
DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity



Abstract
:1. Introduction
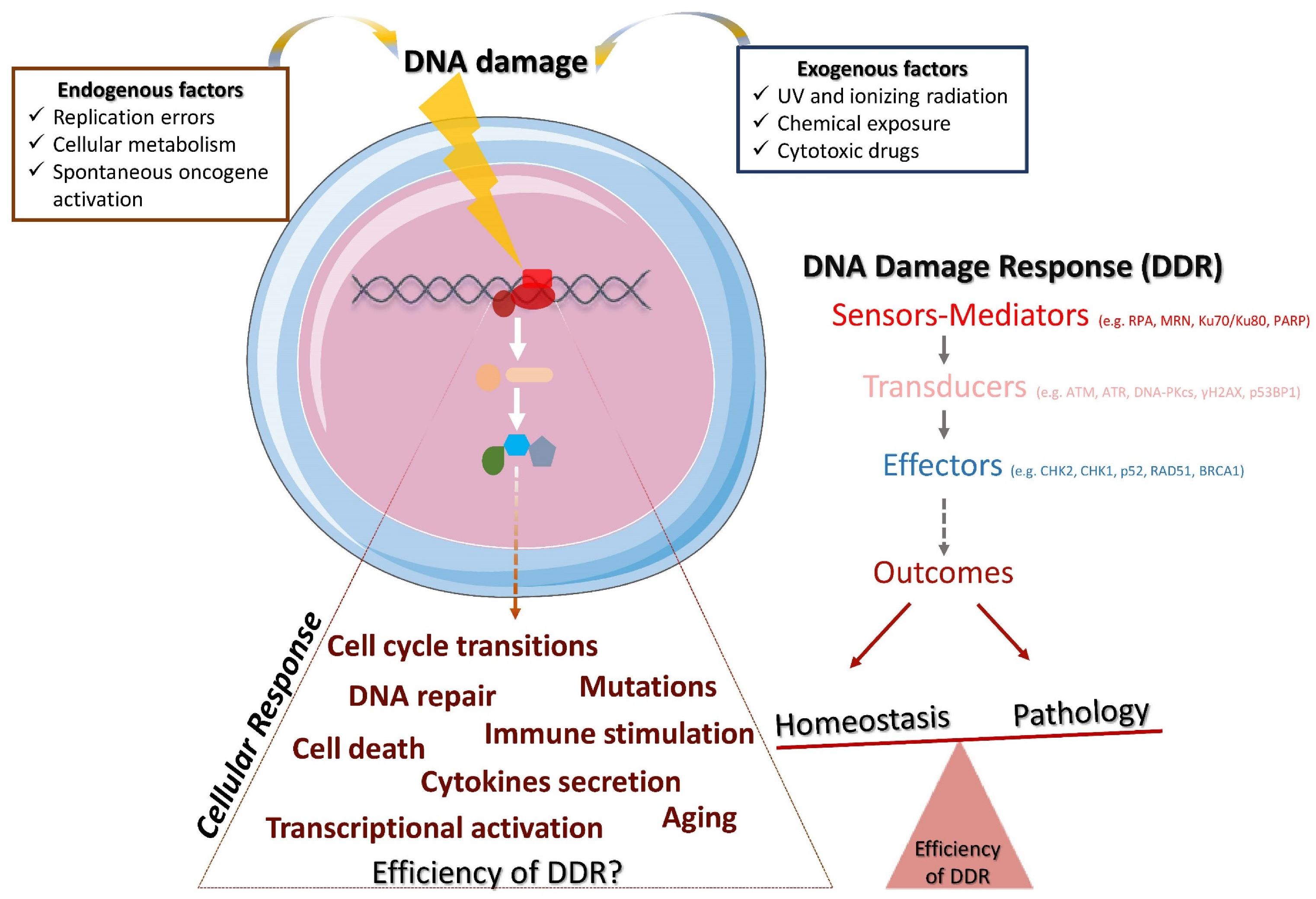
1.1. Definition and Components of DDR
1.2. DNA Repair Pathways
1.3. Triggers of DDR
1.4. DDR and the Immune Response
2. DDR and Immunity: A Dubious Relationship That May Culminate in Autoimmunity
The Early Steps and Important Findings
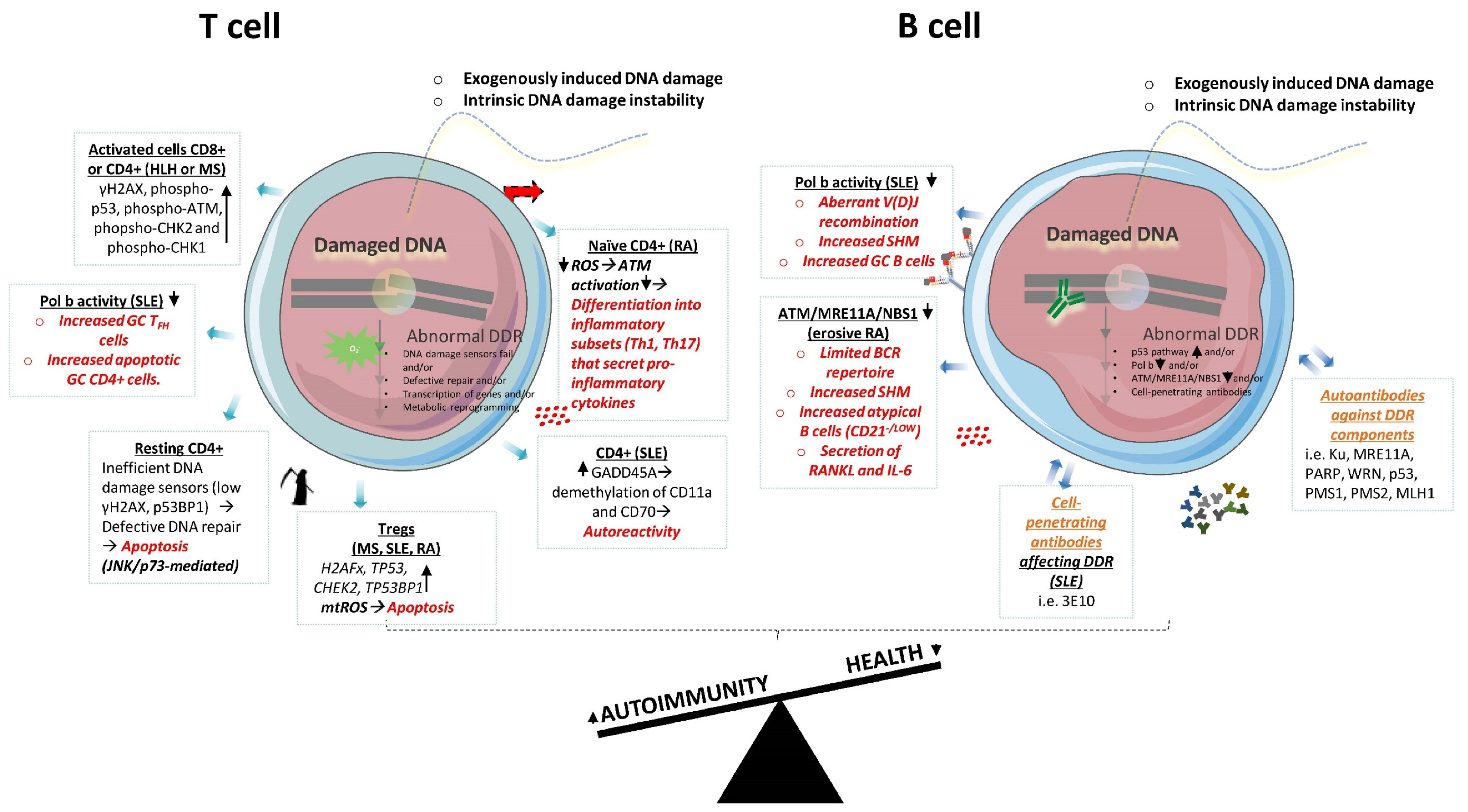
3. Linking T Cell DDR with Autoimmunity
4. Linking B Cell DDR with Autoimmunity
5. Aberrant DDR in Innate Cells May Exacerbate Aberrant Immune Responses in Adaptive Cells: The Case of Dendritic Cells (DCs)
6. The Role of DDR in Other Cells of the Adaptive Immunity: NK Cells, γδ Τ and NKT Cells
7. DDR and Cytokines in Autoimmunity
7.1. DDR May Lead to Exaggerated Cytokine Production and Promote Autoimmune Inflammation
7.2. Cell Free DNA may Induce Cytokine Production
8. Therapeutic Manipulation of DDR in Autoimmunity
8.1. Lessons Learnt from Cancer
8.2. DDR Targeting in Autoimmune Diseases
9. Synthesis and Concluding Remarks
Open Questions
- 1.
- Comprehensive understanding of the DDR cascade in B and T cells in autoimmune diseases:
- (a)
- Do DDR alterations in lymphocytes drive their autoreactive phenotype (e.g., plasmablast formation, antibody production, inflammatory cytokine secretion)?
- (b)
- How do these alterations imprint on the inflammatory cascade driven by autoreactive B and T cells?
- (c)
- Does reversing the DDR aberrancies in lymphocytes restore homeostasis, leading to the remission of autoimmunity?
- 2.
- Delineation of the DDR molecular pathways in lymphocytes in autoimmunity:
- (a)
- Combine genomic, proteomic and epigenetic technologies to uncover new checkpoint molecules on the DDR aberrancies in autoreactive lymphocytes, which will provide further insights for future therapeutic interventions.
- (b)
- Considering that metabolism orchestrates B and T cells functions, revealing a potential crosstalk of metabolic and DDR pathways operating in autoreactive lymphocytes may provide further knowledge for the disturbed self-tolerance.
- (c)
- Can early DDR events drive therapeutic decisions in autoimmunity?
- 3.
- Do the DDR pathways function differently in multi-organ damages in systemic autoimmunity (e.g., SLE, AGS, RA) compared to organ-specific autoimmunity (e.g., MS, psoriasis, type 1 diabetes)? What are the distinct pathways involved in each disease?
- 4.
- Do current therapeutic regimens affect the DDR in adaptive immune cells? Do they deregulate the DDR? For instance, JAK inhibitors that are used for cytokine targeting may attenuate the DDR and increase DNA damage cargo [118], and thus influence the resistance to therapy.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Molecule | Detailed name or function |
| AGO1 | Protein argonaute-1 |
| APEX1 | Apurinic/apyrimidinic endodeoxyribonuclease 1 protein |
| ATM | Ataxia telangiectasia-mutated protein kinase |
| ATR | Ataxia telangiectasia- and Rad3-related protein kinase |
| AURKA | Aurora kinase A |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BRCA2 | Breast cancer type 2 susceptibility protein |
| CHK1 | Checkpoint protein kinase 1 |
| CHK2 | Checkpoint protein kinase 2 |
| DNA-PKcs | Catalytic subunit of DNA-dependent protein kinase |
| GADD45A | Growth arrest and DNA damage-inducible alpha gene |
| H2AFx | H2A histone family member X |
| HMGB1 | High-mobility group protein B1 |
| IFIT5 | Interferon-induced protein with tetratricopeptide repeats 5 |
| IFN-γ | Interferon gamma 1 |
| IL-6 | Interleukin 6 |
| Ku | Dimeric protein complex: heterodimer of two polypeptides, Ku70 and Ku80 |
| Ku70 | Protein encoded by the XRCC6 gene |
| Ku80 | Protein encoded by the XRCC5 gene |
| MAPKAPK3 | MAP kinase-activated protein kinase 3 |
| MDM2 | E3 ubiquitin–protein ligase |
| MLH1 | DNA mismatch repair protein |
| MRE11A | Meiotic recombination 11 homolog; double-strand break repair protein |
| MRN | Complex-interacting protein complex consisting of MRE11A, RAD51 and NBS1 |
| NBS1 | Nijmegen breakage syndrome protein 1; DNA repair and telomere maintenance protein |
| p53 | Tumor protein encoded by TP53 gene |
| p53BP1 | Tumor protein p53 binding protein 1 encoded by TP53BP1 gene |
| PADI4 | Protein arginine deiminase type-4 |
| PARP | Poly (ADP-ribose) polymerase family of proteins |
| PMS1 | Protein homolog 1; mismatch repair system component |
| PMS2 | Protein homolog 2; mismatch repair system component |
| POLB | DNA polymerase beta |
| RAD51 | DNA repair protein homolog 1 |
| RAG | Recombination-activating gene enzyme |
| RANKL | Receptor activator of nuclear factor kappa-Β ligand protein; member of the tumor necrosis factor (TNF) cytokine family |
| RGS3 | Regulator of G-protein signaling 3 |
| RPA | Replication protein A |
| TNF-α | Tumor necrosis factor alpha cytokine |
| TREX1 | Three-prime repair exonuclease 1 |
| UBE2S | Ubiquitin-conjugating enzyme E2 S |
| VRK1 | Vaccinia related kinase 1 |
| WEE1 | G2 checkpoint kinase |
| WRN | Werner syndrome ATP-dependent helicase |
| γH2AX | Phosphorylated form of histone H2AX protein encoded by H2AFx gene |
| The UniProt Consortium: UniProt: the universal protein knowledgebase in 2021 [119]; GeneCards: the human gene database [120] | |
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nat. Cell Biol. 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2010, 3, a000745. [Google Scholar] [CrossRef] [PubMed]
- Stratigopoulou, M.; Van Dam, T.P.; Guikema, J.E.J. Base Excision Repair in the Immune System: Small DNA Lesions with Big Consequences. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nat. Cell Biol. 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.-B.S.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nat. Cell Biol. 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.-B.S.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Agami, R.; Bernards, R. Distinct Initiation and Maintenance Mechanisms Cooperate to Induce G1 Cell Cycle Arrest in Response to DNA Damage. Cell 2000, 102, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Neves-Costa, A.; Moita, L.F. Modulation of inflammation and disease tolerance by DNA damage response pathways. FEBS J. 2016, 284, 680–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y. DNA damage: A trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat. Rev. Immunol. 2006, 6, 261–270. [Google Scholar] [CrossRef]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Moretton, A.; Loizou, J.I. Interplay between Cellular Metabolism and the DNA Damage Response in Cancer. Cancers 2020, 12, 2051. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Bednarski, J.J.; Sleckman, B.P. At the intersection of DNA damage and immune responses. Nat. Rev. Immunol. 2019, 19, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Lee, B.J.; Zha, S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair 2020, 94, 102874. [Google Scholar] [CrossRef] [PubMed]
- Methot, S.; Di Noia, J. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv. Immunol. 2017, 133, 37–87. [Google Scholar] [CrossRef]
- Oster, S.; Aqeilan, R.I. Programmed DNA Damage and Physiological DSBs: Mapping, Biological Significance and Perturbations in Disease States. Cells 2020, 9, 1870. [Google Scholar] [CrossRef]
- Hoeijmakers, J. (Jan) DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409. [Google Scholar] [CrossRef] [Green Version]
- Chatzinikolaou, G.; Karakasilioti, I.; Garinis, G.A. DNA damage and innate immunity: Links and trade-offs. Trends Immunol. 2014, 35, 429–435. [Google Scholar] [CrossRef]
- Heylmann, D.; Ponath, V.; Kindler, T.; Kaina, B. Comparison of DNA repair and radiosensitivity of different blood cell populations. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Heylmann, D.; Rödel, F.; Kindler, T.; Kaina, B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1846, 121–129. [Google Scholar] [CrossRef]
- Ioannidou, A.; Goulielmaki, E.; Garinis, G.A. DNA Damage: From Chronic Inflammation to Age-Related Deterioration. Front. Genet. 2016, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Travers, P.; Walport, M. Immunobiology. In The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Bashir, S.; Harris, G.; Denman, M.A.; Blake, D.R.; Winyard, P. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann. Rheum. Dis. 1993, 52, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, B.; Scharff, M.D. Somatic mutation of the T15 heavy chain gives rise to an antibody with autoantibody specificity. Proc. Natl. Acad. Sci. USA 1984, 81, 5841–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, G.; Cramp, W.; Edwards, J.; George, A.; Sabovljev, S.; Hart, L.; Hughes, G.; Denman, A.; Yatvin, M. Radiosensitivity of Peripheral Blood Lymphocytes in Autoimmune Disease. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1985, 47, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Harris, G.; Lawley, P.; Asbery, L.; Denman, A.; Hylton, W. Defective repair of 06-methylguanine in autoimmune diseases. Lancet 1982, 320, 952–956. [Google Scholar] [CrossRef]
- Yang, Y.-G.; Lindahl, T.; Barnes, D.E. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.; Newman, W.G.; Dean, J.; Patrick, T.; Parmar, R.; Flintoff, K.; Robins, P.; Harvey, S.; Hollis, T.; O’Hara, A.; et al. Heterozygous Mutations in TREX1 Cause Familial Chilblain Lupus and Dominant Aicardi-Goutières Syndrome. Am. J. Hum. Genet. 2007, 80, 811–815. [Google Scholar] [CrossRef]
- Rice, G.; Patrick, T.; Parmar, R.; Taylor, C.F.; Aeby, A.; Aicardi, J.; Artuch, R.; Montalto, S.A.; Bacino, C.A.; Barroso, B.; et al. Clinical and Molecular Phenotype of Aicardi-Goutières Syndrome. Am. J. Hum. Genet. 2007, 81, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.; Maagdenberg, A.M.J.M.V.D.; Jen, J.C.; Kavanagh, D.; Bertram, P.; Spitzer, D.; Liszewski, M.K.; Barilla-LaBarca, M.-L.; Terwindt, G.M.; Kasai, Y.; et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 2007, 39, 1068–1070. [Google Scholar] [CrossRef]
- Nikolopoulos, D.; Fanouriakis, A.; Boumpas, D.T. Update on the pathogenesis of central nervous system lupus. Curr. Opin. Rheumatol. 2019, 31, 669–677. [Google Scholar] [CrossRef]
- Gall, A.; Treuting, P.; Elkon, K.B.; Loo, Y.-M.; Gale, M.; Barber, G.N.; Stetson, D.B. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity 2012, 36, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21CDKN1A in the DNA damage response. Mutat. Res. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef]
- Dutto, I.; Sukhanova, M.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Scovassi, A.I.; Lavrik, O.; Prosperi, E. p21CDKN1A Regulates the Binding of Poly(ADP-Ribose) Polymerase-1 to DNA Repair Intermediates. PLoS ONE 2016, 11, e0146031. [Google Scholar] [CrossRef] [PubMed]
- Daszkiewicz, L.; Vázquez-Mateo, C.; Rackov, G.; Ballesteros-Tato, A.; Weber, K.; Madrigal-Avilés, A.; Di Pilato, M.; Fotedar, A.; Fotedar, R.; Flores, J.M.; et al. Distinct p21 requirements for regulating normal and self-reactive T cells through IFN-γ production. Sci. Rep. 2015, 5, 7691. [Google Scholar] [CrossRef] [Green Version]
- Balomenos, D.; Martín-Caballero, J.; García, M.I.; Prieto, I.; Flores, J.M.; Serrano, M.; Martínez-A, C. The cell cycle inhibitor p21 controls T-cell proliferation and sex-linked lupus development. Nat. Med. 2000, 6, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Lawson, B.R.; Baccala, R.; Song, J.; Croft, M.; Kono, D.H.; Theofilopoulos, A.N. Deficiency of the Cyclin Kinase Inhibitor p21(WAF-1/CIP-1) Promotes Apoptosis of Activated/Memory T Cells and Inhibits Spontaneous Systemic Autoimmunity. J. Exp. Med. 2004, 199, 547–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senejani, A.G.; Liu, Y.; Kidane, D.; Maher, S.E.; Zeiss, C.J.; Park, H.-J.; Kashgarian, M.; McNiff, J.M.; Zelterman, D.; Bothwell, A.L.M.; et al. Mutation of POLB causes lupus in mice. Cell Rep. 2014, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Herkela, J.; Mimranb, A.; Erez, N.; Kamb, N.; Lohse, A.W.; Hermanna, E.M.-; Rotterc, V.; Cohen, I.R. Autoimmunity to the p53 Protein is a Feature of Systemic Lupus Erythematosus (SLE) Related to Anti-DNA Antibodies. J. Autoimmun. 2001, 17, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, L.; Bao, D.; Wang, L.; Zhao, H.; Lian, Y.; Yan, M.; Mohan, C.; Li, Q.-Z. Novel Autoantibodies Related to Cell Death and DNA Repair Pathways in Systemic Lupus Erythematosus. Genom. Proteom. Bioinform. 2019, 17, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Fujii, H.; Colmegna, I.; Oishi, H.; Goronzy, J.J.; Weyand, C.M. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J. Exp. Med. 2009, 206, 1435–1449. [Google Scholar] [CrossRef]
- Schild-Poulter, C.; Su, A.; Shih, A.; Kelly, O.P.; Fritzler, M.J.; Goldstein, R.; Hache, R.J.G. Association of autoantibodies with Ku and DNA repair proteins in connective tissue diseases. Rheumatolgy 2007, 47, 165–171. [Google Scholar] [CrossRef] [Green Version]
- McNally, J.P.; Millen, S.H.; Chaturvedi, V.; Lakes, N.; Terrell, C.E.; Elfers, E.E.; Carroll, K.; Hogan, S.P.; Andreassen, P.R.; Kanter, J.; et al. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E4782–E4791. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Xie, Y.; Ge, Y.; Nie, X.; Tao, J.; Zhao, Y. Resting T cells are hypersensitive to DNA damage due to defective DNA repair pathway. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namas, R.; Renauer, P.; Ognenovski, M.; Tsou, P.-S.; Sawalha, A.H. Histone H2AX phosphorylation as a measure of DNA double-strand breaks and a marker of environmental stress and disease activity in lupus. Lupus Sci. Med. 2016, 3, e000148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhao, M.; Yin, H.; Gao, F.; Wu, X.; Luo, Y.; Zhao, S.; Zhang, X.; Su, Y.; Hu, N.; et al. Overexpression of the growth arrest and DNA damage-induced 45α gene contributes to autoimmunity by promoting DNA demethylation in lupus T cells. Arthritis Rheum. 2010, 62, 1438–1447. [Google Scholar] [CrossRef] [Green Version]
- Salvador, J.M.; Hollander, M.; Nguyen, A.T.; Kopp, J.B.; Barisoni, L.; Moore, J.K.; Ashwell, J.D.; Fornace, A.J.F., Jr. Mice Lacking the p53-Effector Gene Gadd45a Develop a Lupus-Like Syndrome. Immunity 2002, 16, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Goronzy, J.J.; Weyand, C.M. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol. Med. 2010, 2, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, Y.; Hohensinner, P.; Ju, J.; Wen, Z.; Goodman, S.B.; Zhang, H.; Goronzy, J.J.; Weyand, C.M. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity 2016, 45, 903–916. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef] [Green Version]
- Alissafi, T.; Kalafati, L.; Lazari, M.; Filia, A.; Kloukina, I.; Manifava, M.; Lim, J.-H.; Alexaki, V.I.; Ktistakis, N.T.; Doskas, T.; et al. Mitochondrial Oxidative Damage Underlies Regulatory T Cell Defects in Autoimmunity. Cell Metab. 2020, 32, 591–604.e7. [Google Scholar] [CrossRef]
- Lai, F.P.L.; Tsukada, Y.; Ichikawa, H.; Dunster, K.; Sentry, J.W.; Toh, B.-H. Autoantibody to DNA Excision Repair Enzyme hMYH in a Patient with Rheumatic Disease. Clin. Immunol. 2001, 99, 291–297. [Google Scholar] [CrossRef]
- Casciola-Rosen, L.A.; Pluta, A.F.; Plotz, P.H.; Cox, A.E.; Morris, S.; Wigley, F.M.; Petri, M.; Gelber, A.C.; Rosen, A. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum. 2001, 44, 389–396. [Google Scholar] [CrossRef]
- Noble, P.W.; Bernatsky, S.; Clarke, A.E.; Isenberg, D.A.; Ramsey-Goldman, R.; Hansen, J.E. DNA-damaging autoantibodies and cancer: The lupus butterfly theory. Nat. Rev. Rheumatol. 2016, 12, 429–434. [Google Scholar] [CrossRef]
- Sheng, Y.-J.; Gao, J.-P.; Li, J.; Han, J.-W.; Xu, Q.; Hu, W.-L.; Pan, T.-M.; Cheng, Y.-L.; Yu, Z.-Y.; Ni, C.; et al. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology 2010, 50, 682–688. [Google Scholar] [CrossRef] [Green Version]
- Zeller, T.; Wild, P.; Szymczak, S.; Rotival, M.; Schillert, A.; Castagne, R.; Maouche, S.; Germain, M.; Lackner, K.; Rossmann, H.; et al. Genetics and Beyond—The Transcriptome of Human Monocytes and Disease Susceptibility. PLoS ONE 2010, 5, e10693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, K.A.; Chen, J.W.; Schickel, J.-N.; Isnardi, I.; Yamakawa, N.; Vega-Loza, A.; Anolik, J.H.; Gatti, R.A.; Gelfand, E.W.; Montgomery, R.R.; et al. Impaired ATM activation in B cells is associated with bone resorption in rheumatoid arthritis. Sci. Transl. Med. 2019, 11, eaaw4626. [Google Scholar] [CrossRef]
- Erttmann, S.F.; Härtlova, A.; Sloniecka, M.; Raffi, F.A.; Hosseinzadeh, A.; Edgren, T.; Rofougaran, R.; Resch, U.; Fällman, M.; Ek, T.; et al. Loss of the DNA Damage Repair Kinase ATM Impairs Inflammasome-Dependent Anti-Bacterial Innate Immunity. Immunity 2016, 45, 106–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, R.C.; Pettijohn, K.; Fike, F.; Wang, J.; Nahas, S.A.; Tunuguntla, R.; Hu, H.; Gatti, R.A.; McCurdy, D. Defective DNA double-strand break repair in pediatric systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 568–578. [Google Scholar] [CrossRef]
- Scharer, C.D.; Blalock, E.L.; Mi, T.; Barwick, B.G.; Jenks, S.A.; Deguchi, T.; Cashman, K.S.; Neary, B.E.; Patterson, D.; Hicks, S.L.; et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 2019, 20, 1071–1082. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licandro, G.; Khor, H.L.; Beretta, O.; Lai, J.; Derks, H.; Laudisi, F.; Conforti-Andreoni, C.; Qian, H.L.; Teng, G.G.; Ricciardi-Castagnoli, P.; et al. The NLRP3 inflammasome affects DNA damage responses after oxidative and genotoxic stress in dendritic cells. Eur. J. Immunol. 2013, 43, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Li, H.; Niu, J.; Wu, S.; Xue, G.; Yao, X.; Guo, Q.; Wan, N.; Abliz, P.; Yang, G.; et al. Hyperactivation of the NLRP3 Inflammasome in Myeloid Cells Leads to Severe Organ Damage in Experimental Lupus. J. Immunol. 2016, 198, 1119–1129. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef]
- So, E.Y.; Ouchi, T. Translational initiation regulated by ATM in dendritic cells development. Cell Death Dis. 2014, 5, e1418. [Google Scholar] [CrossRef] [Green Version]
- Petersone, L.; Edner, N.M.; Ovcinnikovs, V.; Heuts, F.; Ross, E.M.; Ntavli, E.; Wang, C.J.; Walker, L.S.K. T Cell/B Cell Collaboration and Autoimmunity: An Intimate Relationship. Front. Immunol. 2018, 9, 1941. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, K.S.C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nat. Cell Biol. 2017, 542, 110–114. [Google Scholar] [CrossRef]
- Mitkin, N.A.; Muratova, A.M.; Sharonov, G.V.; Korneev, K.V.; Sviriaeva, E.N.; Mazurov, D.; Schwartz, A.M.; Kuprash, D.V. p63 and p73 repress CXCR5 chemokine receptor gene expression in p53-deficient MCF-7 breast cancer cells during genotoxic stress. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2017, 1860, 1169–1178. [Google Scholar] [CrossRef]
- Shao, L. DNA Damage Response Signals Transduce Stress From Rheumatoid Arthritis Risk Factors Into T Cell Dysfunction. Front. Immunol. 2018, 9, 3055. [Google Scholar] [CrossRef] [Green Version]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun. Rev. 2018, 17, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Saunders, M.E.; Jett, B.D. Chapter 11-NK, γδ T and NKT cells. In Primer to the Immune Response, 2nd ed.; Newnes: Oxford, UK, 2014; pp. 247–268. [Google Scholar] [CrossRef]
- Wu, L.; Kaer, L. Natural Killer T Cells and Autoimmune Disease. Curr. Mol. Med. 2009, 9, 4–14. [Google Scholar] [CrossRef]
- Johansson, S.; Berg, L.; Hall, H.; Höglund, P. NK cells: Elusive players in autoimmunity. Trends Immunol. 2005, 26, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, A.; Lünemann, J.D.; Münz, C. Regulatory NK-Cell Functions in Inflammation and Autoimmunity. Mol. Med. 2009, 15, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Karo, J.M.; Schatz, D.G.; Sun, J.C. The RAG Recombinase Dictates Functional Heterogeneity and Cellular Fitness in Natural Killer Cells. Cell 2014, 159, 94–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugmann, S.D.; Lee, A.I.; Shockett, P.E.; Villey, I.J.; Schatz, D.G. The RAG Proteins and V(D)J Recombination: Complexes, Ends, and Transposition. Annu. Rev. Immunol. 2000, 18, 495–527. [Google Scholar] [CrossRef] [PubMed]
- Schlissel, M.S. Regulating antigen-receptor gene assembly. Nat. Rev. Immunol. 2003, 3, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Geier, C.B.; Farmer, J.R.; Foldvari, Z.; Ujhazi, B.; Steininger, J.; Sleasman, J.W.; Parikh, S.; Dilley, M.A.; Pai, S.-Y.; Henderson, L.; et al. Vasculitis as a Major Morbidity Factor in Patients With Partial RAG Deficiency. Front. Immunol. 2020, 11, 574738. [Google Scholar] [CrossRef]
- Farmer, J.R.; Foldvari, Z.; Ujhazi, B.; De Ravin, S.S.; Chen, K.; Bleesing, J.J.; Schuetz, C.; Al-Herz, W.; Abraham, R.S.; Joshi, A.Y.; et al. Outcomes and Treatment Strategies for Autoimmunity and Hyperinflammation in Patients with RAG Deficiency. J. Allergy Clin. Immunol. Pract. 2019, 7, 1970–1985.e4. [Google Scholar] [CrossRef]
- Chen, K.; Wu, W.; Mathew, D.; Zhang, Y.; Browne, S.K.; Rosen, L.B.; McManus, M.P.; Pulsipher, M.A.; Yandell, M.; Bohnsack, J.F.; et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J. Allergy Clin. Immunol. 2014, 133, 880–882.e10. [Google Scholar] [CrossRef] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR–dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [Green Version]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nat. Cell Biol. 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Groh, V.; Bruhl, A.; El-Gabalawy, H.; Nelson, J.L.; Spies, T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2003, 100, 9452–9457. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.K.; Sumariwalla, P.F.; McCann, F.E.; Amjadi, P.; Chang, C.; McNamee, K.; Tornehave, D.; Haase, C.; Agersø, H.; Stennicke, V.W.; et al. Blockade of NKG2D ameliorates disease in mice with collagen-induced arthritis: A potential pathogenic role in chronic inflammatory arthritis. Arthritis Rheum. 2011, 63, 2617–2629. [Google Scholar] [CrossRef] [PubMed]
- Hüe, S.; Mention, J.-J.; Monteiro, R.C.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A Direct Role for NKG2D/MICA Interaction in Villous Atrophy during Celiac Disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Meresse, B.; Chen, Z.; Ciszewski, C.; Tretiakova, M.; Bhagat, G.; Krausz, T.N.; Raulet, D.H.; Lanier, L.L.; Groh, V.; Spies, T.; et al. Coordinated Induction by IL15 of a TCR-Independent NKG2D Signaling Pathway Converts CTL into Lymphokine-Activated Killer Cells in Celiac Disease. Immunity 2004, 21, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.-D.; Takeda, K.; Akira, S.; Sarvetnick, N.; Ljunggren, H.-G. IL-18 Directs Autoreactive T Cells and Promotes Autodestruction in the Central Nervous System Via Induction of IFN-γ by NK Cells. J. Immunol. 2000, 165, 3099–3104. [Google Scholar] [CrossRef] [Green Version]
- Van Belle, T.L.; Von Herrath, M.G. The role of the activating receptor NKG2D in autoimmunity. Mol. Immunol. 2009, 47, 8–11. [Google Scholar] [CrossRef]
- Ogasawara, K.; Hamerman, J.A.; Ehrlich, L.R.; Bour-Jordan, H.; Santamaria, P.; Bluestone, J.A.; Lanier, L.L. NKG2D Blockade Prevents Autoimmune Diabetes in NOD Mice. Immunity 2004, 20, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Swann, J.B.; Uldrich, A.P.; Van Dommelen, S.; Sharkey, J.; Murray, W.K.; Godfrey, D.I.; Smyth, M.J. Type I natural killer T cells suppress tumors caused by p53 loss in mice. Blood 2009, 113, 6382–6385. [Google Scholar] [CrossRef] [Green Version]
- Shabrish, S.; Mittra, I. Cytokine Storm as a Cellular Response to dsDNA Breaks: A New Proposal. Front. Immunol. 2021, 12, 622738. [Google Scholar] [CrossRef] [PubMed]
- Mittra, I.; Samant, U.; Sharma, S.; Raghuram, G.V.; Saha, T.; Tidke, P.; Pancholi, N.; Gupta, D.; Prasannan, P.; Gaikwad, A.; et al. Cell-free chromatin from dying cancer cells integrate into genomes of bystander healthy cells to induce DNA damage and inflammation. Cell Death Discov. 2017, 3, 17015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würtele, H.; Little, K.C.E.; Chartrand, P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003, 10, 1791–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvvuri, B.; Lood, C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 2019, 10, 502. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.M.; Kunkel, H.G. Characteristics of a soluble nuclear antigen precipitating with sera of patients with systemic lupus ery-thematosus. J Immunol. 1966, 96, 464–471. [Google Scholar]
- Liu, X.; Fang, L.; Guo, T.B.; Mei, H.; Zhang, J.Z. Drug targets in the cytokine universe for autoimmune disease. Trends Immunol. 2013, 34, 120–128. [Google Scholar] [CrossRef]
- Bird, L. Targeting cytokines in disease. Nat. Immunol. 2016, 17, S17. [Google Scholar] [CrossRef]
- Gunderson, C.C.; Moore, K.N. Olaparib: An oral PARP-1 and PARP-2 inhibitor with promising activity in ovarian cancer. Futur. Oncol. 2015, 11, 747–757. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; DeMaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef] [PubMed]
- Falcke, S.E.; Rühle, P.F.; Deloch, L.; Fietkau, R.; Frey, B.; Gaipl, U.S. Clinically Relevant Radiation Exposure Differentially Impacts Forms of Cell Death in Human Cells of the Innate and Adaptive Immune System. Int. J. Mol. Sci. 2018, 19, 3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernatsky, S.; Ramsey-Goldman, R.; Labrecque, J.; Joseph, L.; Boivin, J.-F.; Petri, M.; Zoma, A.; Manzi, S.; Urowitz, M.B.; Gladman, D.; et al. Cancer risk in systemic lupus: An updated international multi-centre cohort study. J. Autoimmun. 2013, 42, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, D.; Kenu, E.; Isenberg, D. Cancer complicating systemic lupus erythematosus—A dichotomy emerging from a nested case-control study. Lupus 2013, 22, 919–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goobie, G.C.; Bernatsky, S.; Ramsey-Goldman, R.; Clarke, A.E. Malignancies in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2015, 27, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Qiu, L.-J.; Hu, L.-F.; Cen, H.; Zhang, M.; Wen, P.-F.; Wang, X.-S.; Pan, H.-F.; Ye, D.-Q. Lung, liver, prostate, bladder malignancies risk in systemic lupus erythematosus: Evidence from a meta-analysis. Lupus 2014, 23, 284–292. [Google Scholar] [CrossRef]
- Hansen, J.E.; Chan, G.; Liu, Y.; Hegan, D.C.; Dalal, S.; Dray, E.; Kwon, Y.; Xu, Y.; Xu, X.; Peterson-Roth, E.; et al. Targeting Cancer with a Lupus Autoantibody. Sci. Transl. Med. 2012, 4, 157ra142. [Google Scholar] [CrossRef] [Green Version]
- Weisbart, R.H.; Gera, J.F.; Chan, G.; Hansen, J.E.; Li, E.; Cloninger, C.; Levine, A.J.; Nishimura, R.N. A Cell-Penetrating Bispecific Antibody for Therapeutic Regulation of Intracellular Targets. Mol. Cancer Ther. 2012, 11, 2169–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddig, A.; Voss, L.; Guttek, K.; Roggenbuck, D.; Feist, E.; Reinhold, D. Impact of Different JAK Inhibitors and Methotrexate on Lymphocyte Proliferation and DNA Damage. J. Clin. Med. 2021, 10, 1431. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Cell Type | Autoimmune Context | DDR Mechanism | Proposed Therapeutic Targeting | Reference | |
|---|---|---|---|---|---|
| Main adaptive immune cells | CD8+ T | HLH | Increased γH2AX/phospho-p53, phospho-ATM, and phospho-CHK1/2 (activated healthy T cells) | PPCA therapy eliminated only pathological CD8+ T cells | [52] |
| CD8+ T | SLE | Increased γH2AX/defective repair after induced DNA damage | [54] | ||
| CD4+ T | MS | Increased γH2AX/phospho-p53, phospho-ATM, and phospho-CHK1/2 (activated healthy T cells) | PPCA therapy eliminated only pathological CD4+ T cells | [52] | |
| CD4+ T | SLE | Increased γH2AX/defective repair after induced DNA damage | [54] | ||
| CD4+ T | SLE | Elevated GADD45A mRNA expression and global DNA hypomethylation | Silencing of GADD45A increased DNA methylation and reduced T cell autoreactivity | [55] | |
| CD4+ T | RA | ATM and G2/M checkpoint insufficiencies due to ROS production deficiency | Pharmacologic interventions that restore ROS production (e.g., Menadione) | [59] | |
| CD4+ T resting | not assessed/p53 deficiencies | Unrepaired DNA upon induced DNA damage: a) deficient accumulation of 53BP and γH2AX foci and b) increased apoptosis via JNK/p73 pathway (non-DDR pathway) | [53] | ||
| CD4+ T follicular | SLE | Polb decreased expression generated a lupus phenotype and increased GC CD4+ T follicular cells which were mainly apoptotic (CD4+). | [47] | ||
| Tregs | MS, SLE, RA | Increased H2AFx, TP53, CHK2, TP53BP1/oxidative stress(mtROS)/cell death | Treg-specific scavenging of mtROS in vivo restrained DDR, reduced cell death and autoimmune responses | [60] | |
| B | SLE | Polb decreased expression generated a lupus phenotype, altered V(D)J, increased SHM and increased GC B cells. | [47] | ||
| B naïve | RA with bone erosion | Low ATM, MRE11A, NBS1 | [66] | ||
| CD20+ B | RA or RA with bone erosion | Low phospho-ATM limited BCR, increased immunogenic B cells and secretion of RANKL and IL-6 cytokines. | Anti-CD20+ B cell–depleting biologic therapy (i.e., rituximab) | [66] | |
| B lymphoblastoid cell lines | SLE | Ineffective DNA repair mechanisms | [68] | ||
| B subsets (rN, T3, aN, SM, DN2) | SLE | p53 pathway upregulated (all subsets); G2/M pathway upregulated (all subsets-DN2); G2/M pathway downregulated (DN2) | [69] | ||
| Oher immune cells interacting with adaptive immune cells | DCs | not assessed/high NLRP3 and Caspase 1 | Reduced DDR and p53 induced apoptosis upon attenuation of Nlrp3 and Caspase 1 | Decrease Nlrp3 and Caspase 1 expression | [71] |
| DCs | not assessed/ATM expression defects | ATM inhibition delayed maturation, increased apoptosis and reduced T-cell activation. | [74] | ||
| NK | not assessed/RAG deficiencies | RAG deficiency increased γH2AX and reduced DNA-PKcs, Ku80, CHK2 and ATM expression levels. | [84] | ||
| NK, γδ Τ, ΝΚΤ, T subsets | not assessed/aberrant expression of NKG2D receptor and ligands | Chronic activation of DDR as occurs in tumor cells may account for NKG2D ligand overexpression. | [91] |
| 1. | Assess alterations in the DDR and provide mechanistic insights for pathogenic cell populations during autoimmunity. |
| 2. | Link the DDR molecules’ expression with specific cell behavior during autoimmune response. |
| 3. | Investigate the effects of the DDR on immune cell interactions. |
| 4. | Explore both the activated and total forms of the DDR proteins in autoimmunity. |
| 5. | Distinguish between the beneficial and the detrimental outcomes of the DDR during immune responses. |
| 6. | Examine the perturbation of the DDR components towards the suppression of distinct autoimmune responses. |
| 7. | Generate animal models for autoimmune diseases that allow cell-targeted therapeutic approaches of the DDR. |
| 8. | Extrapolate existing in vitro and in vivo research to autoimmune human diseases. |
| 9. | Explore the potential therapeutic benefit of targeting the DDR pathway in cancers for autoimmunity and the role of the DDR-affecting cell-penetrating autoantibodies in autoimmunity to engineer therapeutic antibodies. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manolakou, T.; Verginis, P.; Boumpas, D.T. DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity. Int. J. Mol. Sci. 2021, 22, 5842. https://doi.org/10.3390/ijms22115842
Manolakou T, Verginis P, Boumpas DT. DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity. International Journal of Molecular Sciences. 2021; 22(11):5842. https://doi.org/10.3390/ijms22115842
Chicago/Turabian StyleManolakou, Theodora, Panayotis Verginis, and Dimitrios T. Boumpas. 2021. "DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity" International Journal of Molecular Sciences 22, no. 11: 5842. https://doi.org/10.3390/ijms22115842
APA StyleManolakou, T., Verginis, P., & Boumpas, D. T. (2021). DNA Damage Response in the Adaptive Arm of the Immune System: Implications for Autoimmunity. International Journal of Molecular Sciences, 22(11), 5842. https://doi.org/10.3390/ijms22115842