. Cell Physiol. 2019, 317, C481–C491. [Google Scholar] [CrossRef]
- Zigdon, H.; Meshcheriakova, A.; Farfel-Becker, T.; Volpert, G.; Sabanay, H.; Futerman, A.H. Altered lysosome distribution is an early neuropathological event in neurological forms of Gaucher disease. FEBS Lett. 2017, 591, 774–783. [Google Scholar] [CrossRef] [Green Version]
- Uusi-Rauva, K.; Kyttala, A.; van der Kant, R.; Vesa, J.; Tanhuanpaa, K.; Neefjes, J.; Olkkonen, V.M.; Jalanko, A. Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell. Mol. Life Sci. CMLS 2012, 69, 2075–2089. [Google Scholar] [CrossRef]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Yang, D.S.; Lee, J.H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhou, B.; Lin, M.Y.; Wang, S.; Foust, K.D.; Sheng, Z.H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Erie, C.; Sacino, M.; Houle, L.; Lu, M.L.; Wei, J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur. J. Neurosci. 2015, 42, 1941–1951. [Google Scholar] [CrossRef] [Green Version]
- Heuser, J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J. Cell Biol. 1989, 108, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wu, W.K.; Gu, C.; Zhong, D.; Zhao, X.; Kong, Y.; Lin, Q.; Chan, M.T.; Zhou, Z.; Liu, S. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget 2016, 7, 14693–14707. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Norred, W.P.; Bacon, C.W.; Riley, R.T.; Merrill, A.H., Jr. Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J. Biol. Chem. 1991, 266, 14486–14490. [Google Scholar] [PubMed]
- Samuelsson, K.; Zetterstrom, R. Ceramides in a patient with lipogranulomatosis (Farber’s disease) with chronic course. Scand. J. Clin. Lab. Investig. 1971, 27, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Lucki, N.C.; Bandyopadhyay, S.; Wang, E.; Merrill, A.H.; Sewer, M.B. Acid ceramidase (ASAH1) is a global regulator of steroidogenic capacity and adrenocortical gene expression. Mol. Endocrinol. 2012, 26, 228–243. [Google Scholar] [CrossRef] [Green Version]
- Kao, L.P.; Morad, S.A.F.; Davis, T.S.; MacDougall, M.R.; Kassai, M.; Abdelmageed, N.; Fox, T.E.; Kester, M.; Loughran, T.P., Jr.; Abad, J.L.; et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL-60 cells. J. Lipid Res. 2019, 60, 1590–1602. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef] [PubMed]
- White-Gilbertson, S.; Lu, P.; Norris, J.S.; Voelkel-Johnson, C. Genetic and pharmacological inhibition of acid ceramidase prevents asymmetric cell division by neosis. J. Lipid Res. 2019, 60, 1225–1235. [Google Scholar] [CrossRef]
- Beckmann, N.; Kadow, S.; Schumacher, F.; Gothert, J.R.; Kesper, S.; Draeger, A.; Schulz-Schaeffer, W.J.; Wang, J.; Becker, J.U.; Kramer, M.; et al. Pathological manifestations of Farber disease in a new mouse model. Biol. Chem. 2018, 399, 1183–1202. [Google Scholar] [CrossRef]
- Yu, F.P.S.; Molino, S.; Sikora, J.; Rasmussen, S.; Rybova, J.; Tate, E.; Geurts, A.M.; Turner, P.V.; McKillop, W.M.; Medin, J.A. Hepatic pathology and altered gene transcription in a murine model of acid ceramidase deficiency. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 1572–1592. [Google Scholar] [CrossRef]
- Alayoubi, A.M.; Wang, J.C.; Au, B.C.; Carpentier, S.; Garcia, V.; Dworski, S.; El-Ghamrasni, S.; Kirouac, K.N.; Exertier, M.J.; Xiong, Z.J.; et al. Systemic ceramide accumulation leads to severe and varied pathological consequences. EMBO Mol. Med. 2013, 5, 827–842. [Google Scholar] [CrossRef]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanni, N.; Fruscione, F.; Ferlazzo, E.; Striano, P.; Robbiano, A.; Traverso, M.; Sander, T.; Falace, A.; Gazzerro, E.; Bramanti, P.; et al. Impairment of ceramide synthesis causes a novel progressive myoclonus epilepsy. Ann. Neurol. 2014, 76, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Couttas, T.A.; Kain, N.; Suchowerska, A.K.; Quek, L.E.; Turner, N.; Fath, T.; Garner, B.; Don, A.S. Loss of ceramide synthase 2 activity, necessary for myelin biosynthesis, precedes tau pathology in the cortical pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2016, 43, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Mosbech, M.B.; Olsen, A.S.; Neess, D.; Ben-David, O.; Klitten, L.L.; Larsen, J.; Sabers, A.; Vissing, J.; Nielsen, J.E.; Hasholt, L.; et al. Reduced ceramide synthase 2 activity causes progressive myoclonic epilepsy. Ann. Clin. Transl. Neurol. 2014, 1, 88–98. [Google Scholar] [CrossRef]
- Godeiro Junior, C.O.; Vale, T.C.; Afonso, C.O.M.; Kok, F.; Pedroso, J.L.; Barsottini, O.G. Progressive Myoclonic Epilepsy Type 8 Due to CERS1 Deficiency: A Novel Mutation with Prominent Ataxia. Mov. Disord. Clin. Pract. 2018, 5, 330–332. [Google Scholar] [CrossRef] [Green Version]
- Dyment, D.A.; Sell, E.; Vanstone, M.R.; Smith, A.C.; Garandeau, D.; Garcia, V.; Carpentier, S.; Le Trionnaire, E.; Sabourdy, F.; Beaulieu, C.L.; et al. Evidence for clinical, genetic and biochemical variability in spinal muscular atrophy with progressive myoclonic epilepsy. Clin. Genet. 2014, 86, 558–563. [Google Scholar] [CrossRef]
- Gan, J.J.; Garcia, V.; Tian, J.; Tagliati, M.; Parisi, J.E.; Chung, J.M.; Lewis, R.; Baloh, R.; Levade, T.; Pierson, T.M. Acid ceramidase deficiency associated with spinal muscular atrophy with progressive myoclonic epilepsy. Neuromuscul. Disord. NMD 2015, 25, 959–963. [Google Scholar] [CrossRef]
- Giraldez, B.G.; Guerrero-Lopez, R.; Ortega-Moreno, L.; Verdu, A.; Carrascosa-Romero, M.C.; Garcia-Campos, O.; Garcia-Munozguren, S.; Pardal-Fernandez, J.M.; Serratosa, J.M. Uniparental disomy as a cause of spinal muscular atrophy and progressive myoclonic epilepsy: Phenotypic homogeneity due to the homozygous c.125C>T mutation in ASAH1. Neuromuscul. Disord. NMD 2015, 25, 222–224. [Google Scholar] [CrossRef]
- Rubboli, G.; Veggiotti, P.; Pini, A.; Berardinelli, A.; Cantalupo, G.; Bertini, E.; Tiziano, F.D.; D’Amico, A.; Piazza, E.; Abiusi, E.; et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy: A rare condition caused by mutations in ASAH1. Epilepsia 2015, 56, 692–698. [Google Scholar] [CrossRef]
- He, X.; Okino, N.; Dhami, R.; Dagan, A.; Gatt, S.; Schulze, H.; Sandhoff, K.; Schuchman, E.H. Purification and characterization of recombinant, human acid ceramidase. Catalytic reactions and interactions with acid sphingomyelinase. J. Biol. Chem. 2003, 278, 32978–32986. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Sankaram, M.B.; Thompson, T.E. Interaction of cholesterol with various glycerophospholipids and sphingomyelin. Biochemistry 1990, 29, 10670–10675. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Golub, T.; Wacha, S.; Caroni, P. Spatial and temporal control of signaling through lipid rafts. Curr. Opin. Neurobiol. 2004, 14, 542–550. [Google Scholar] [CrossRef]
- Tsui-Pierchala, B.A.; Encinas, M.; Milbrandt, J.; Johnson, E.M., Jr. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002, 25, 412–417. [Google Scholar] [CrossRef]
- Sot, J.; Goni, F.M.; Alonso, A. Molecular associations and surface-active properties of short- and long-N-acyl chain ceramides. Biochim. Biophys. Acta 2005, 1711, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Montes, L.R.; Ruiz-Arguello, M.B.; Goni, F.M.; Alonso, A. Membrane restructuring via ceramide results in enhanced solute efflux. J. Biol. Chem. 2002, 277, 11788–11794. [Google Scholar] [CrossRef] [Green Version]
- Pinto, S.N.; Silva, L.C.; de Almeida, R.F.; Prieto, M. Membrane domain formation, interdigitation, and morphological alterations induced by the very long chain asymmetric C24:1 ceramide. Biophys. J. 2008, 95, 2867–2879. [Google Scholar] [CrossRef] [Green Version]
- Westerlund, B.; Grandell, P.M.; Isaksson, Y.J.; Slotte, J.P. Ceramide acyl chain length markedly influences miscibility with palmitoyl sphingomyelin in bilayer membranes. Eur. Biophys. J. EBJ 2010, 39, 1117–1128. [Google Scholar] [CrossRef]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiznerowicz, M.; Trono, D. Conditional suppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 2003, 77, 8957–8961. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Bedia, C.; Casas, J.; Garcia, V.; Levade, T.; Fabrias, G. Synthesis of a novel ceramide analogue and its use in a high-throughput fluorogenic assay for ceramidases. Chembiochem 2007, 8, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Bedia, C.; Camacho, L.; Abad, J.L.; Fabrias, G.; Levade, T. A simple fluorogenic method for determination of acid ceramidase activity and diagnosis of Farber disease. J. Lipid Res. 2010, 51, 3542–3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawlitta, D.; Oomens, C.W.; Baaijens, F.P.; Bouten, C.V. Evaluation of a continuous quantification method of apoptosis and necrosis in tissue cultures. Cytotechnology 2004, 46, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Krishan, A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J. Cell Biol. 1975, 66, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Bielawski, J.; Pierce, J.S.; Snider, J.; Rembiesa, B.; Szulc, Z.M.; Bielawska, A. Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol. Biol. 2009, 579, 443–467. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyriakou, K.; W. Lederer, C.; Kleanthous, M.; Drousiotou, A.; Malekkou, A. Acid Ceramidase Depletion Impairs Neuronal Survival and Induces Morphological Defects in Neurites Associated with Altered Gene Transcription and Sphingolipid Content. Int. J. Mol. Sci. 2020, 21, 1607. https://doi.org/10.3390/ijms21051607
Kyriakou K, W. Lederer C, Kleanthous M, Drousiotou A, Malekkou A. Acid Ceramidase Depletion Impairs Neuronal Survival and Induces Morphological Defects in Neurites Associated with Altered Gene Transcription and Sphingolipid Content. International Journal of Molecular Sciences. 2020; 21(5):1607. https://doi.org/10.3390/ijms21051607
Chicago/Turabian StyleKyriakou, Kalia, Carsten W. Lederer, Marina Kleanthous, Anthi Drousiotou, and Anna Malekkou. 2020. "Acid Ceramidase Depletion Impairs Neuronal Survival and Induces Morphological Defects in Neurites Associated with Altered Gene Transcription and Sphingolipid Content" International Journal of Molecular Sciences 21, no. 5: 1607. https://doi.org/10.3390/ijms21051607