
Phosphinotripeptidic Inhibitors of Leucylaminopeptidases

 , , , and
, , , and
Abstract
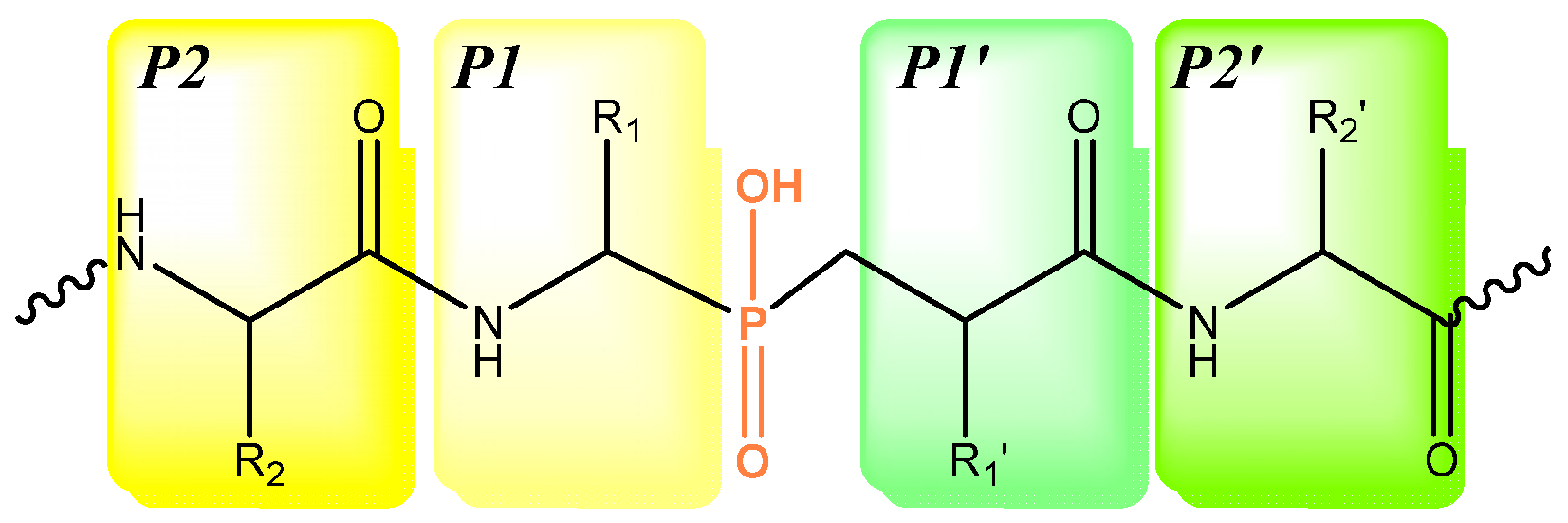
:1. Introduction
2. Results and Discussions
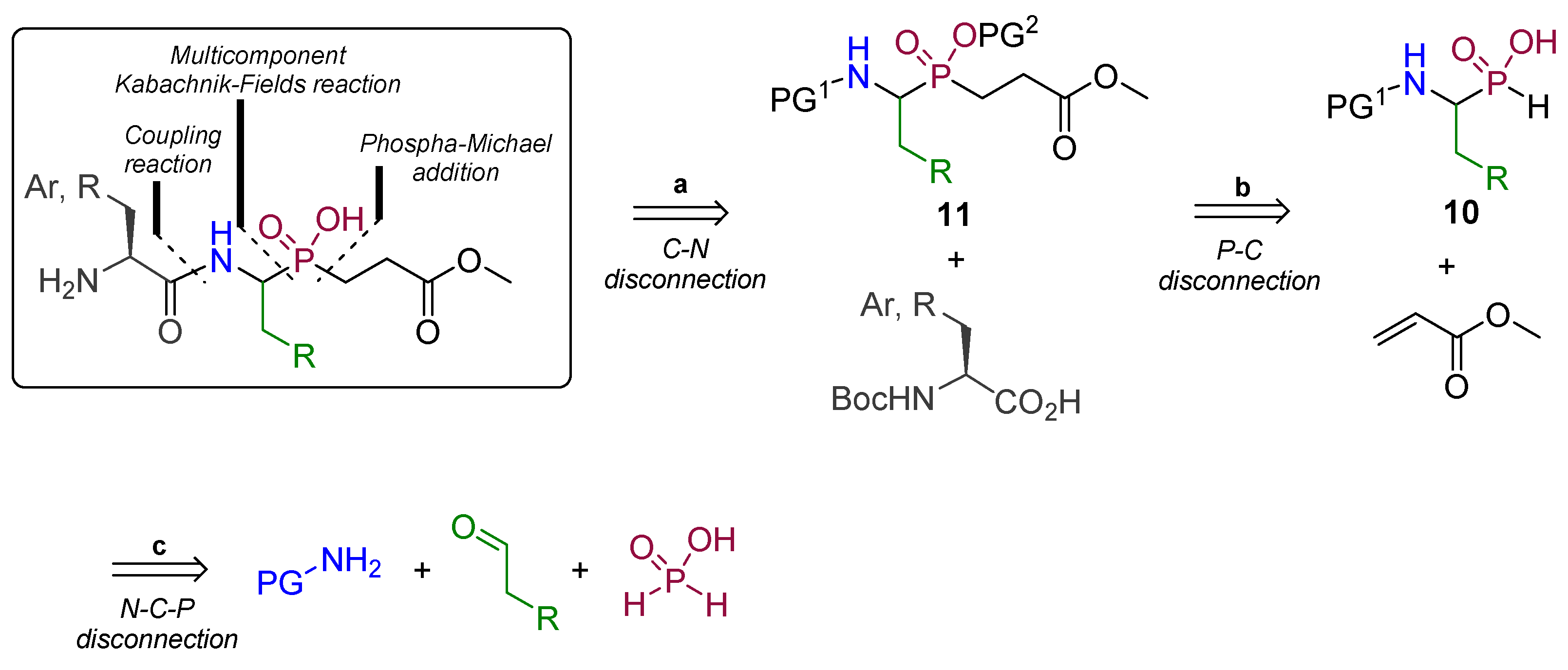
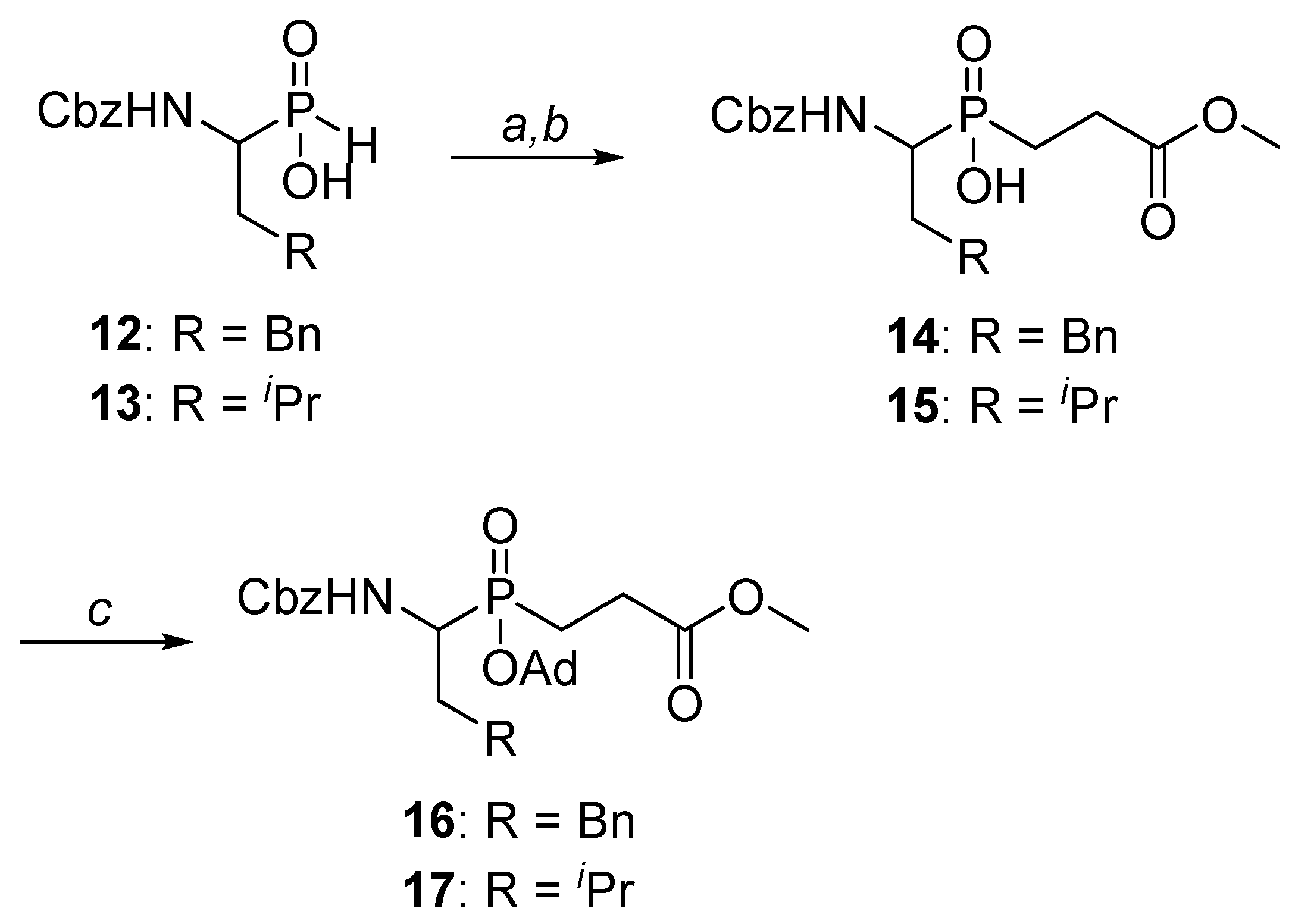
2.1. Chemistry
2.2. Enzyme Inhibition
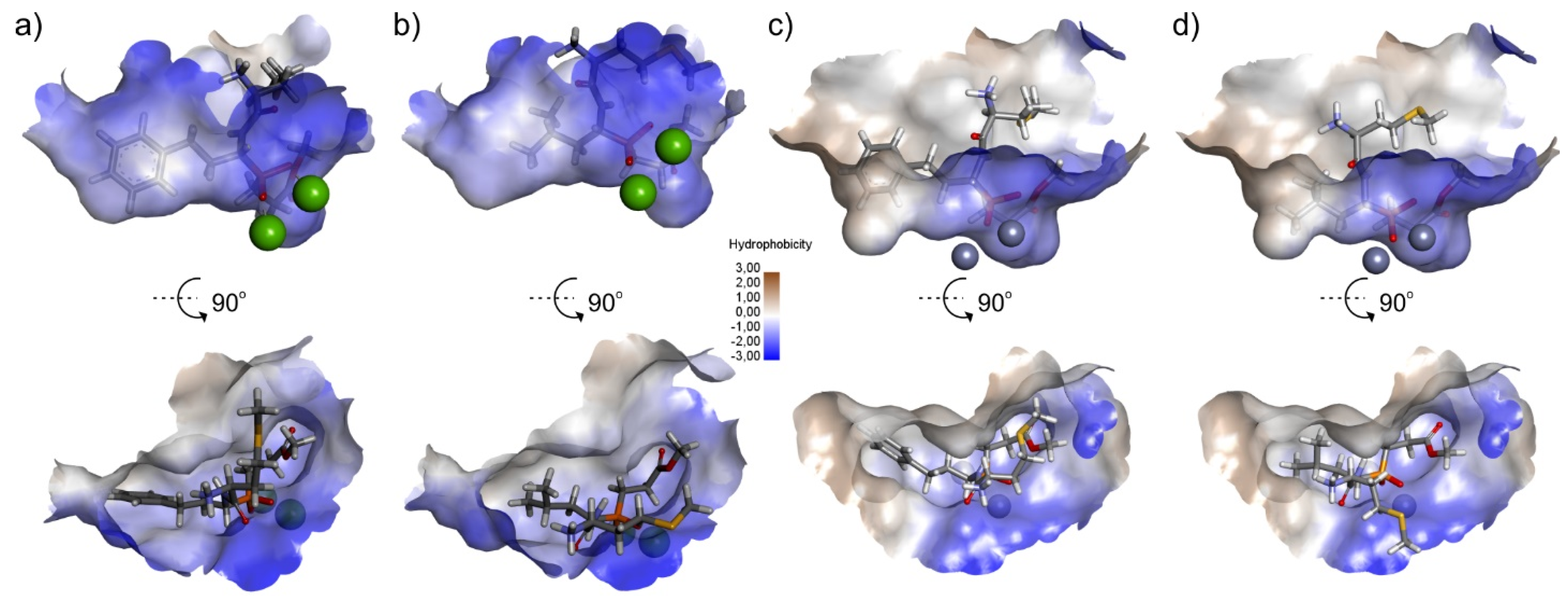
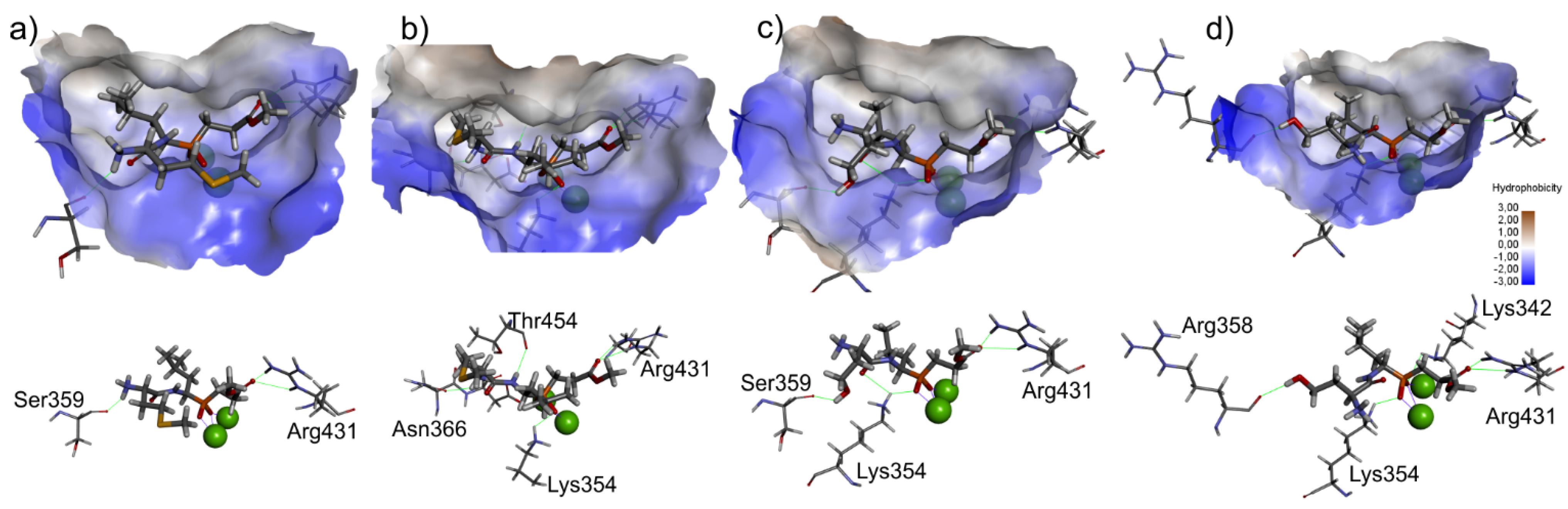
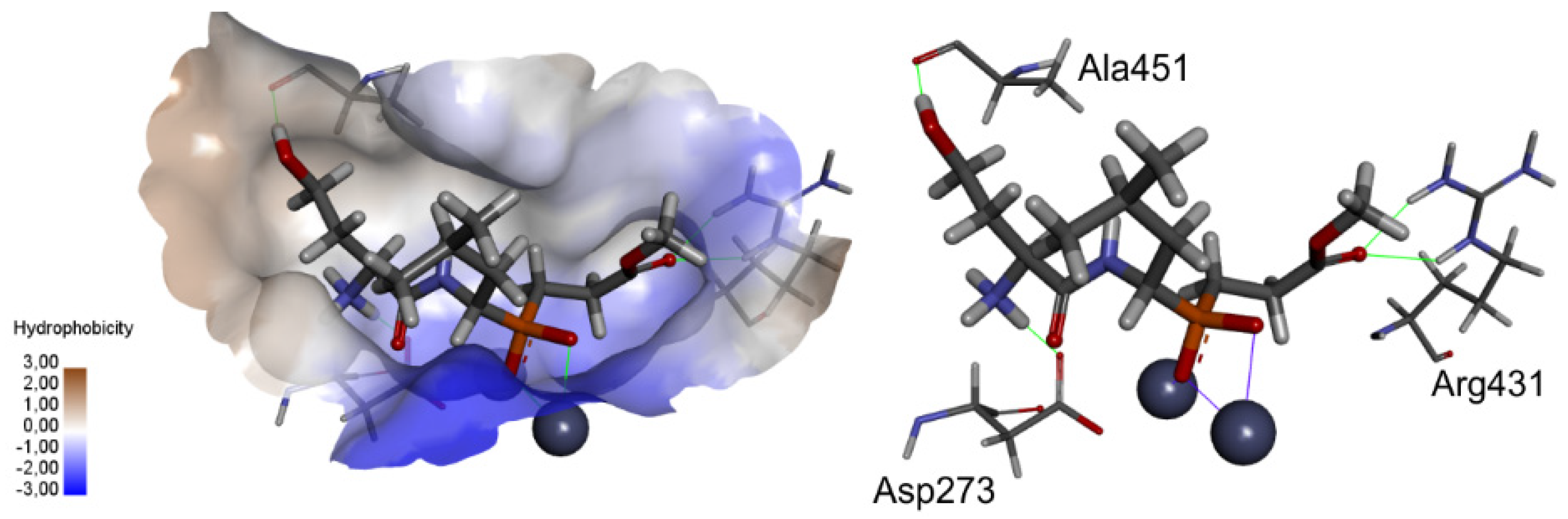
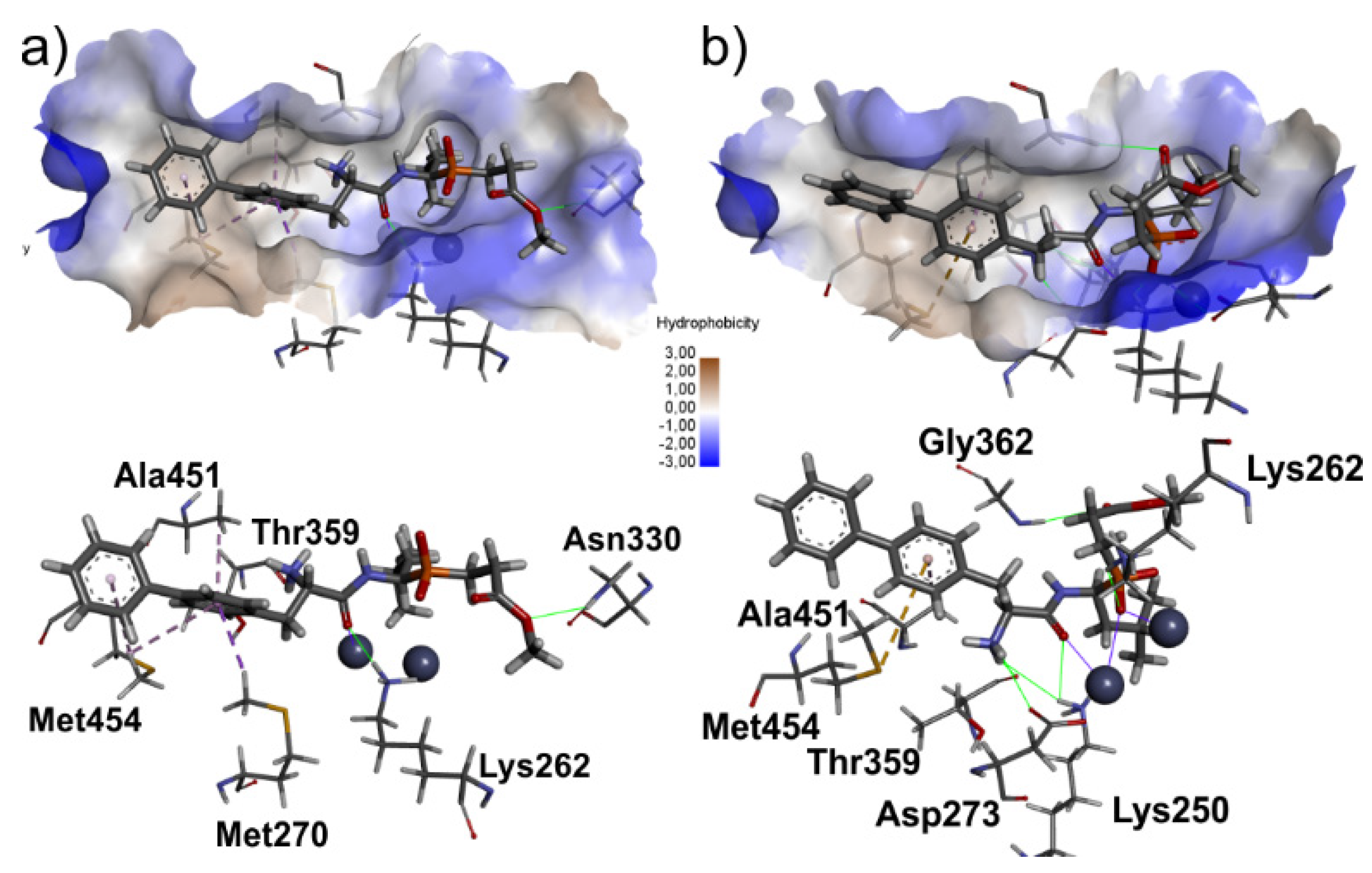
2.3. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. [1-(N-Benzyloxycarbonylamino)-3-phenylpropyl]phosphinic Acid (12)
3.1.2. [1-(N-Benzyloxycarbonylamino)-3-methylbutyl]phosphinic Acid (13)
3.1.3. General Procedure for the Synthesis of 14 and 15
Methyl 3-[1-N-(benzyloxycarbonylamino)-3-phenylpropyl]hydroxyphosphinyl propanoate (14)
Methyl 3-[1-N-(benzyloxycarbonylamino)-3-methylbutyl]hydroxyphosphinyl propanoate (15)
3.1.4. General Procedure for the Protection of Hydroxyphosphinyl Group in 14 and 15
Methyl 3-[(1-N-(benzyloxycarbonylamino)-3-phenylpropyl)adamantyloxyphoshinyl]propanoate (16)
Methyl 3-[(1-N-(benzyloxycarbonylamino)-3-methylbutyl)adamantyloxyphoshinyl]propanoate (17)
3.1.5. General Procedure for the Coupling Reaction of Building Blocks 16 and 17 Involving HOBt and EDC
Methyl 3-[(adamantan-1-yloxy)-(1-((S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butan-amido)-3-phenylpropyl)phosphinyl]propanoate (20)
Methyl 3-[(adamantan-1-yloxy)(1-((S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butan-amido)-3-methylbutyl)phosphinyl]propanoate (21)
Methyl 3-[(adamantan-1-yloxy)(1-((S)-2-(tert-butoxycarbonylamino)-4-((tert-butyldime-thylsilyl)-oxy)butanamido)-3-methylbutyl)phosphinyl]propanoate (22)
Methyl 3-[(adamantan-1-yloxy)(1-((S)-2-(tert-butoxycarbonylamino)-4-(hydroxybutan-amido)-3-methylbutyl)phosphinyl]propanoate (23)
Methyl 3-[(adamantan-1′-yloxy)-((1-((S)-3-([1,1′-biphenyl]-4-yl)-2-(tert-butoxycarbonyl-amino)propanamido)-3-phenylpropyl)phosphinyl]propanoate (26)
Methyl 3-[(adamantan-1-yloxy)-((1-((S)-3-([1,1′-biphenyl]-4-yl)-2-(tert-butoxycarbonyl-amino)propanamido)-3-methylbutyl)phosphinyl]propanoate (27)
Methyl 3-[(adamantan-1-yloxy)-((1-((S)-3-(4-benzoylphenyl)-2-(tert-butoxycarbonyl-amino)-propanamido)-3-phenylpropyl)phosphinyl]propanoate (28)
Methyl 3-[(adamantan-1-yloxy)-((1-((S)-3-(4-benzoylphenyl)-2-(tert-butoxycarbony)-amino)-propanamido)-3′-methylbutyl)phosphinyl]propanoate (29)
3.1.6. General Procedure for Removal of the C-Terminal Methyl Ester Group
3-[(Adamantan-1-yloxy)(1-((S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)-3-phenylpropyl)phosphinyl]propanoic acid (24)
3-[(Adamantan-1-yloxy)(1-((S)-2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)-3-methylbutyl)phosphinyl]propanoic acid (25)
3.1.7. General Procedure for Removal of Boc and Ad Protecting Groups
Methyl 3-[(1-((S)-2-amino-4-(methylthio)butanamido)-3-phenylpropyl)phosphinyl]propanoate (1)
Methyl 3-[(1-((S)-2-amino-4-(methylthio)butanamido)-3-methylbutyl)phosphinyl]propanoate (2)
Methyl 3-[(1-((S)-2-amino-4-(hydroxybutanamido)-3-methylbutyl)phosphinyl]propanoate (3)
3-[(1-((S)-2-Amino)-4-(methylthio)butanamido)-3-phenylpropyl)-phosphinyl]propanoic acid (4)
3-[(1-((S)-2-Amino-4-(methylthio)butanamido)-3-methylbutyl)phosphinyl]propanoic acid (5)
Methyl 3-[(1-((S)-3-([1,1′-biphenyl]-4-yl)-2-aminopropanamido)-3-phenylpropyl)phosphinyl]-propanoate (6)
Methyl 3-[(1-((S)-3-([1,1′-biphenyl]-4-yl)-2-aminopropanamido)-3-methylbutyl)phosphinyl]-propanoate (7)
Methyl 3-[(1-((S)-2-amino-3-(4-benzoylphenyl)propanamido)-3-phenylpropyl)phosphinyl]-propanoate (8)
Methyl 3-[(1-((S)-2-amino-3-(4-benzoylphenyl)propanamido)-3-methylbutyl)phosphinyl]-propanoate (9)
3.2. Inhibitor Activity
3.2.1. Enzyme Preparation
3.2.2. Kinetic Assays
3.2.3. Kinetic Calculation
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Yiotakis, A.; Georgiadis, D.; Matziari, M.; Makaritis, A.; Dive, V. Phosphinic Peptides: Synthetic Approaches and Biochemical Evaluation as Zn-Metalloprotease Inhibitors. Curr. Org. Chem. 2004, 8, 1135–1158. [Google Scholar] [CrossRef]
- Baylis, E.K.; Campbell, C.D.; Dingwall, J.G. 1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J. Chem. Soc. Perkin Trans. 1984, 1, 2845–2853. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J. Leucine Aminopeptidase as a Target for Inhibitor Design. MiniRev. Med. Chem. 2001, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Fowler, J.H.; Walling, L.L. Leucine aminopeptidases: Diversity in structure and function. Biol. Chem. 2006, 387, 1535–1544. [Google Scholar] [CrossRef]
- Cappiello, M.; Lazzarotti, A.; Buono, F.; Scaloni, A.; D’Ambrosio, C.; Amodeo, P.; Méndez, B.L.; Pelosi, P.; Del Corso, A.; Mura, U. New role for leucyl aminopeptidase in glutathione turnover. Biochem. J. 2004, 378, 35–44. [Google Scholar] [CrossRef]
- Carroll, R.K.; Robison, T.M.; Rivera, F.E.; Davenport, J.E.; Jonsson, I.-M.; Florczyk, D.; Tarkowski, A.; Potempa, J.; Koziel, J.; Shaw, L.N. Identification of an intracellular M17 family leucine aminopeptidase that is required for virulence in Staphylococcus aureus. Microbes Infect. 2012, 14, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Pulido-Cejudo, G.; Conway, B.; Proulx, P.; Brown, R.; Izaguirre, C.A. Bestatin-mediated inhibition of leucine aminopeptidase may hinder HIV infection. Antivir. Res. 1997, 36, 167–177. [Google Scholar] [CrossRef]
- Beninga, J.; Rock, K.L.; Goldberg, A.L. Interferon-γ Can Stimulate Post-proteasomal Trimming of the N Terminus of an Antigenic Peptide by Inducing Leucine Aminopeptidase. J. Biol. Chem. 1998, 273, 18734–18742. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Daims, M.; Lee, J.; Surgenor, T. Identification and quantification of leucine aminopeptidase in aged normal and cataractous human lenses and ability of bovine lens LAP to cleave bovine crystallins. Curr. Eye Res. 1982, 2, 47–56. [Google Scholar] [CrossRef]
- Umezawa, H. Screening of Small Molecular Microbial Products Modulating Immune Responses and Bestatin. In Cancer Chemo- and Immunopharmacology: 2: Immunopharmacology, Relations, and General Problems; Mathé, G., Muggia, F.M., Eds.; Springer: Berlin/Heidelberg, Germany, 1980; pp. 115–125. [Google Scholar]
- Gupta, S.K.; Aziz, M.; Khan, A.A. Serum leucine aminopeptidase estimation: A sensitive prognostic indicator of invasiveness in breast carcinoma. Indian J. Pathol. Microbiol. 1989, 32, 301–305. [Google Scholar]
- Bartling, D.; Weiler, E.W. Leucine aminopeptidase from Arabidopsis thaliana. Molecular evidence for a phylogenetically conserved enzyme of protein turnover in higher plants. JBIC J. Biol. Inorg. Chem. 1992, 205, 425–431. [Google Scholar] [CrossRef]
- DeSimone, M.; Krüger, M.; Wessel, T.; Wehofsky, M.; Hoffmann, R.; Wagner, E. Purification and characterization of an aminopeptidase from the chloroplast stroma of barley leaves by chromatographic and electrophoretic methods. J. Chromatogr. B Biomed. Sci. Appl. 2000, 737, 285–293. [Google Scholar] [CrossRef]
- Thayer, S.S.; Choe, H.T.; Rausser, S.; Huffaker, R.C. Characterization and Subcellular Localization of Aminopeptidases in Senescing Barley Leaves. Plant Physiol. 1988, 87, 894–897. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, N.; Amano, T.; Satoh, M.; Shioi, Y. Leucine aminopeptidase from etiolated barley seedlings: Characterization and partial purification of isoforms. Plant Sci. 2005, 168, 575–581. [Google Scholar] [CrossRef]
- Chien, H.-C.R.; Lin, L.-L.; Chao, S.-H.; Chen, C.-C.; Wang, W.-C.; Shaw, C.-Y.; Tsai, Y.-C.; Hu, H.-Y.; Hsu, W.-H. Purification, characterization, and genetic analysis of a leucine aminopeptidase from Aspergillus sojae. Biochim. Biophys. Acta Gene Struct. Expr. 2002, 1576, 119–126. [Google Scholar] [CrossRef]
- Grembecka, J.; Mucha, A.; Cierpicki, A.T.; Kafarski, P. The Most Potent Organophosphorus Inhibitors of Leucine Aminopeptidase. Structure-Based Design, Chemistry, and Activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-Guided, Single-Point Modifications in the Phosphinic Dipeptide Structure Yield Highly Potent and Selective Inhibitors of Neutral Aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef]
- Vassiliou, S.; Xeilari, M.; Yiotakis, A.; Grembecka, J.; Pawełczak, M.; Kafarski, P.; Mucha, A. A synthetic method for diversification of the P1′ substituent in phosphinic dipeptides as a tool for exploration of the specificity of the S1′ binding pockets of leucine aminopeptidases. Bioorganic Med. Chem. 2007, 15, 3187–3200. [Google Scholar] [CrossRef]
- Mucha, A.; Kunert, A.; Grembecka, J.; Pawełczak, M.; Kafarski, P. A phosphonamidate containing aromatic N-terminal amino group as inhibitor of leucine aminopeptidase—Design, synthesis and stability. Eur. J. Med. Chem. 2006, 41, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, K.; Talma, M.; Oszywa, B.; Pawełczak, M.; Kafarski, P.; Mucha, A. N-Benzyl Residues as the P1′ Substituents in Phosphorus-Containing Extended Transition State Analog Inhibitors of Metalloaminopeptidases. Molecules 2020, 25, 4334. [Google Scholar] [CrossRef]
- Talma, M.; Mucha, A. P1′ Residue-Oriented Virtual Screening for Potent and Selective Phosphinic (Dehydro) Dipeptide Inhibitors of Metallo-Aminopeptidases. Biomolecules 2020, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals 2019, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H. A new group of organophosphorus compounds. Chem. Ber. 1948, 81, 477–483. [Google Scholar] [CrossRef]
- Carter, H.E.; Frank, R.L.; Johnston, H.W. Carbobenzoxy Chloride and Derivatives. Org. Synth. 2003, 3, 13. [Google Scholar] [CrossRef]
- Matziari, A.Y.M.; Nasopoulou, M. Active methylene phosphinic peptides: A new diversification approach. Org. Lett. 2006, 8, 2317–2319. [Google Scholar] [CrossRef]
- Yiotakis, V.; Vassiliou, A.; Jirácek, S.; Dive, J. Protection of the Hydroxyphosphinyl Function of Phosphinic Dipeptides by Adamantyl. Application to the Solid-Phase Synthesis of Phosphinic Peptides. J. Org. Chem. 1996, 61, 6601–6605. [Google Scholar] [CrossRef] [PubMed]
- Campagne, J.-M.; Coste, J.; Guillou, L.; Heitz, A.; Jouin, P. Solid phase synthesis of phosphinic peptides. Tetrahedron Lett. 1993, 34, 4181–4184. [Google Scholar] [CrossRef]
- Chen, H.; Roques, B.P.; Fournié-Zaluski, M.-C. Design of the first highly potent and selective aminopeptidase N (EC 3.4.11.2) inhibitor. Bioorganic Med. Chem. Lett. 1999, 9, 1511–1516. [Google Scholar] [CrossRef]
- Caldwell, C.G.; Sahoo, S.P.; Polo, S.A.; Eversole, R.R.; Lanza, T.J.; Mills, S.G.; Niedzwiecki, L.M.; Izquierdo-Martin, M.; Chang, B.C.; Harrison, R.K.; et al. Phosphinic acid inhibitors of matrix metalloproteinases. Bioorganic Med. Chem. Lett. 1996, 6, 323–328. [Google Scholar] [CrossRef]
- Mucha, A.; Pawełczak, M.; Hurek, J.; Kafarski, P.; Paweł, M. Synthesis and activity of phosphinic tripeptide inhibitors of cathepsin C. Bioorganic Med. Chem. Lett. 2004, 14, 3113–3116. [Google Scholar] [CrossRef]
- Kende, A.S.; Dong, H.-Q.; Liu, X.; Ebetino, F.H. A useful synthesis of the Phe-Arg phosphinic acid dipeptide isostere. Tetrahedron Lett. 2002, 43, 4973–4976. [Google Scholar] [CrossRef]
- Miller, D.J.; Hammond, S.M.; Anderluzzi, D.; Bugg, T.D.H. Aminoalkylphosphinate inhibitors of D-Ala-D-Ala adding enzyme. J. Chem. Soc. Perkin Trans. 1998, 1, 131–142. [Google Scholar] [CrossRef]
- Allen, M.C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J.M. Renin inhibitors. Synthesis of transition-state analog inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem. 1989, 32, 1652–1661. [Google Scholar] [CrossRef]
- Pícha, J.; Buděšínský, M.; Fiedler, P.; Sanda, M.; Jiráček, J. Synthesis of α-carboxyphosphinopeptides derived from norleucine. Amino Acids 2010, 39, 1265–1280. [Google Scholar] [CrossRef] [PubMed]
- Buchardt, J.; Ferreras, M.; Krog-Jensen, C.; Delaissé, J.-M.; Foged, N.T.; Meldal, M. Phosphinic Peptide Matrix Metalloproteinase-9 Inhibitors by Solid-Phase Synthesis Using a Building Block Approach. Chem. A Eur. J. 1999, 5, 2877–2884. [Google Scholar] [CrossRef]
- Anwer, M.K.; Spatola, A.F. An Advantageous Method for the Rapid Removal of Hydrogenolysable Protecting Groups under Ambient Conditions; Synthesis of Leucine-enkephalin. Synthesis 1980, 1980, 929–932. [Google Scholar] [CrossRef]
- Kline, T.; Andersen, N.H.; Harwood, E.A.; Bowman, J.; Malanda, A.; Endsley, S.; Erwin, A.L.; Doyle, M.; Fong, S.; Harris, A.L.; et al. Potent, Novel in Vitro Inhibitors of thePseudomonasaeruginosaDeacetylase LpxC. J. Med. Chem. 2002, 45, 3112–3129. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.X.Y.; Zou, X.-M.; Niu, Y.; Xu, F.-R.; Mou, K.; Zhou, B.; Wang, C.; Li, Y.-J.; Yang, G.-Y. Synthesis of acyclic analogs of syringolin A as potential 20S proteasome inhibitors. J. Chin. Pharm. Sci. 2010, 19, 423–435. [Google Scholar]
- Strancar, K.; Blanot, D.; Gobec, S. Design, synthesis and structure–activity relationships of new phosphinate inhibitors of MurD. Bioorganic Med. Chem. Lett. 2006, 16, 343–348. [Google Scholar] [CrossRef]
- Hug, G.L.; Bobrowski, K.; Pogocki, D.; Hörner, G.; Marciniak, B.; Hoerner, G. Conformational Influence on the Type of Stabilization of Sulfur Radical Cations in Cyclic Peptides. Chem. Phys. Chem. 2007, 8, 2202–2210. [Google Scholar] [CrossRef]
- Georgiadis, D.; Dive, V.; Yiotakis, A. Synthesis and Comparative Study on the Reactivity of Peptidyl-Type Phosphinic Esters: Intramolecular Effects in the Alkaline and Acidic Cleavage of Methyl β-Carboxyphosphinates. J. Org. Chem. 2001, 66, 6604–6610. [Google Scholar] [CrossRef]
- Koubeissi, A.; Raad, I.; Ettouati, L.; Guilet, D.; Dumontet, C.; Paris, J. Inhibition of P-glycoprotein-mediated multidrug efflux by aminomethylene and ketomethylene analogs of reversins. Bioorganic Med. Chem. Lett. 2006, 16, 5700–5703. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: Broad applications or therapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Oszywa, B.; Makowski, M.; Pawełczak, M. Purification and partial characterization of aminopeptidase from barley (Hordeum vulgare L.) seeds. Plant Physiol. Biochem. 2013, 65, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Doi, H. Substrate specificity of Aminopeptidase from Japanese classified barley flour. J. Food Biochem. 2006, 30, 292–301. [Google Scholar] [CrossRef]
- Oszywa, B.; Pawełczak, M.; Kafarski, P. The influence of α-aminophosphonic acids on the activity of aminopeptidase from barley seeds—an approach to determine the enzyme specificity. Acta Physiol. Plant. 2015, 37, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Volz, K.W.; Lipscomb, W.N.; Takemoto, L.J. Leucine aminopeptidase from bovine lens and hog kidney. Comparison using immunological techniques, electron microscopy, and X-ray diffraction. J. Biol. Chem. 1984, 259, 14757–14761. [Google Scholar] [CrossRef]
- Taylor, A.; Surgenor, T.; Thomson, D.K.; Graham, R.J.; Oettgen, H. Comparison of leucine aminopeptidase from human lens, beef lens and kidney, and hog lens and kidney. Exp. Eye Res. 1984, 38, 217–229. [Google Scholar] [CrossRef]
- Sträter, N.; Lipscomb, W.N. Transition State Analog L-Leucinephosphonic Acid Bound to Bovine Lens Leucine Aminopeptidase: X-ray Structure at 1.65.ANG. Resolution in a New Crystal Form. Biochemistry 1995, 34, 9200–9210. [Google Scholar] [CrossRef]
- Duprez, K.; Scranton, M.A.; Walling, L.L.; Fan, L. Structure of tomato wound-induced leucine aminopeptidase sheds light on substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 1649–1658. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Noble, F.; Mothé, A.; Meudal, H.; Coric, P.; Danascimento, S.; Roques, B.P.; George, P.; Fournié-Zaluski, M.-C. Phosphinic derivatives as new dual enkephalin-degrading enzyme inhibitors: Synthesis, biological properties, and antinociceptive activities. J. Med. Chem. 2000, 43, 1398–1408. [Google Scholar] [CrossRef]
- Spackman, D.H.; Smith, E.L.; Brown, D.M. Leucine aminopeptidase. IV. Isolation and properties of the enzyme from swine kidney. J. Biol. Chem. 1955, 212, 255–269. [Google Scholar] [CrossRef]
- Ledeme, N.; Hennon, G.; Vincent-Fiquet, O.; Plaquet, R. Purification and enzymatic properties of an l-leucine aminopeptidase from swine liver. Biochim. Biophys. Acta Enzym. 1981, 660, 262–270. [Google Scholar] [CrossRef]
- Lejczak, B.; Kafarski, P.; Zygmunt, J. Inhibition of aminopeptidases by aminophosphonates. Biochem. 1989, 28, 3549–3555. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Isley, T.C.; Wolfeden, R. Alfa-Aminoaldehydes: Transition State Analogue Inhibitors of Leucine Aminopeptidase. Biochemistry 1982, 21, 4177–4180. [Google Scholar] [CrossRef] [PubMed]
- Frisch, D.J.F.M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | KI [μM] | ||
|---|---|---|---|---|
| SsLAP | HvLAP | |||
| 1 | Met-hPheP[CH2]Gly-OMe | 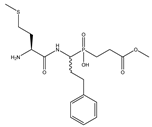 | 532 ± 46 | 38 ± 9 |
| 2 | Met-LeuP[CH2]Gly-OMe | 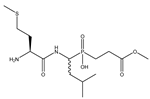 | 1090 ± 73 | 649 ± 43 |
| 3 | Ser-LeuP[CH2]Gly-OMe |  | 14 ± 1 | 121 ± 19 |
| 4 | Met-hPheP[CH2]Gly | 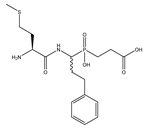 | 229 ± 40 | 228 ± 10 |
| 5 | Met-LeuP[CH2]Gly | 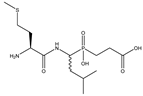 | 3618 ± 433 | 1035 ± 102 |
| 6 | Bip-hPheP[CH2]Gly-OMe | 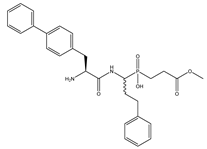 | 175 ± 3 | 878 ± 70 |
| 7 | Bip-LeuP[CH2]Gly-OMe | 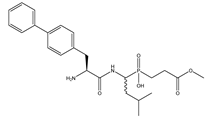 | 130 ± 14 | 1303 ± 84 |
| 8 | Bpa-hPheP[CH2]Gly-OMe | 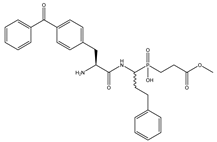 | 95 ± 9 | 159 ± 25 |
| 9 | Bpa-LeuP[CH2]Gly-OMe | 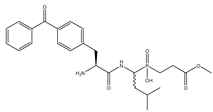 | 507 ± 31 | 797 ± 76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jewgiński, M.; Haremza, K.; de los Santos, J.M.; Es Sbai, Z.; Oszywa, B.; Pawełczak, M.; Palacios, F.; Latajka, R. Phosphinotripeptidic Inhibitors of Leucylaminopeptidases. Int. J. Mol. Sci. 2021, 22, 5090. https://doi.org/10.3390/ijms22105090
Jewgiński M, Haremza K, de los Santos JM, Es Sbai Z, Oszywa B, Pawełczak M, Palacios F, Latajka R. Phosphinotripeptidic Inhibitors of Leucylaminopeptidases. International Journal of Molecular Sciences. 2021; 22(10):5090. https://doi.org/10.3390/ijms22105090
Chicago/Turabian StyleJewgiński, Michał, Kinga Haremza, Jesús M. de los Santos, Zouhair Es Sbai, Bartosz Oszywa, Małgorzata Pawełczak, Francisco Palacios, and Rafał Latajka. 2021. "Phosphinotripeptidic Inhibitors of Leucylaminopeptidases" International Journal of Molecular Sciences 22, no. 10: 5090. https://doi.org/10.3390/ijms22105090