
Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
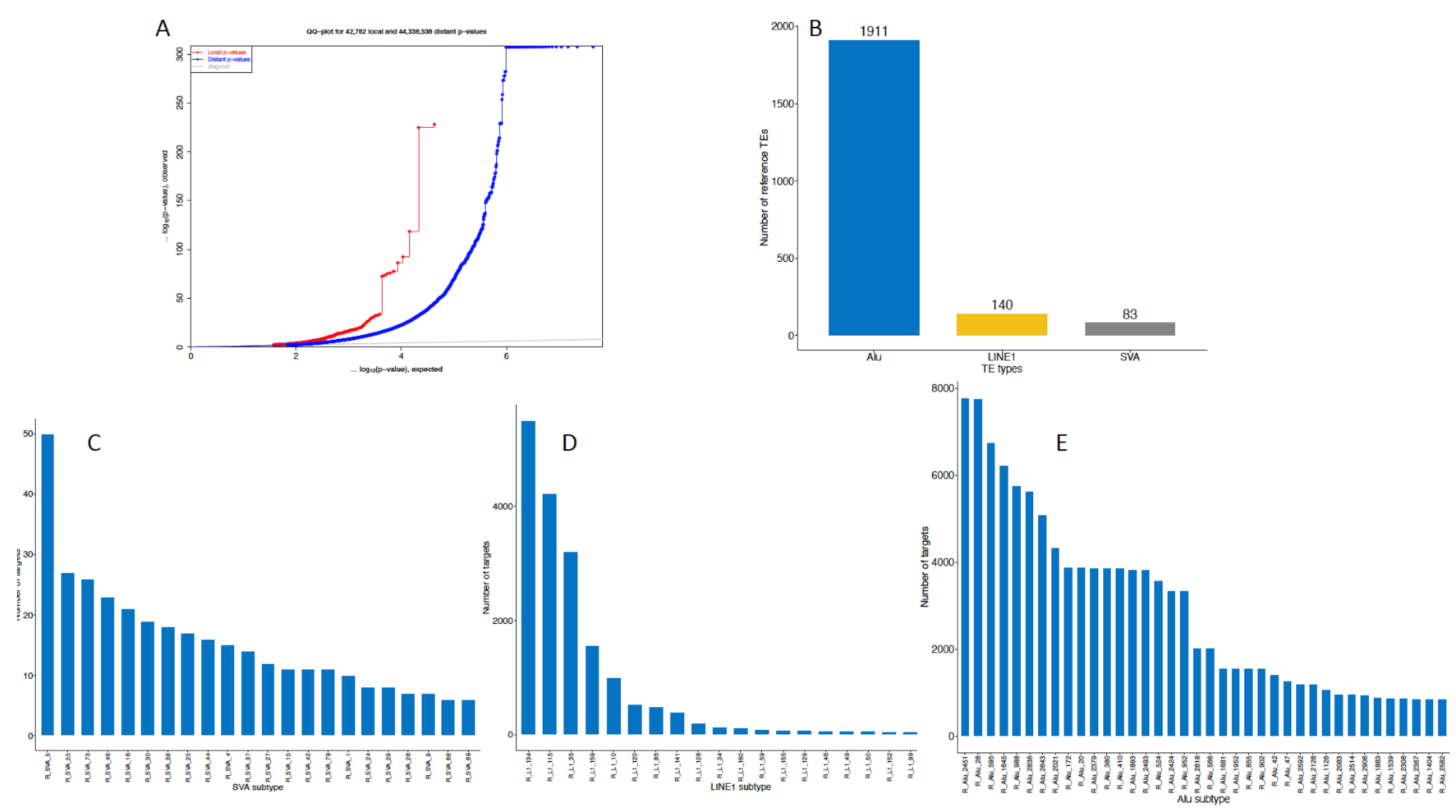
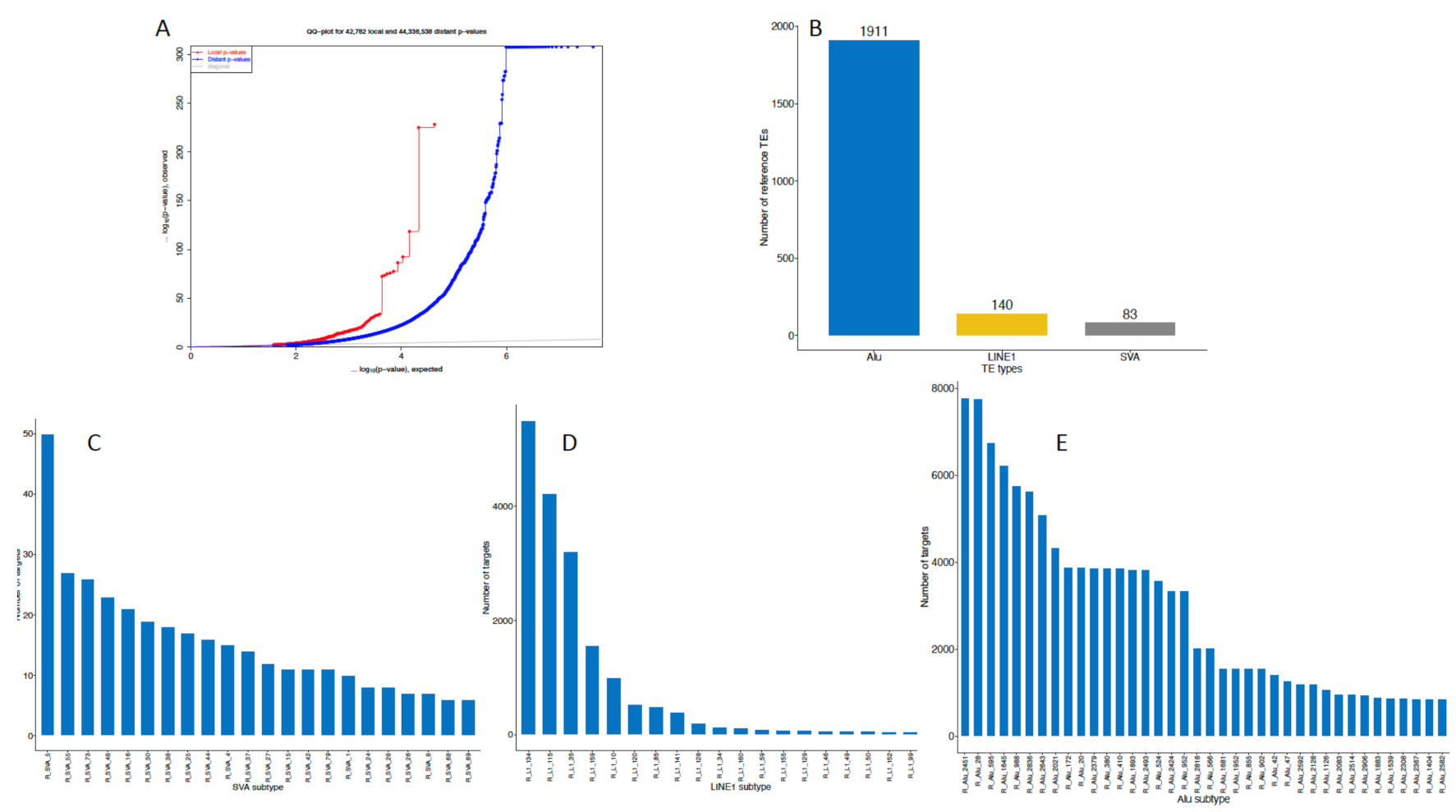
2.1. General eQTL Effects of the TEs
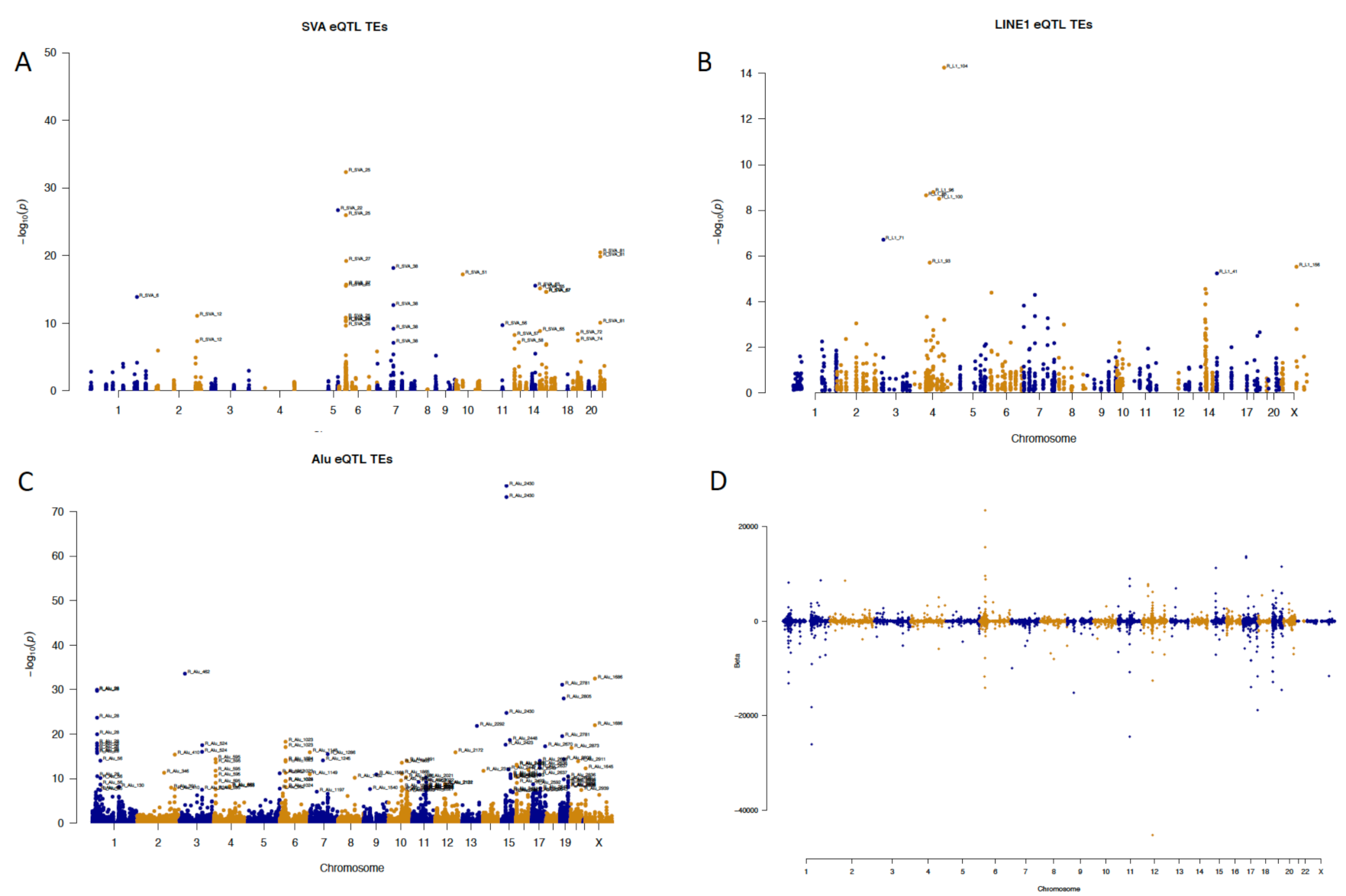
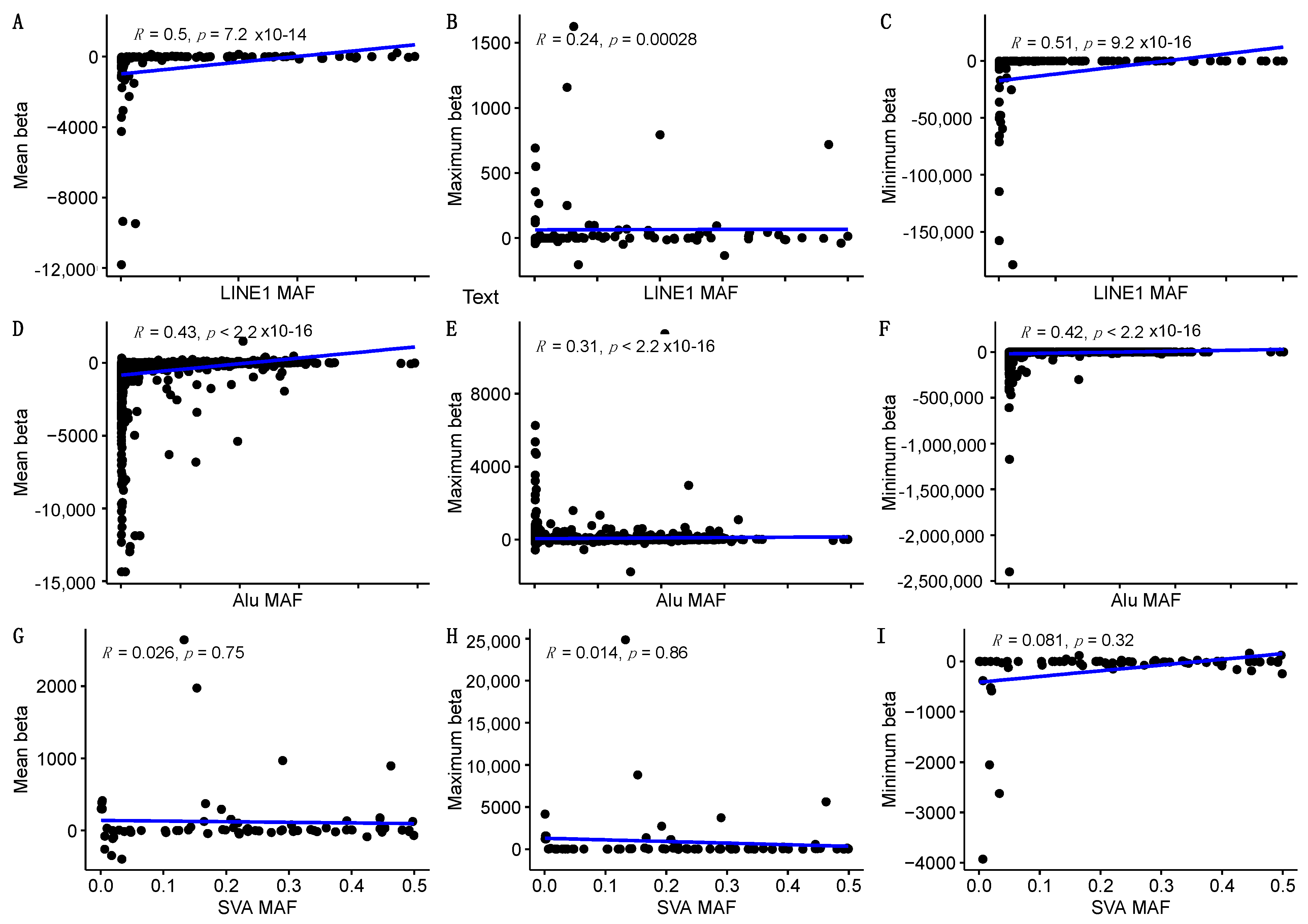
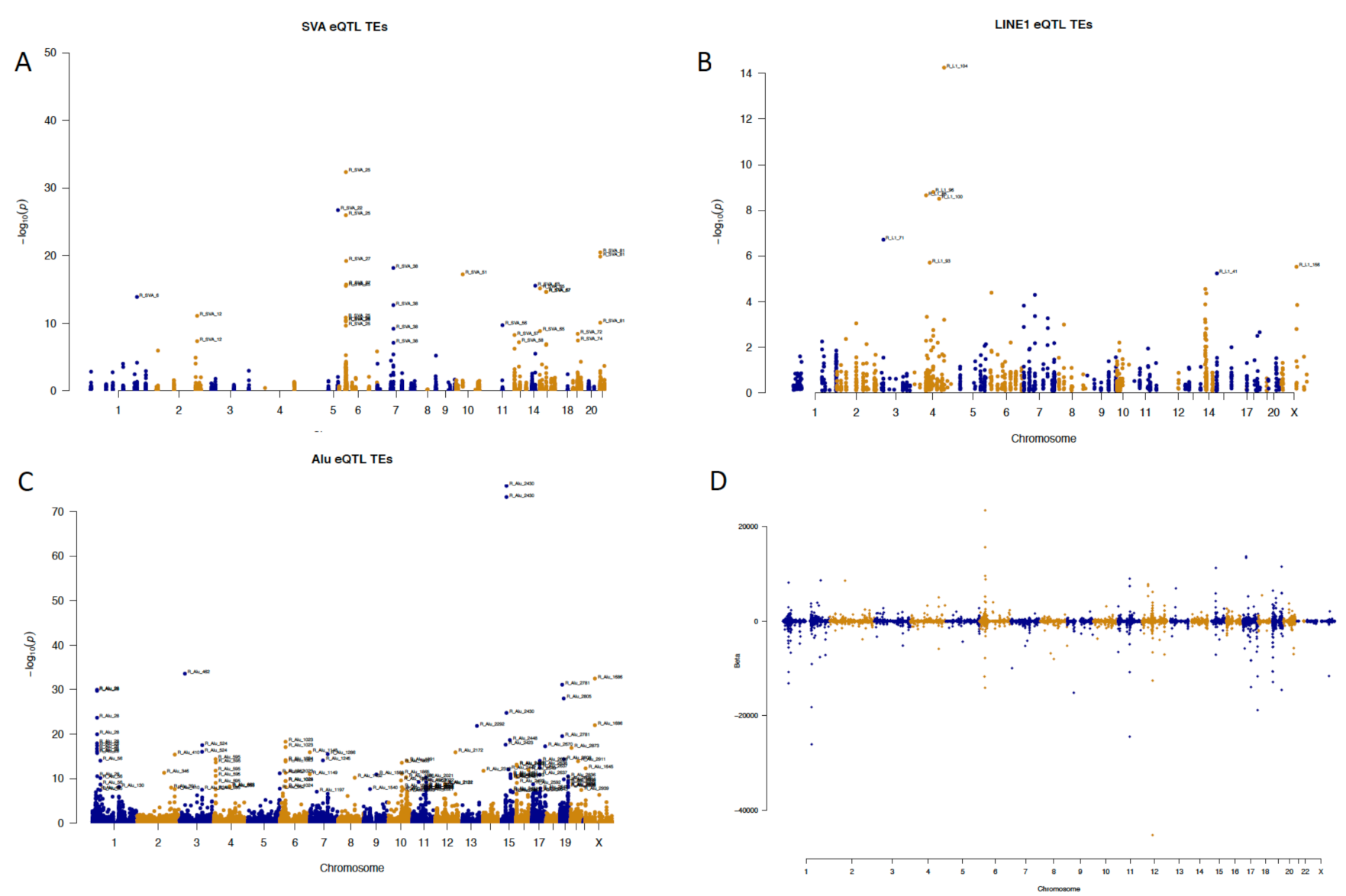
2.2. Element-Specific eQTL Effects in the Genome
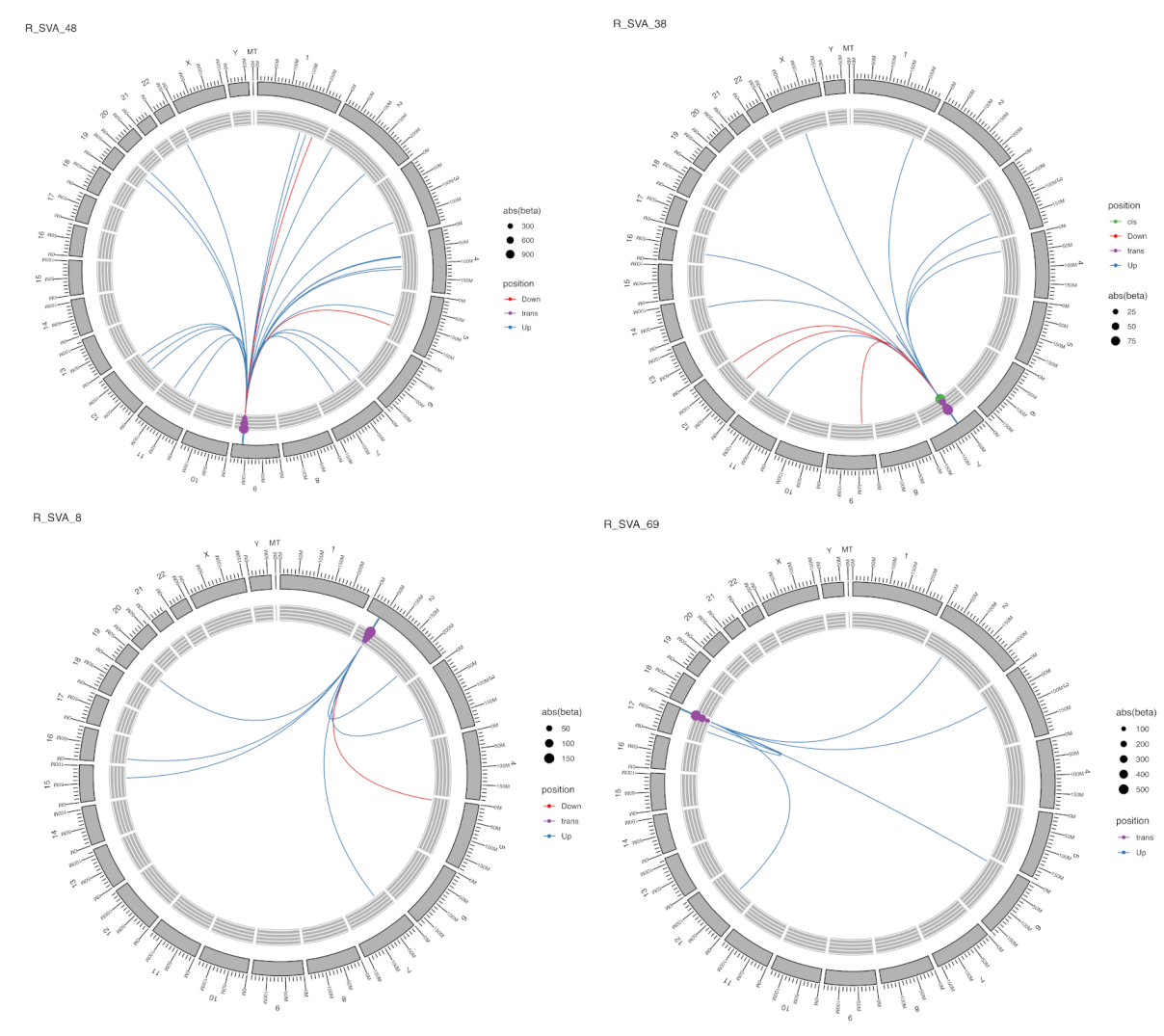
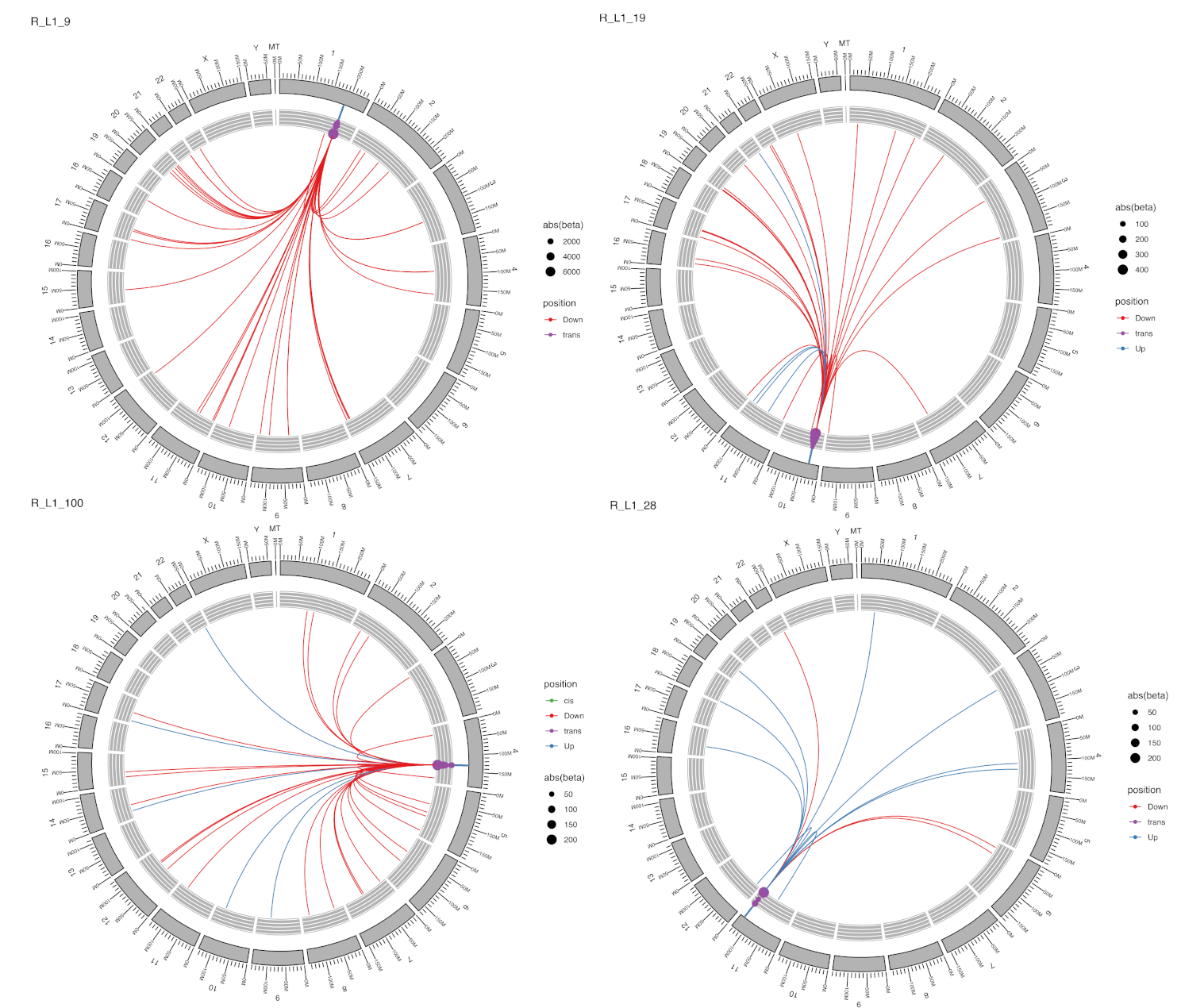
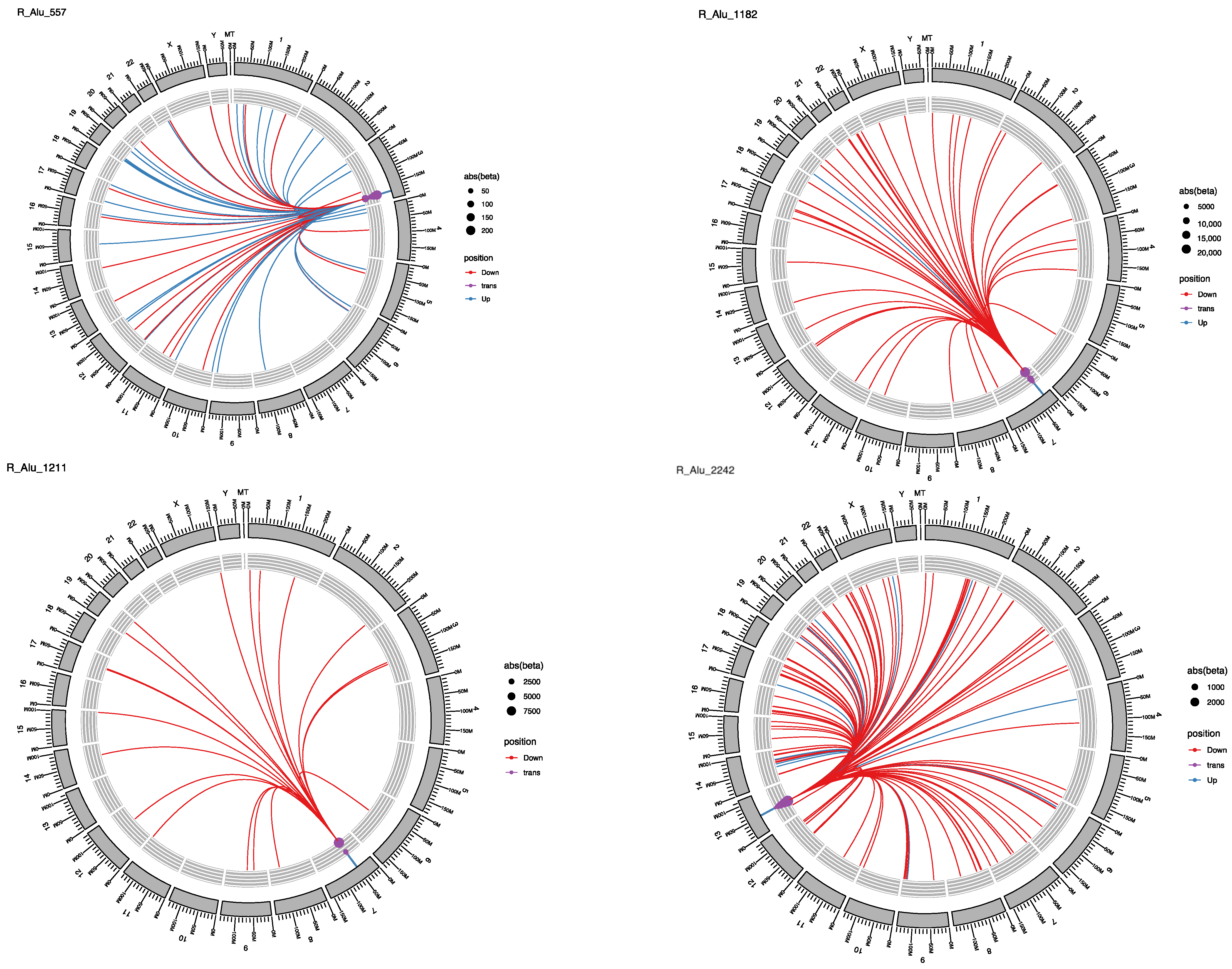
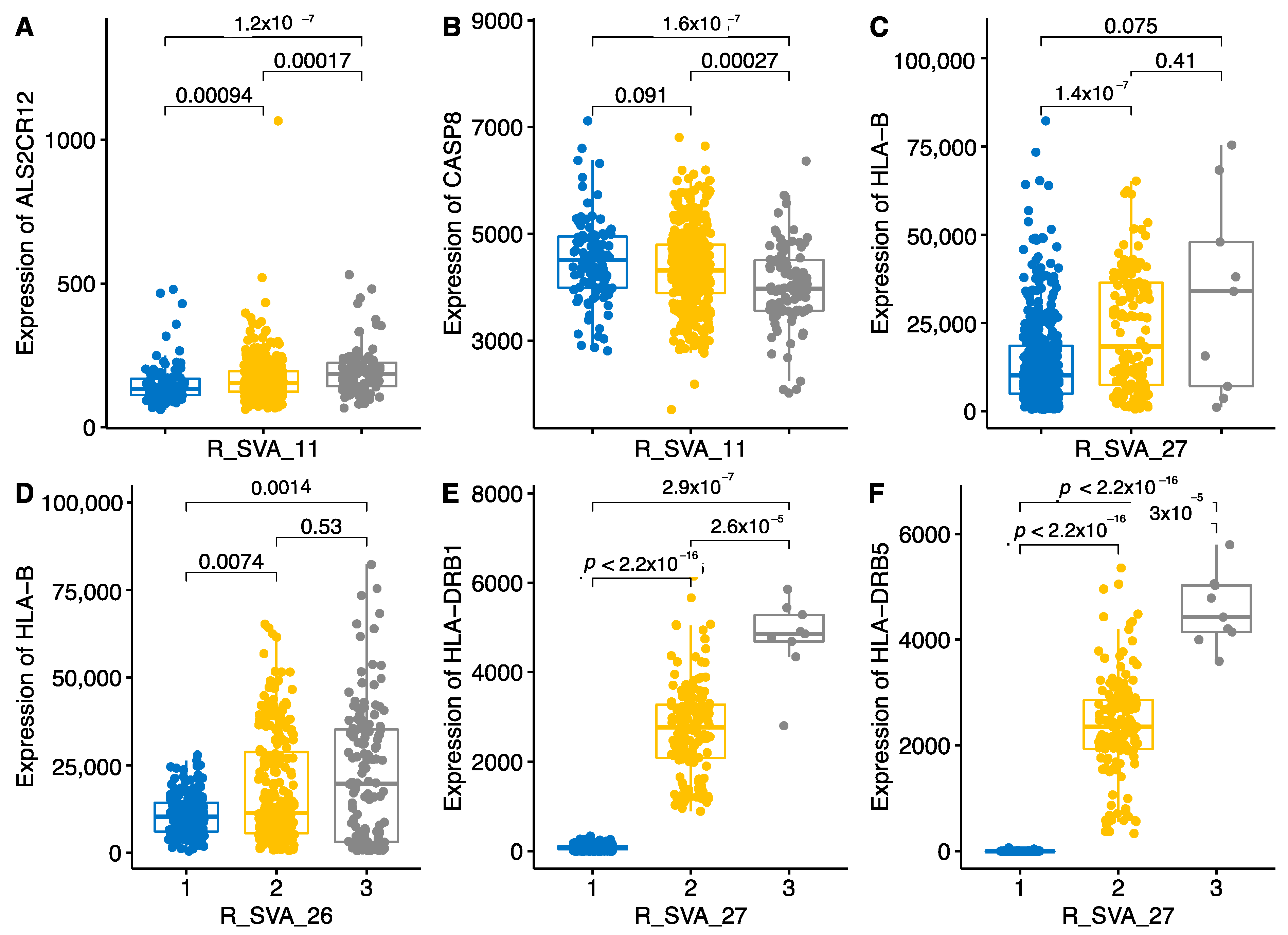
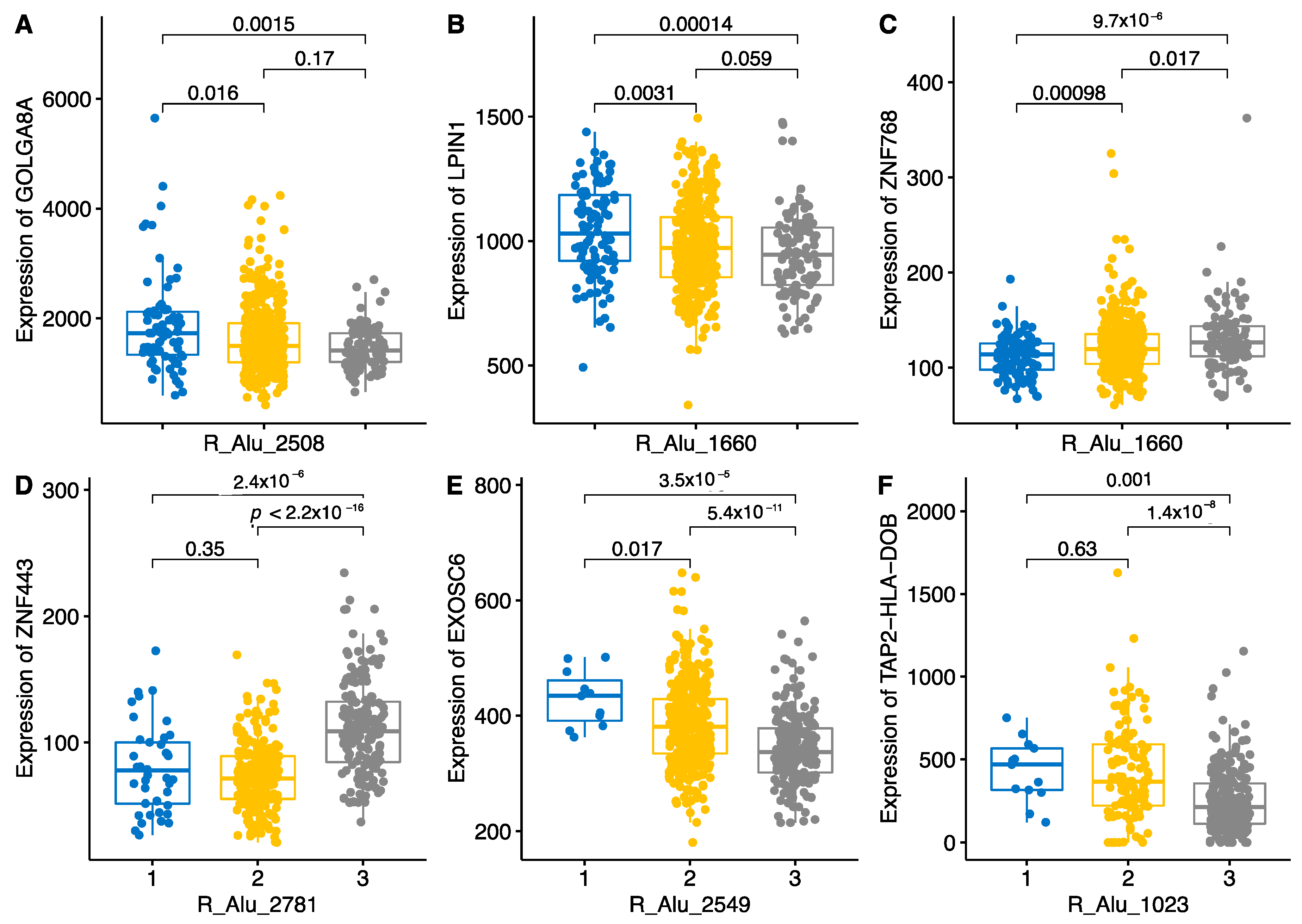
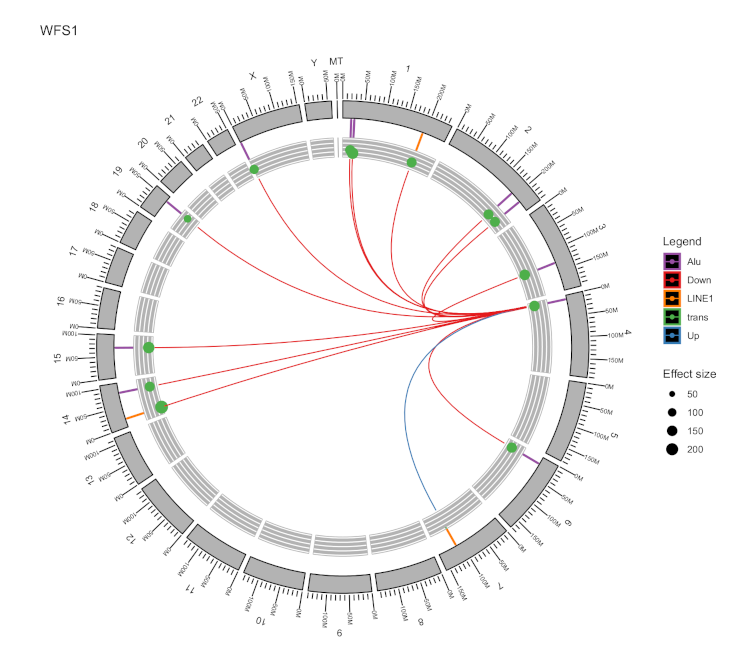
2.3. Locus-Specific eQTL Effects of the Selected TEs
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. eQTL Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Piegu, B.; Bire, S.; Arensburger, P.; Bigot, Y. A survey of transposable element classification systems—A call for a fundamental update to meet the challenge of their diversity and complexity. Mol. Phylogenet. Evol. 2015, 86, 90–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.R.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.; Smit, A.; Boissinot, S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006, 16, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Middelberg, R.P.; Ferreira, M.A.; Henders, A.K.; Heath, A.C.; Madden, P.A.; Montgomery, G.W.; Martin, N.G.; Whitfield, J.B. Genetic variants in LPL, OASL and TOMM40/APOE-C1-C2-C4 genes are associated with multiple cardiovascular-related traits. BMC Med. Genet. 2011, 12, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. Active human retrotransposons: Variation and disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Aneichyk, T.; Hendriks, W.T.; Yadav, R.; Shin, D.; Gao, D.; Vaine, C.A.; Collins, R.L.; Domingo, A.; Currall, B.; Stortchevoi, A.; et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172, 897–909 e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.M.; Masson, E.; Macek, M., Jr.; Raguénès, O.; Piskackova, T.; Fercot, B.; Fila, L.; Cooper, D.N.; Audrézet, M.P.; Férec, C. Detection of two Alu insertions in the CFTR gene. J. Cyst. Fibros. 2008, 7, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. An Increased Burden of Highly Active Retrotransposition Competent L1s Is Associated with Parkinson’s Disease Risk and Progression in the PPMI Cohort. Int. J. Mol. Sci 2020, 21, 6562. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Lättekivi, F.; Planken, A.; Reimann, E.; Kurvits, L.; Kadastik-Eerme, L.; Kasterpalu, K.M.; Bubb, V.J.; Quinn, J.P.; Kõks, S.; et al. Analysis of repetitive element expression in the blood and skin of patients with Parkinson’s disease identifies differential expression of satellite elements. Sci. Rep. 2019, 9, 4369. [Google Scholar] [CrossRef] [PubMed]
- Lättekivi, F.; Kõks, S.; Keermann, M.; Reimann, E.; Prans, E.; Abram, K.; Silm, H.; Kõks, G.; Kingo, K. Transcriptional landscape of human endogenous retroviruses (HERVs) and other repetitive elements in psoriatic skin. Sci. Rep. 2018, 8, 4358. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.L.; Lopez, A.I.; Iacoangeli, A.; Bubb, V.J.; Smith, B.; Troakes, C.; Alahmady, N.; Koks, S.; Schumann, G.G.; Al-Chalabi, A.; et al. Frequency and methylation status of selected retrotransposition competent L1 loci in amyotrophic lateral sclerosis. Mol. Brain 2020, 13, 154. [Google Scholar] [CrossRef]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, V.; Wang, T. Transposable Element Mediated Innovation in Gene Regulatory Landscapes of Cells: Re-Visiting the "Gene-Battery" Model. Bioessays 2018, 40, 1700155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koks, S.; Soomets, U.; Paya-Cano, J.L.; Fernandes, C.; Luuk, H.; Plaas, M.; Terasmaa, A.; Tillmann, V.; Noormets, K.; Vasar, E.; et al. Wfs1 gene deletion causes growth retardation in mice and interferes with the growth hormone pathway. Physiol. Genom. 2009, 37, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Koks, S.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P. Transcript Variants of Genes Involved in Neurodegeneration Are Differentially Regulated by the APOE and MAPT Haplotypes. Genes (Basel) 2021, 12, 423. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Koks, S.; Nikopensius, T.; Maron, E.; Altmae, S.; Heinaste, E.; Vabrit, K.; Tammekivi, V.; Hallast, P.; Kurg, A.; et al. Polymorphisms in wolframin (WFS1) gene are possibly related to increased risk for mood disorders. Int. J. Neuropsychopharmacol. 2005, 8, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ivask, M.; Pajusalu, S.; Reimann, E.; Kõks, S. Hippocampus and Hypothalamus RNA-sequencing of WFS1-deficient Mice. Neuroscience 2018, 374, 91–103. [Google Scholar] [CrossRef]
- Ivask, M.; Hugill, A.; Kõks, S. RNA-sequencing of WFS1-deficient pancreatic islets. Physiol. Rep. 2016, 4, e12750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunder, S.; Koks, S.; Koks, G.; Reimann, E.; Hackl, H.; Gruber, R.; Moosbrugger-Martinz, V.; Schmuth, M.; Dubrac, S. Enhanced Expression of Genes Related to Xenobiotic Metabolism in the Skin of Patients with Atopic Dermatitis but Not with Ichthyosis Vulgaris. J. Investig. Dermatol 2018, 138, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Keermann, M.; Koks, S.; Reimann, E.; Prans, E.; Abram, K.; Kingo, K. Transcriptional landscape of psoriasis identifies the involvement of IL36 and IL36RN. BMC Genomics 2015, 16, 322. [Google Scholar] [CrossRef] [Green Version]
- Kingo, K.; Aunin, E.; Karelson, M.; Ratsep, R.; Silm, H.; Vasar, E.; Koks, S. Expressional changes in the intracellular melanogenesis pathways and their possible role in the pathogenesis of vitiligo. J. Dermatol. Sci. 2008, 52, 39–46. [Google Scholar] [CrossRef]
- Lill, M.; Koks, S.; Soomets, U.; Schalkwyk, L.C.; Fernandes, C.; Lutsar, I.; Taba, P. Peripheral blood RNA gene expression profiling in patients with bacterial meningitis. Front. Neurosci. 2013, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Jordan, I.K. Transposable element activity, genome regulation and human health. Curr. Opin. Genet. Dev. 2018, 49, 25–33. [Google Scholar] [CrossRef]
- Wang, L.; Rishishwar, L.; Marino-Ramirez, L.; Jordan, I.K. Human population-specific gene expression and transcriptional network modification with polymorphic transposable elements. Nucleic. Acids Res. 2017, 45, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Reimann, E.; Kingo, K.; Karelson, M.; Reemann, P.; Loite, U.; Sulakatko, H.; Keermann, M.; Raud, K.; Abram, K.; Vasar, E.; et al. The mRNA expression profile of cytokines connected to the regulation of melanocyte functioning in vitiligo skin biopsy samples and peripheral blood mononuclear cells. Hum. Immunol. 2012, 73, 393–398. [Google Scholar] [CrossRef]
- Koks, G.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. At the dawn of the transcriptomic medicine. Exp. Biol. Med. (Maywood) 2021, 246, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Kurvits, L.; Lättekivi, F.; Reimann, E.; Kadastik-Eerme, L.; Kasterpalu, K.M.; Kõks, S.; Taba, P.; Planken, A. Transcriptomic profiles in Parkinson’s disease. Exp. Biol. Med. (Maywood) 2021, 246, 584–595. [Google Scholar] [CrossRef]
- Schalkwyk, L.C.; Fernandes, C.; Nash, M.W.; Kurrikoff, K.; Vasar, E.; Koks, S. Interpretation of knockout experiments: The congenic footprint. Genes Brain Behav. 2007, 6, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, X.D.; Nguyen, H.G.; Trinh, L.H.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, V.Q.; Nguyen, V.H.; Le, N.T.N.; et al. Analysis of the Expression of Repetitive DNA Elements in Osteosarcoma. Front. Genet. 2017, 8, 193. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Hammell, M. Analysis of RNA-Seq Data Using TEtranscripts. Methods Mol. Biol. 2018, 1751, 153–167. [Google Scholar]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef] [Green Version]
- Kõks, S.; Kõks, G. The Role of Human Endogenous Retroviruses (HERVs) in the Pathologies of the Nervous System. In Molecular-Genetic and Statistical Techniques for Behavioral and Neural Research; Gerlai, R.T., Ed.; Academic Press: San Diego, CA, USA, 2018; Chapter 22; pp. 519–533. [Google Scholar]
- González-Navarro, H.; Dogan, S.; Tuna, B.G.; Potter, P.K.; Koks, G.; Koks, S. Aging in Rodents. In Encyclopedia of Biomedical Gerontology; Rattan, S.I.S., Ed.; Academic Press: Oxford, UK, 2020; pp. 73–87. [Google Scholar]
- Panda, K.; Slotkin, R.K. Long-Read cDNA Sequencing Enables a "Gene-Like" Transcript Annotation of Transposable Elements. Plant Cell 2020, 32, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, O.; Geary, B.; Savage, A.L.; Billingsley, K.J.; Bubb, V.J.; Quinn, J.P. The Role of SINE-VNTR-Alu (SVA) Retrotransposons in Shaping the Human Genome. Int. J. Mol. Sci. 2019, 20, 5977. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.L.; Bubb, V.J.; Breen, G.; Quinn, J.P. Characterisation of the potential function of SVA retrotransposons to modulate gene expression patterns. BMC Evol. Biol. 2013, 13, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrozziello, T.; Dios, A.M.; Mueller, K.A.; Vaine, C.A.; Hendriks, W.T.; Glajch, K.E.; Mills, A.N.; Mangkalaphiban, K.; Penney, E.B.; Ito, N.; et al. SVA insertion in X-linked Dystonia Parkinsonism alters histone H3 acetylation associated with TAF1 gene. PLoS ONE 2020, 15, e0243655. [Google Scholar] [CrossRef]
- Marshall, J.N.; Lopez, A.I.; Pfaff, A.L.; Koks, S.; Quinn, J.P.; Bubb, V.J. Variable number tandem repeats - Their emerging role in sickness and health. Exp. Biol. Med. (Maywood) 2021. [Google Scholar] [CrossRef]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Pittard, W.S.; Mills, R.E.; Genomes Project, C.; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Shabalin, A.A. Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Cook, D.; Lawrence, M. ggbio: An R package for extending the grammar of graphics for genomic data. Genome Biol. 2012, 13, R77. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koks, S.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P. Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome. Int. J. Mol. Sci. 2021, 22, 6319. https://doi.org/10.3390/ijms22126319
Koks S, Pfaff AL, Bubb VJ, Quinn JP. Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome. International Journal of Molecular Sciences. 2021; 22(12):6319. https://doi.org/10.3390/ijms22126319
Chicago/Turabian StyleKoks, Sulev, Abigail L. Pfaff, Vivien J. Bubb, and John P. Quinn. 2021. "Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome" International Journal of Molecular Sciences 22, no. 12: 6319. https://doi.org/10.3390/ijms22126319
APA StyleKoks, S., Pfaff, A. L., Bubb, V. J., & Quinn, J. P. (2021). Expression Quantitative Trait Loci (eQTLs) Associated with Retrotransposons Demonstrate their Modulatory Effect on the Transcriptome. International Journal of Molecular Sciences, 22(12), 6319. https://doi.org/10.3390/ijms22126319