
A Highly Versatile Polymer Network Based on Liquid Crystalline Dendrimers

Abstract
1. Introduction
2. Results and Discussion
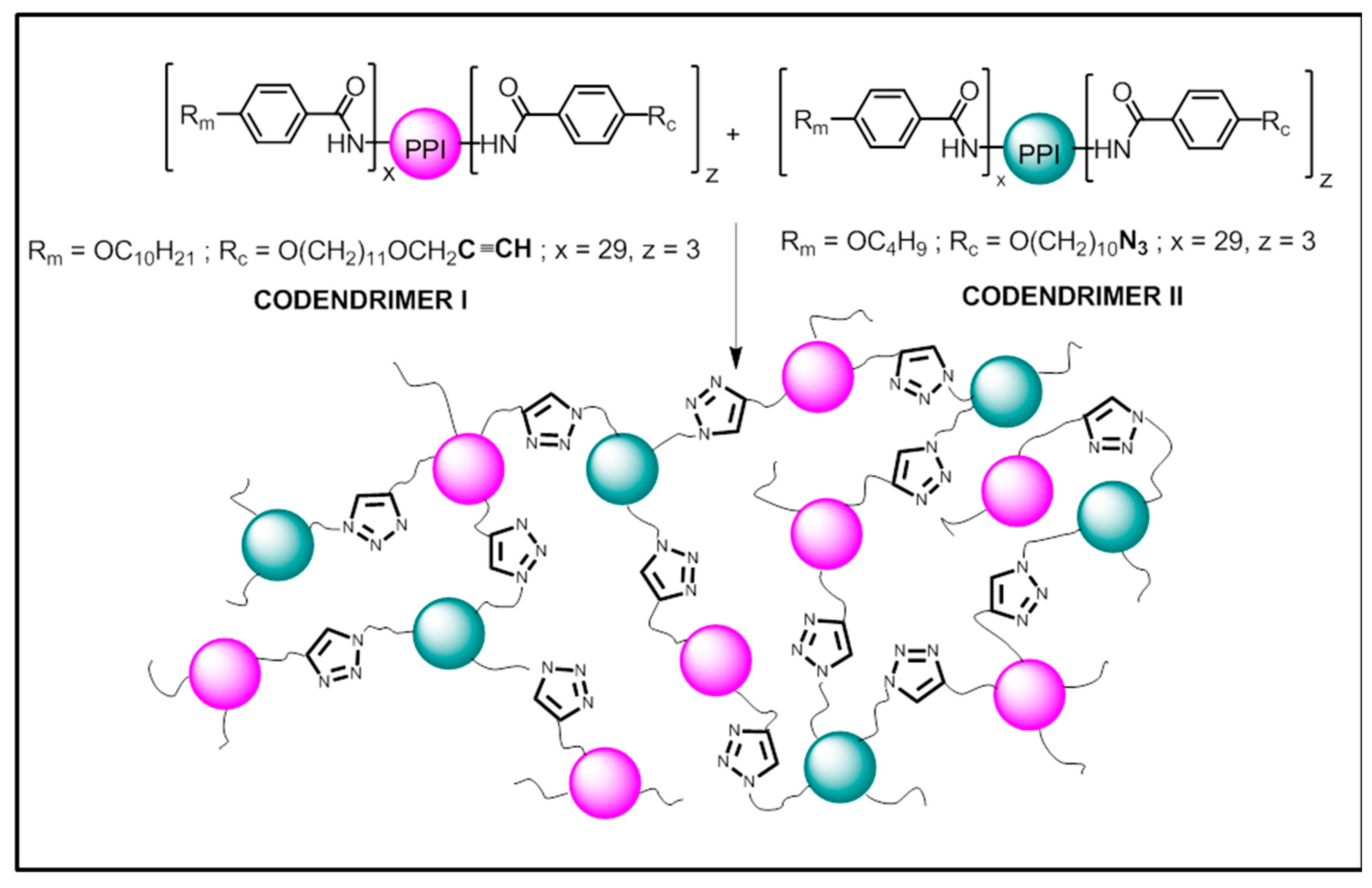
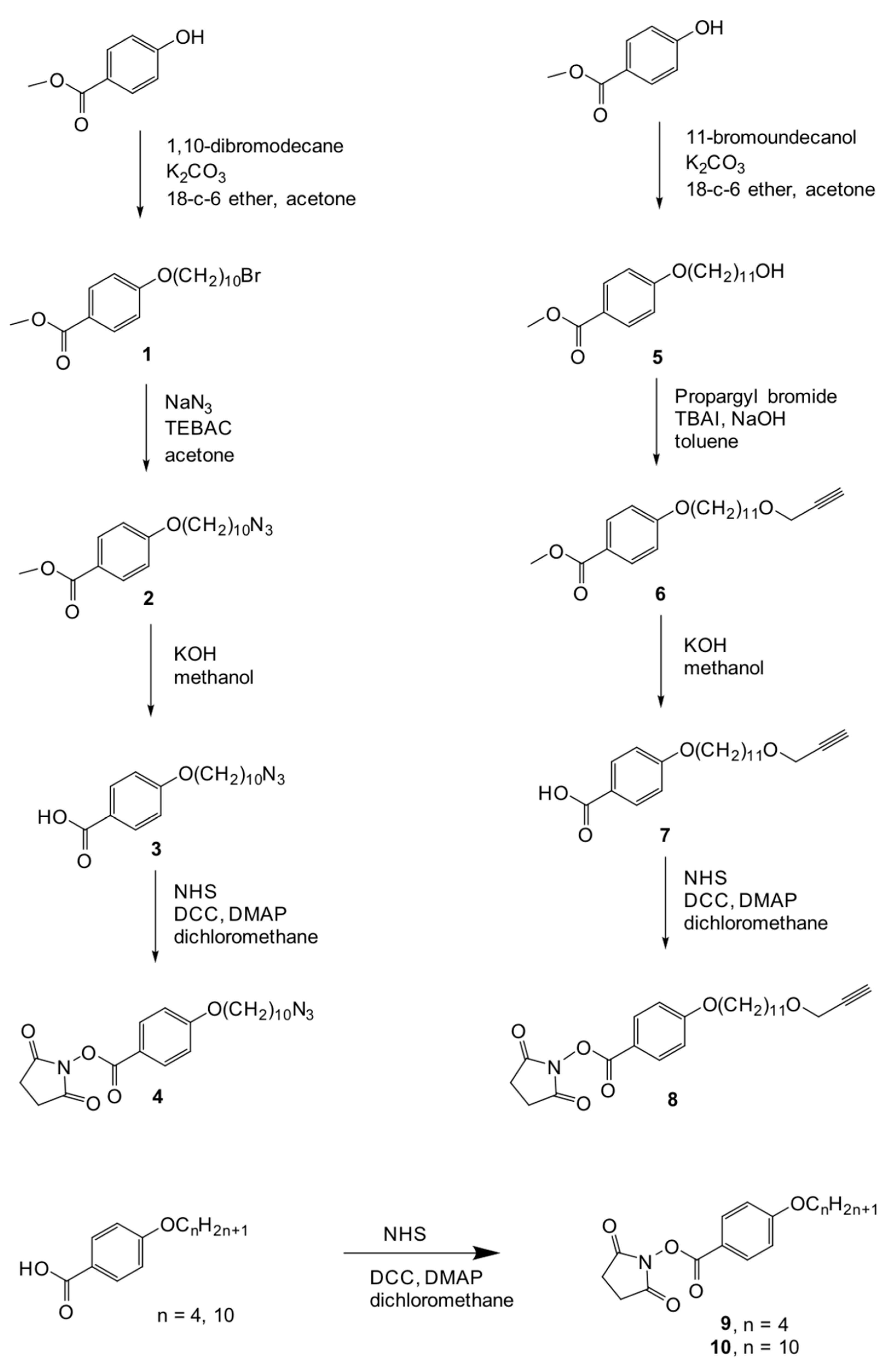
2.1. Synthesis
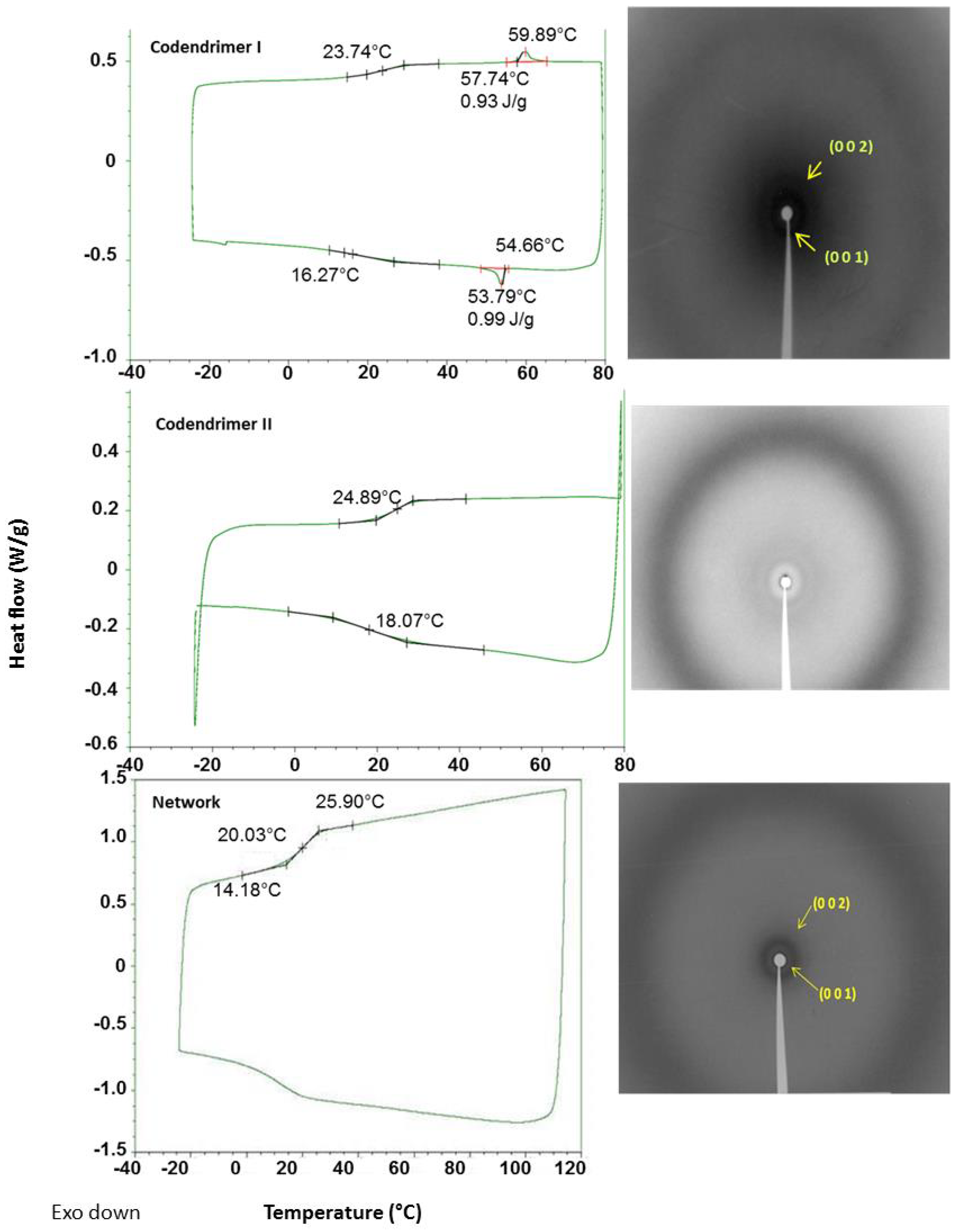
2.2. Thermal Properties
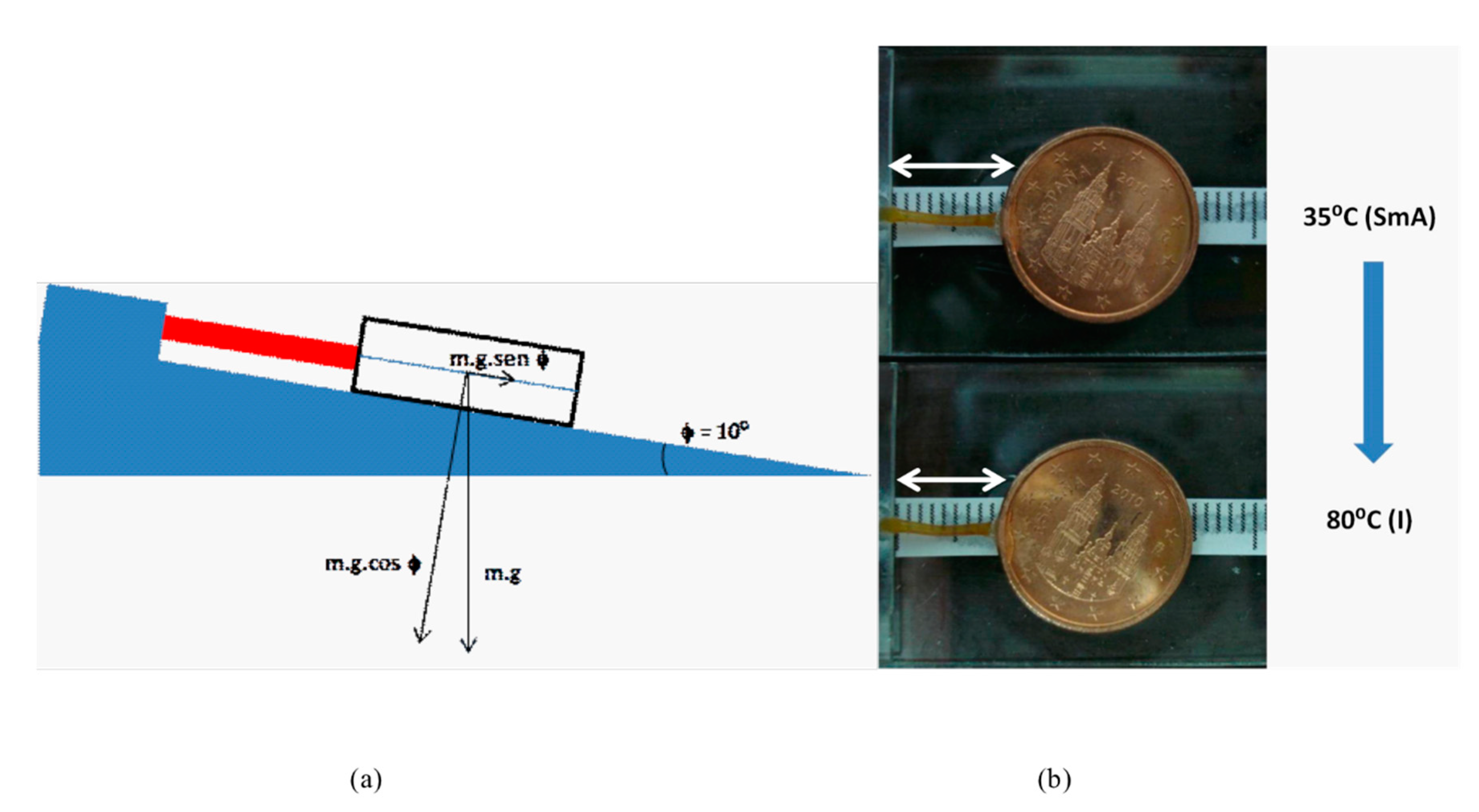
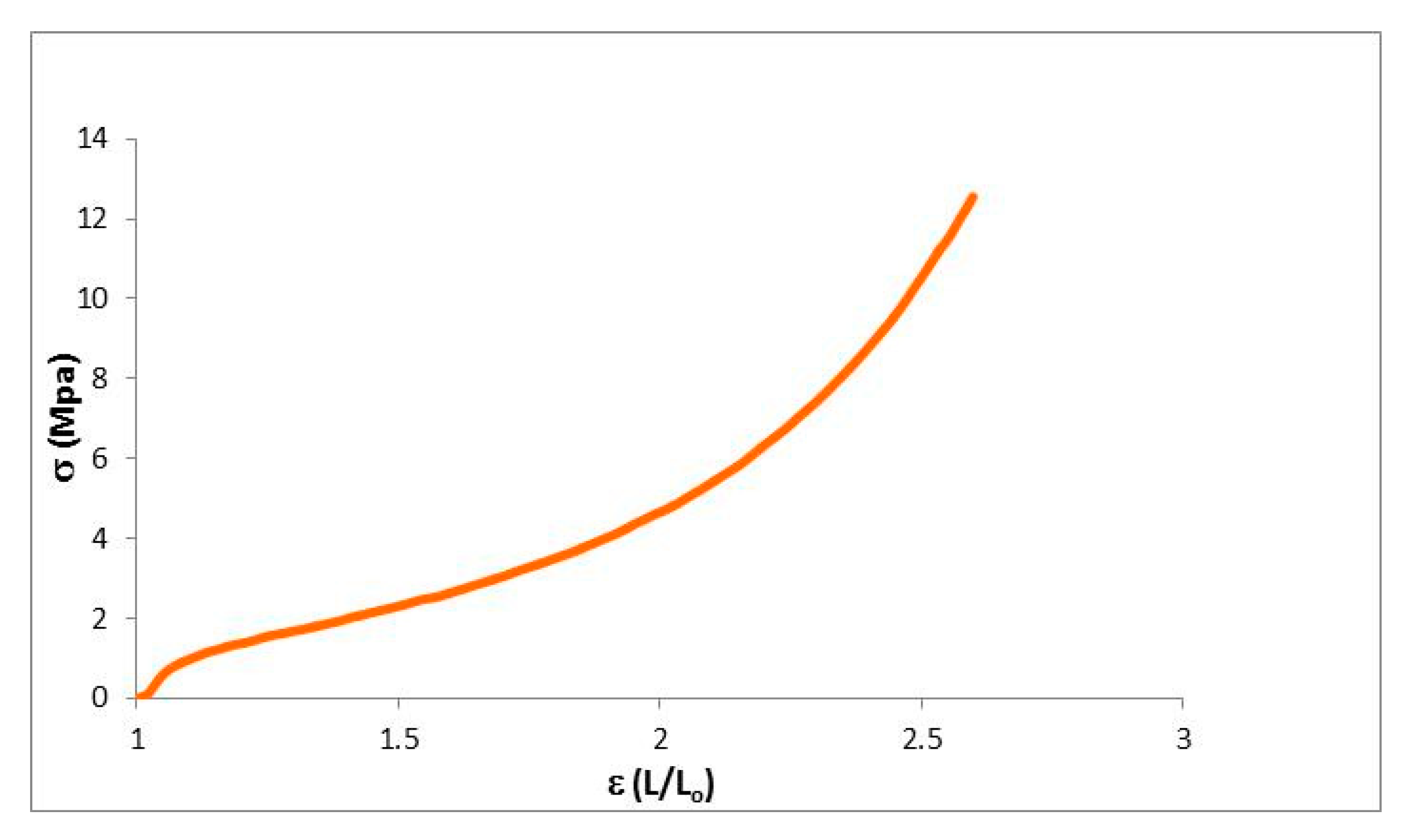
2.3. Mechanical Properties
2.4. Microstructure and Encapsulation Studies
2.4.1. Sorption of Solvents
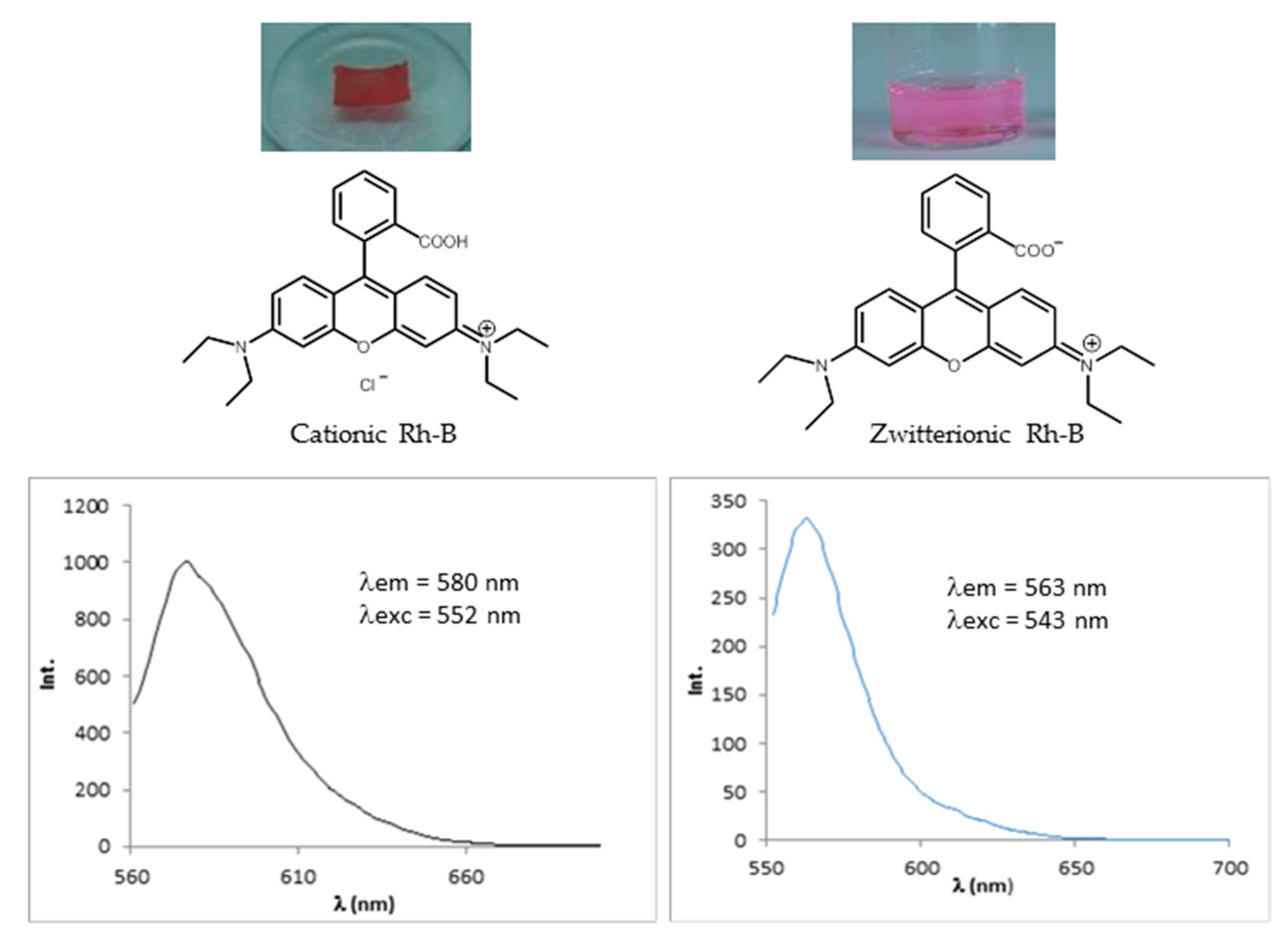
2.4.2. Encapsulation of Dyes
Rhodamine B
Disperse Red 1 and β-Carotene
3. Materials and Methods
3.1. Synthesis and Characterization of the Codendrimers
3.2. Preparation of the Dendrimeric Network
3.3. General Method for the Encapsulation/Release of Dyes
3.4. Techniques
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Liu, J.; Lin, S.; Zhao, X. Hydrogel Machines. Mater. Today 2020, 36, 102–124. [Google Scholar] [CrossRef]
- Yang, C.; Suo, Z. Hydrogel Ionotronics. Nat. Rev. Mater. 2018, 3, 125–142. [Google Scholar] [CrossRef]
- Bay, L.; West, K.; Sommer-Larsen, P.; Skaarup, S.; Benslimane, M. A conducting polymer artificial muscle with 12% linear strain. Adv. Mater. 2003, 15, 310–313. [Google Scholar] [CrossRef]
- Chortos, A.; Hajiesmaili, E.; Morales, J.; Clarke, D.R.; Lewis, J.A. 3D Printing of Interdigitated Dielectric Elastomer Actuators. Adv. Funct. Mater. 2020, 30, 1907375. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, J.; Ikeda, T.; Jiang, L. Bio-inspired liquid crystal actuator materials. J. Mater. Chem. C 2019, 7, 3413–3428. [Google Scholar] [CrossRef]
- Jiang, H.; Li, C.; Huang, X. Actuators based on liquid crystalline elastomer materials. Nanoscale 2013, 5, 5225–5240. [Google Scholar] [CrossRef]
- Pei, Z.; Yang, Y.; Chen, Q.; Terentjev, E.M.; Wei, Y.; Ji, Y. Mouldable liquid-crystalline elastomer actuators with exchangeable covalent bonds. Nat. Mater. 2014, 13, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ohm, C.; Brehmer, M.; Zentel, R. Liquid crystalline elastomers as actuators and sensors. Adv. Mater. 2010, 22, 3366–3387. [Google Scholar] [CrossRef]
- de Haan, L.T.; Verjans, J.M.N.; Broer, D.J.; Bastiaansen, C.W.M.; Schenning, A.P.H.J. Humidity-Responsive Liquid Crystalline Polymer Actuators with an Asymmetry in the Molecular Trigger That Bend, Fold, and Curl. J. Am. Chem. Soc. 2014, 136, 10585–10588. [Google Scholar] [CrossRef]
- Li, S.; Tu, Y.Q.; Bai, H.D.; Hibi, Y.; Wiesner, L.W.; Pan, W.Y.; Wang, K.Y.; Giannelis, E.P.; Shepherd, R.F. Simple Synthesis of Elastomeric Photomechanical Switches That Self-Heal. Macromol. Rapid Commun. 2019, 40, 1800815. [Google Scholar] [CrossRef]
- McBride, M.K.; Gong, T.; Nair, D.P.; Bowman, C.N. Photo-mediated copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) “click” reactions for forming polymer networks as shape memory materials. Polymer 2014, 55, 5880–5884. [Google Scholar] [CrossRef] [PubMed]
- de Haan, L.T.; Schenning, A.P.H.J.; Broer, D.J. Programmed morphing of liquid crystal networks. Polymer 2014, 55, 5885–5896. [Google Scholar] [CrossRef]
- Ube, T.; Ikeda, T. Photomobile Polymer Materials with Crosslinked Liquid-Crystalline Structures: Molecular Design, Fabrication, and Functions. Angew. Chem. Int. Ed. 2014, 53, 10290–10299. [Google Scholar] [CrossRef] [PubMed]
- Schattling, P.; Jochum, F.D.; Theato, P. Multi-stimuli responsive polymers- the all-in-one talents. Polym. Chem. 2014, 5, 25–36. [Google Scholar] [CrossRef]
- Wang, C.J.; Sim, K.; Chen, J.; Kim, H.; Rao, Z.Y.; Li, Y.H.; Chen, W.Q.; Song, J.Z.; Verduzco, R.; Yu, C.J. Soft Ultrathin Electronics Innervated Adaptive Fully Soft Robots. Adv. Mater. 2018, 30, 1706695. [Google Scholar] [CrossRef]
- de Gennes, P.G. Réflexions sur un type de polymères nématiques. C. R. Acad. Sci. Paris 1975, 281, 101. [Google Scholar]
- Fréchet, J.M.J.; Tomalia, D.A. Dendrimers and other Dendritic Polymers; John Wiley and Sons: Chichester, UK, 2001. [Google Scholar]
- Astruc, D.; Boisselier, E.; Ornelas, C. Dendrimers designed for functions: From Physical, Photophysical, and Supramolecular Properties to Applications in Sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem. Rev. 2010, 110, 1857–1959. [Google Scholar] [CrossRef]
- Rosen, B.M.; Wilson, C.J.; Wilson, D.A.; Peterca, M.; Imam, M.R.; Percec, V. Dendron-mediated self-assembly, disassembly, and self-organization of complex systems. Chem. Rev. 2009, 109, 6275–6540. [Google Scholar] [PubMed]
- Smith, D.K.; Hirst, A.R.; Love, C.S.; Hardy, J.G.; Brignell, S.V.; Huang, B. Self-assembly using dendritic building blocks-towards controllable nanomaterials. Prog. Polym. Sci. 2005, 30, 220–293. [Google Scholar] [CrossRef]
- Donnio, B.; Guillon, D. Liquid crystalline dendrimers and polypedes. Adv. Polym. Sci. 2006, 201, 45–155. [Google Scholar]
- Marcos, M.; Martín-Rapún, R.; Omenat, A.; Serrano, J.L. Highly congested liquid crystal structures: Dendrimers, dendrons, dendronized and hyperbranched polymers. Chem. Soc. Rev. 2007, 36, 1889–1901. [Google Scholar] [CrossRef]
- Barberá, J.; Donnio, B.; Gehringer, L.; Guillon, D.; Marcos, M.; Omenat, A.; Serrano, J.L. Self-organization of nanostructured functional dendrimers. J. Mater. Chem. 2005, 15, 4093–4105. [Google Scholar] [CrossRef]
- Hernández-Ainsa, S.S.; Marcos, M.M.; Serrano, J.L. Handbook of Liquid Crystals; Goodby, J.W., Collings, P.J., Kato, T., Tschierske, C., Gleeson, H., Raynes, P., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2014; Volume 6. [Google Scholar]
- Tomalia, D.A.; Uppuluri, S.; Swanson, D.R.; Li, J. Dendrimers as reactive modules for the synthesis of new structure-controlled, higher-complexity megamers. Pure Appl. Chem. 2000, 72, 2343–2358. [Google Scholar] [CrossRef]
- Dvornic, P.R.; Li, J.; de Leuze-Jalloulli, A.M.; Reeves, S.D.; Owen, M.J. Nanostructured dendrimer-based Networks with hydrophylic polyamidoamine and hydrophobic organosilicon domains. Macromolecules 2002, 35, 9323–9333. [Google Scholar] [CrossRef]
- Sarkar, A.; Carver, P.I.; Zhang, T.; Merrington, A.; Bruza, K.J.; Rousseau, J.L.; Keinath, S.E.; Dvornic, P.R. Dendrimer-based coatings for surface modification of polyamide reverse osmosis membranes. J. Membr. Sci. 2010, 349, 421–428. [Google Scholar] [CrossRef]
- Marcos, M.; Martín-Rapún, R.; Serrano, J.L.; Sánchez-Ferrer, A. Liquid crystalline dendritic networks derived from poly(propylene imine) (PPI) dendrimers. Macromol. Rapid Commun. 2005, 26, 1604–1608. [Google Scholar] [CrossRef]
- Cervera-Procas, R.; Sánchez-Somolinos, C.; Serrano, J.L.; Omenat, A. A Polymer Network Prepared by the Thiol-yne Photocrosslinking of a Liquid Crystalline Dendrimer. Macromol. Rapid Commun. 2013, 34, 498–503. [Google Scholar] [CrossRef]
- Lin, Y.; Sun, J. Vinyl-terminated liquid-crystalline dendrimers based on dendritic polyols and their siloxane-based elastomers. J. Polym. Sci. Polym.Chem. 2013, 51, 71–83. [Google Scholar] [CrossRef]
- Fernández-Barbero, A.; Suárez, I.J.; Sierra-Martín, B.; Fernández-Nieves, A.; de las Nieves, F.J.; Márquez, M.; Rubio-Retama, J.; López-Cabarcos, E. Gels and microgels for nanotechnological applications. Adv. Colloid Interface Sci. 2009, 147–148, 88–108. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tørnoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.H.; Facher, A.; Lattermann, G.; Diele, S. Poly(propyleneimine) dendromesogens with hexagonal columnar mesophase. Adv. Mater. 1997, 9, 398–403. [Google Scholar] [CrossRef]
- Percec, V.; Rudick, J.G.; Peterca, M.; Heiney, P.A. Nanomechanical function from self-organizable dendronized helical polyphenylacetylenes. J. Am. Chem. Soc. 2008, 130, 7503–7508. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.V.; Namdas, E.B. Lasing action of rhodamine B in polyacrylic acid films. Appl. Phys. B 1997, 64, 419–422. [Google Scholar] [CrossRef]
- Fikry, M.; Omar, M.M.; Ismail, L.Z. Effect of host medium on the fluorescence emission intensity of rhodamine B in liquid and solid phase. J. Fluoresc. 2009, 19, 741–746. [Google Scholar] [CrossRef]
- Dubin, P.L.; Edwards, S.L.; Kaplan, J.L.; Mehta, M.S.; Tomalia, D.A.; Xia, J. Carboxylated starburst dendrimers as calibration standards for aqueous size exclusion chromatography. Anal. Chem. 1992, 64, 2344–2347. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Phase Transitions a,b,c | hkl e | dexp f (Å) | dcalc f (Å) | σ g (Å) |
|---|---|---|---|---|---|
| Codendrimer I | g 24 SmA 60 (0.9) I | 001 | 45.3 | 44.9 | 44.9 |
| 002 | 22.2 | 22.5 | |||
| Codendrimer II | g 25 I | ||||
| Network | g 20 SmA 57 d I | 001 | 42.2 | 42.4 | 42.4 |
| 002 | 21.3 | 21.2 |
| Young Module (MPa) | Elastic Deform. (%) | Max. Charge (MPa) | Max. Deform. (%) | |
|---|---|---|---|---|
| Network | 17.4 | 5 | 12.7 | 159.8 |
| Solvent | Network wt (mg) [Wd] | Swollen Network wt (mg) [Ws + Wd] | Swelling Ratio [SR = Ws/Wd] |
|---|---|---|---|
| Dichloromethane | 8.2 | 88.5 | 9.8 |
| Tetrahydrofuran | 3.8 | 36.5 | 8.6 |
| Ethanol | 8.1 | 24.4 | 2.0 |
| Water | 11.1 | 21.4 | 0.9 |
| n-Hexane | 13.5 | 20.8 | 0.5 |
| Network Wt. (mg) | Rh-B/EtOH Concentration | Swollen Network (mg) | Rh-B Desorbed a (μg) | Rh-B/EtOH (in the Network) Concentration |
|---|---|---|---|---|
| 31.6 | 1.25 × 10−4 M | 75.7 | 8.3 | 3.1 × 10−4 M |
| 29.9 | 1.0 × 10−3 M | 72.2 | 68.5 | 2.6 × 10−3 M |
| 29.5 | 1.0 × 10−2 M | 68.1 | 584 | 2.5 × 10−2 M |
| Initial Dye/Solvent Concentration | Dye Desorbed a (μg) | Dye/Solvent (in the Network) Concentration | |
|---|---|---|---|
| Rh-B/EtOH | 1.25 × 10−4 M | 8.3 | 3.1 × 10−4 M |
| DR1/THF | 9.32 × 10−5 M | 5.9 | 10.5 × 10−5 M |
| bC/THF | 2.68 × 10−4 M | 8.0 | 2.21 × 10−4 M |
| Compound | Theoretical a | NMR b | MALDI | GPC c | ||||
|---|---|---|---|---|---|---|---|---|
| Mn (Ratio) | Mn (Ratio) | Mn | Mw | PDI d | Mn | Mw | PDI d | |
| Codendrimer I | 12,050 (29/3) | 11,790 (28.5/2.6) | 11,737 | 11,744 | 1.001 | 5728 | 6258 | 1.09 |
| Codendrimer II | 9528 (29/3) | 9555 (27.6/3.9) | 8949 | 8922 | 1.003 | 4109 | 4743 | 1.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervera-Procas, R.; Serrano, J.-L.; Omenat, A. A Highly Versatile Polymer Network Based on Liquid Crystalline Dendrimers. Int. J. Mol. Sci. 2021, 22, 5740. https://doi.org/10.3390/ijms22115740
Cervera-Procas R, Serrano J-L, Omenat A. A Highly Versatile Polymer Network Based on Liquid Crystalline Dendrimers. International Journal of Molecular Sciences. 2021; 22(11):5740. https://doi.org/10.3390/ijms22115740
Chicago/Turabian StyleCervera-Procas, Ramón, José-Luis Serrano, and Ana Omenat. 2021. "A Highly Versatile Polymer Network Based on Liquid Crystalline Dendrimers" International Journal of Molecular Sciences 22, no. 11: 5740. https://doi.org/10.3390/ijms22115740
APA StyleCervera-Procas, R., Serrano, J.-L., & Omenat, A. (2021). A Highly Versatile Polymer Network Based on Liquid Crystalline Dendrimers. International Journal of Molecular Sciences, 22(11), 5740. https://doi.org/10.3390/ijms22115740