1. Introduction
Human complex I, also called NADH-coenzyme Q oxidoreductase, is the first, largest, and most complicated respiratory complex in the oxidative phosphorylation system (OXPHOS), which is not only critical in catalyzing NADH oxidation to transfer two electrons to ubiquinone but also in coupling proton translocation for ATP generation [
1,
2]. In addition, it is the major source of reactive oxygen species (ROS) production [
2,
3,
4]. This protein complex has an L-shaped structure containing a peripheral arm protruding in the matrix and a membrane arm embedded in the inner membrane of mitochondria [
5]. It is composed of 45 (44 unique) subunits with a molecular size of about 1000 kDa [
5,
6,
7]. Among them, 7 subunits (ND1–6 and ND4L) are encoded by mitochondrial DNA (mtDNA) and 37 subunits (NDUFV1–3, NDUFS1–8, NDUFA1–13, NDUFB1–11, NDUFAB1, and NDUFC1–2) are encoded by nuclear DNA (nDNA) [
1,
8]. Due to its dual origins, structural complexity, and important roles in OXPHOS, complex I deficiency has been linked to a large variety of pathological conditions such as fatal infantile lactic acidosis (FILA), Leigh syndrome [
9,
10], Leber hereditary optic neuropathy (LHON), and mitochondrial encephalomyopathy with lactic acidosis (MELAS) [
8], and is associated with the pathogenesis of many devastating neurodegenerative disorders including Parkinson’s disease [
11,
12].
NADH dehydrogenase (ubiquinone) Fe-S protein 8 (NDUFS8) is a nuclear-encoded subunit of human complex I. This protein and the other 13 proteins (NDUFS1–3, NDUFS7, NDUFV1–2, ND1–6, and ND4L) are considered the core subunits of complex I, which are highly conserved from prokaryotic to eukaryotic cells. NDUFS8 contains two tetranuclear iron-sulfur clusters (N6a and N6b) and plays a key role in electron transfer in complex I. NDUFS8 deficiency caused by missense mutations in
NDUFS8 gene was found as the first molecular genetic link between a nuclear-encoded subunit of complex I and Leigh syndrome, which is a severe neurological disorder characterized by bilaterally symmetrical necrotic lesions in the subcortical areas of the brain [
13]. In addition, a novel mutation in this gene was found in children with encephalomyopathy [
14]. The discoveries from recent studies also indicated that
NDUFS8 gene is one of eight OXPHOS component genes that are downregulated in patients with bipolar disorder (BD) [
15].
Defects directly or indirectly affecting the function of OXPHOS are the most frequent causes of mitochondrial diseases. However, due to its dual origins, the complexity of the system, and the diversity of clinical phenotypes, reliable and convenient therapeutic approaches for the treatment of disorders caused by OXPHOS deficiency are still lacking. Most conventional treatments currently available for mitochondrial diseases are only palliative. Enzyme replacement therapy (ERT) [
16], a therapeutic approach for replacing a defective enzyme in patients with a normal one, has recently emerged as a promising approach to treating mitochondria-related disorders. However, to successfully apply this approach in rescuing OXPHOS deficiency, the barrier imposed by plasma membranes that separate the interior of cells from the surrounding environment and thus block the transduction of therapeutic proteins has to be conquered. This limitation can be surmounted by the conjugation of a protein transduction domain ((PTD) also called cell-penetrating peptide (CPP)), which is a short peptide (less than 30 amino acids) capable of crossing cell membranes with no cell-type specificity and thus facilitates the delivery of protein cargoes into cells [
17].
Since the discovery of the cell-penetrating function of PTDs, many PTDs have been identified and designed, including TAT (from human immunodeficiency virus (HIV)-transactivator of transcription protein) [
18,
19], Antp (from
Drosophila homeotic transcription protein antennapedia) [
20], VP22 (from
Herpes simplex virus 1 DNA-binding protein VP22) [
21], and polyarginines (chemically synthesized) [
22]. Among them, TAT peptide and its derivatives are the most adopted and relatively well-characterized. The cationic charges of this peptide were shown to play an essential role in the cellular uptake process, and the minimal sequence with protein transduction function is the amino acid residues 49–57 (RKKRRQRRR) of the original TAT protein [
23]. To deliver cargo proteins to certain organelles with PTDs, the presence of a specific organelle signaling peptide in the PTD-containing fusion proteins is required for proper targeting [
24]. Mitochondrial targeting sequence (MTS) is an N-terminal cleavable signal peptide that can assist with the mitochondrial targeting and entrance of about 60–70% of nuclear-encoded mitochondrial precursor proteins through a presequence import pathway toward the mitochondrial matrix [
25,
26,
27]. This canonical mitochondrial import pathway is driven by both the mitochondrial membrane potential (ΔΨm) and ATP generated via OXPHOS.
Del Gaizo et al. published the first report demonstrating that TAT incorporated in front of an MTS from mitochondrial malate dehydrogenase (mMDH) can achieve plasma membrane crossing and mitochondrial delivery and accumulation of an exogenous enhanced green fluorescent protein (eGFP) protein [
28,
29]. However, while attempting to apply the same concept by the addition of an MTS to exonuclease III (ExoIII) to accomplish the repair of mtDNA damage to the mitochondria of breast cancer cells following oxidative stress, Shokolenko et al. found that the successful delivery of TAT-fused ExoIII into mitochondria occurred only when an MTS was present as the first domain in the N-terminus of the recombinant protein as an MTS–ExoIII–TAT fusion [
30]. The above finding implicated that the presence of TAT preceding an MTS might hamper the mitochondrial targeting of the fusion protein. Nevertheless, Rapoport et al. later succeeded in applying TAT conjugation in front of a nuclear-encoded mitochondrial lipoamide dehydrogenase (TAT–LAD) to deliver the fusion protein into mitochondria to restore pyruvate dehydrogenase complex activity in the mitochondria of patients with LAD deficiency [
31], and in multiple tissues in the LAD-deficiency model mice [
32].
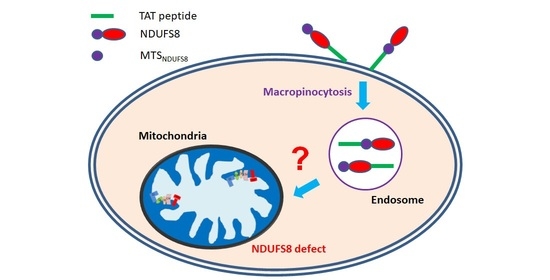
According to the results of the aforementioned studies, the mechanism of delivering proteins to mitochondria by incorporation of TAT with an MTS still remains controversial and elusive. Due to the essential roles of NDUFS8 in complex I function and structure, and the importance of complex I in OXPHOS, we tried to apply the concept of ERT with the combination of the membrane crossing ability of TAT and the mitochondrial targeting capability of MTS in NDUFS8 to develop a novel TAT-mediated protein transduction system to deliver exogenously produced NDUFS8 into mitochondria in this study. The results indicated that, with their native MTS, both exogenously produced TAT-NDUFS8 and NDUFS8-TAT were delivered into mitochondria and processed into the mature forms of NDUFS8. Surprisingly, this delivery system was independent of ΔΨm, which is dramatically different from the result of another endogenously expressed, nuclear-encoded complex I component protein NDUFS3 [
1,
8], which was used for comparison of ΔΨm dependency on crossing the mitochondrial inner membrane in this study. Furthermore, treatment with TAT-NDUFS8 fusion protein partially restored the assembly and functionality of complex I in NDUFS8-deficient cells. Our findings demonstrate the potential application of this protein transduction system for therapeutic treatment of complex I deficiency caused by NDUFS8 defects.
3. Discussion
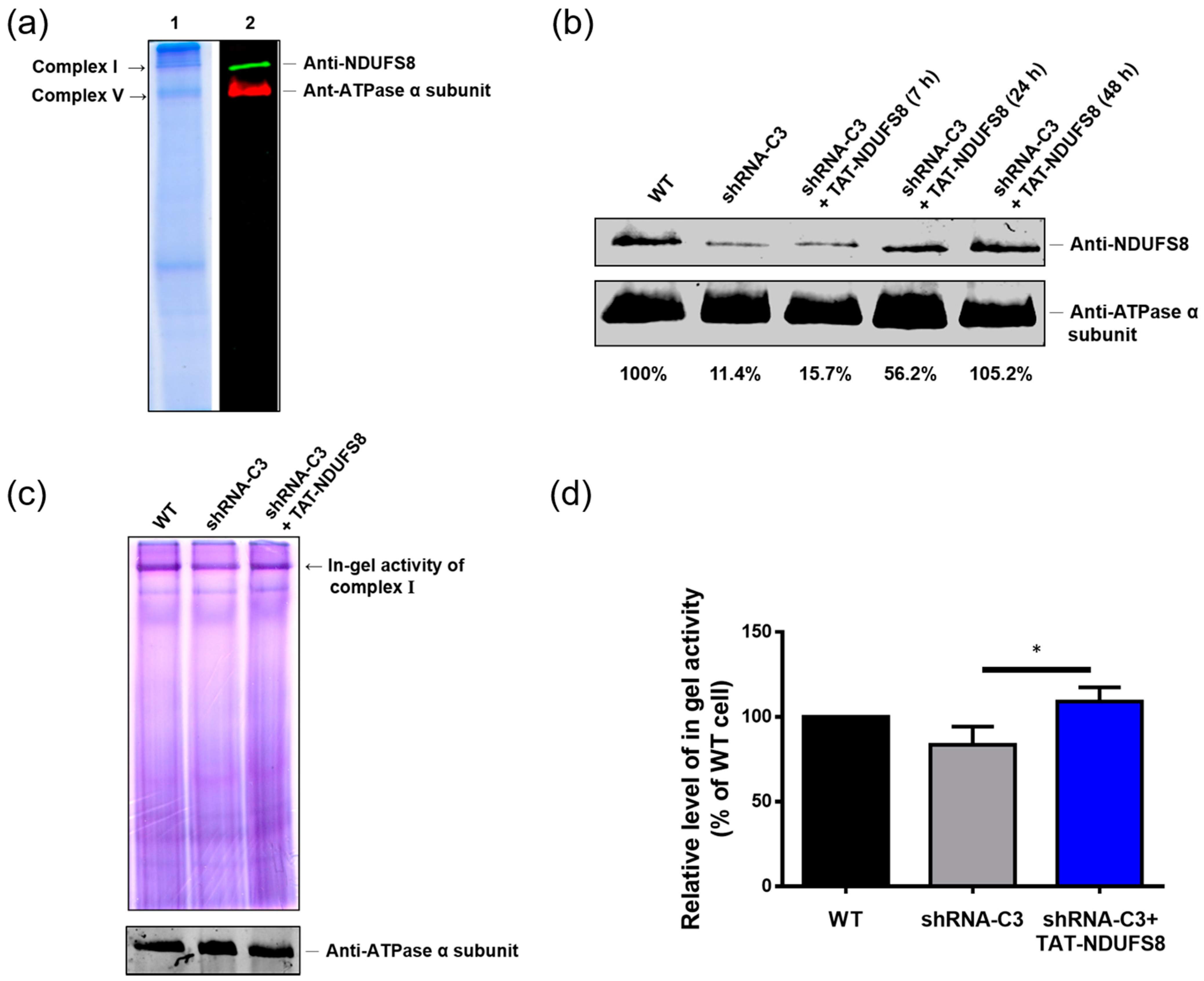
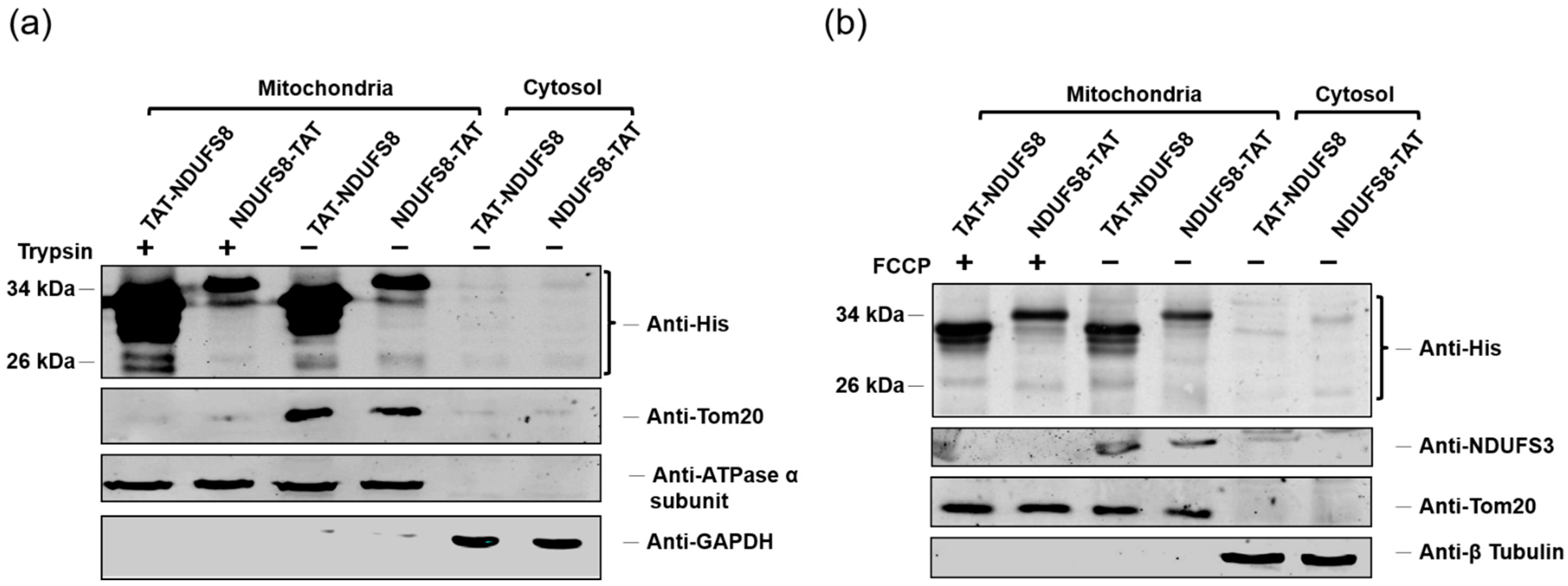
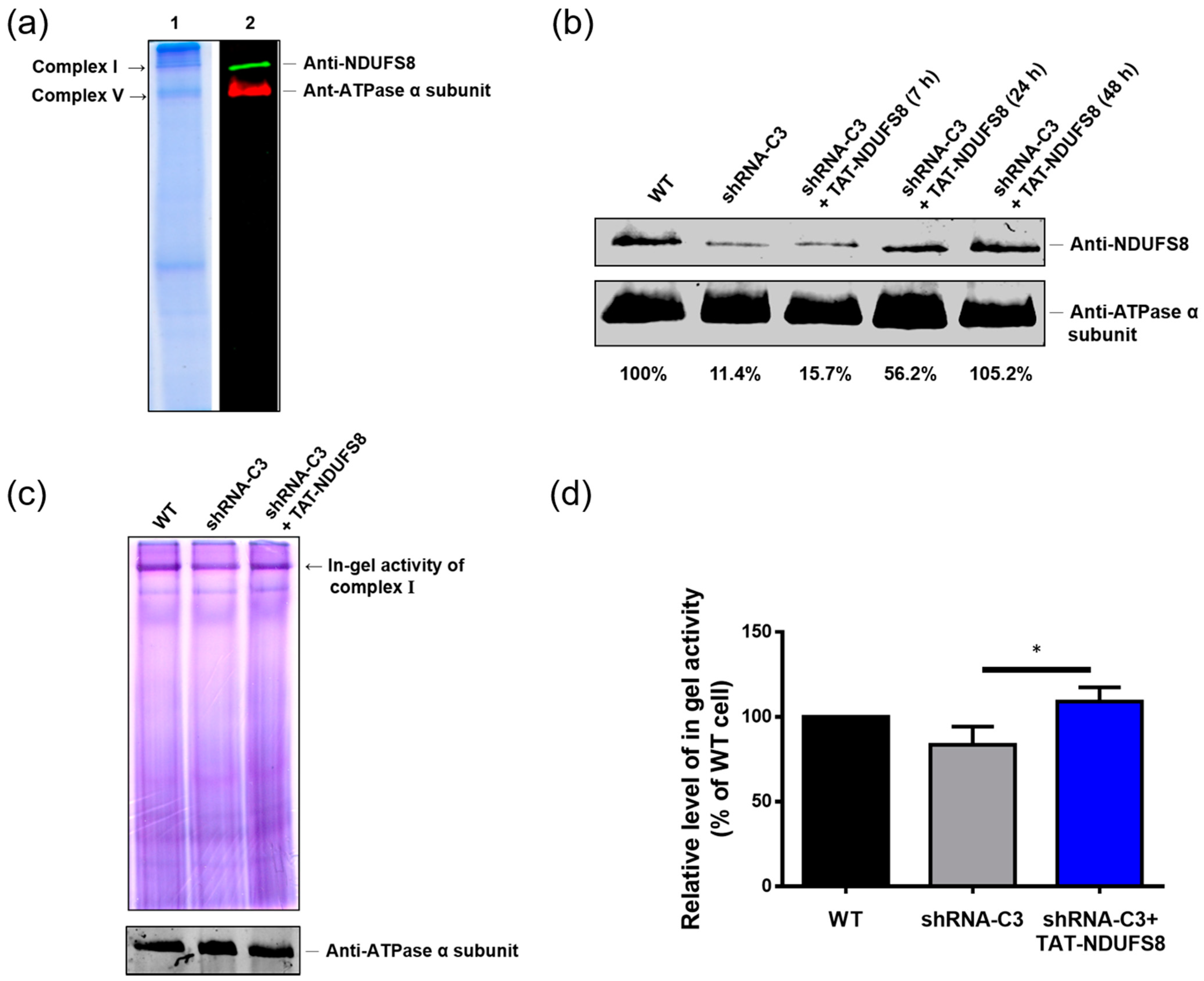
In this study, we generated a protein transduction system using TAT as the PTD to rescue the deficiency caused by the dysfunction of the NDUFS8 subunit in the mitochondrial complex I. According to our results, both exogenously produced TAT-NDUFS8 and NDUFS8-TAT could enter mitochondria; this finding was further confirmed by the results of the trypsin digestion tests. In addition, by including an uncoupler of OXPHOS, FCCP, in the experimental setting, we presented a novel mitochondrial protein import pathway in which the translocation of these two TAT fusion proteins across the inner mitochondrial membrane was independent of ΔΨm. Furthermore, using TAT-NDUFS8 as an example, we showed that after treating exogenously produced TAT-NDUFS8 for 48 h, the level of complex I assembly increased from 11.4% to 105.2% in the NDUFS8 knockdown cell line, shRNA-C3 compared to that of normal cells. Moreover, we found that the activity of complex I in shRNA-C3 cells also increased about 31% and 79% in the in-gel activity assay and the oxygen consumption assay, respectively. These data proved that this TAT-mediated NDUFS8 transduction system can partially restore the activity of complex I in shRNA-C3 cells.
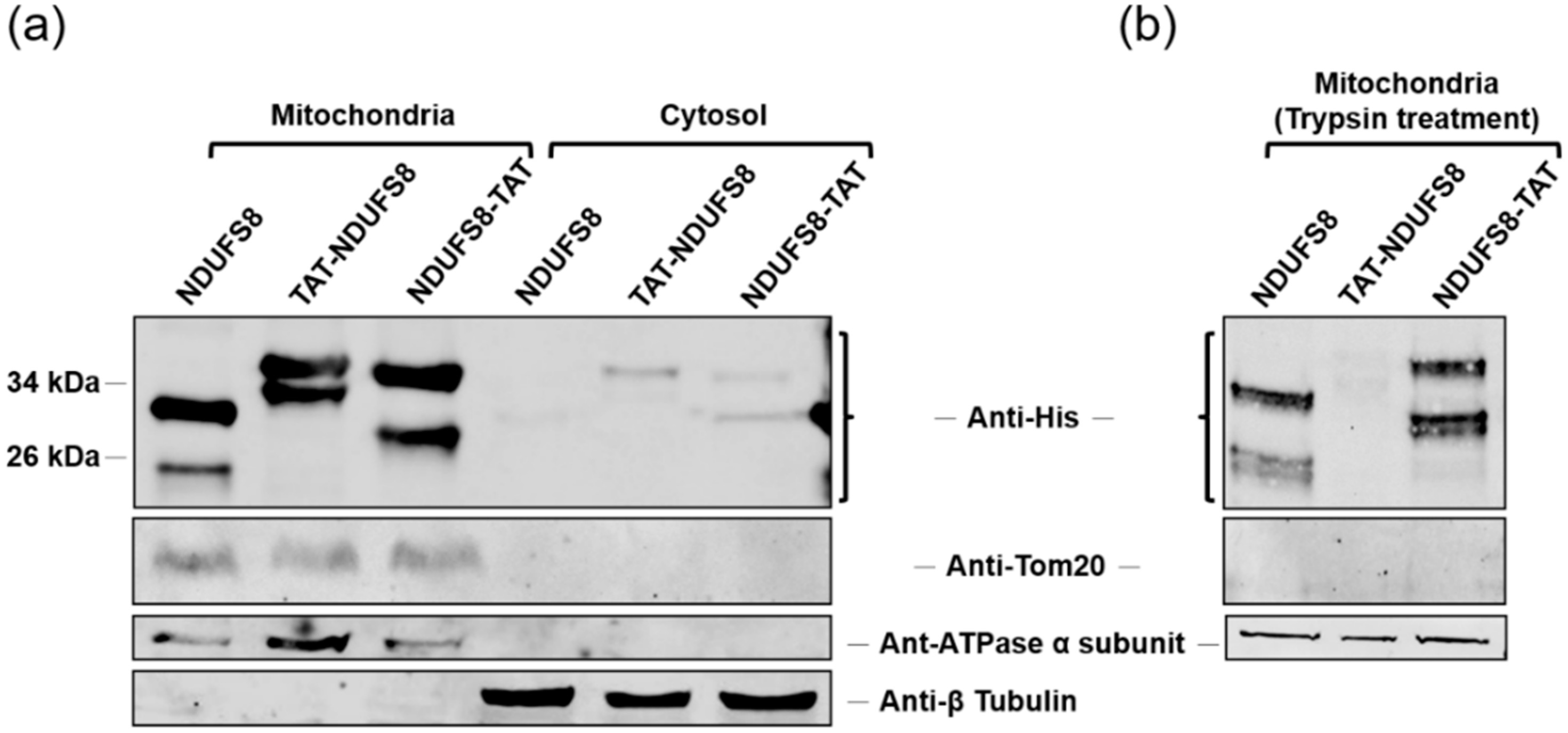
Based on the data presented here, we clearly demonstrate that TAT conjugation plays a pivotal role in bringing exogenously produced NDUFS8 fusion proteins into T-REx-293 cells. When a protein transduction system is applied to deliver a TAT-containing protein into mitochondria, some contradictory findings regarding the relative location of TAT to an MTS and the cargo protein (at the N- or C-terminus) have been reported [
28,
29,
30]. Del Gaizo et al. showed that the incorporation of TAT in front of an MTS from mMDH (MTS
mMDH) and the target protein eGFP provides the advantage of promoting the accumulation of eGFP in mitochondria because after processing by MPPs, TAT-MTS
mMDH is released from the precursor fusion protein and the mature cargo eGFP is formed and retained in the mitochondria [
28,
29]. In contrast, when using ExoIII as the cargo protein, Shokolenko et al. found that only MTS-ExoIII-TAT fusion but not TAT-MTS-ExoIII fusion could be correctly transduced into mitochondria [
30]. Importantly, the authors suggested that the presence of TAT preceding an MTS would impede the mitochondrial targeting of the fusion protein. They emphasized that the successful delivery of TAT fusion proteins into mitochondria occurs only when an MTS is present as the first domain in the N-terminus of the recombinant protein. In our current study, both TAT-NDUFS8 and NDUFS8-TAT fusion proteins were successfully transduced into mitochondria and correctly processed, indicating that the relative location between TAT and MTS (also the cargo) does not affect the mitochondrial targeting of NDUFS8. The discrepancy between ours and others’ results seem to imply that the importance of the location of TAT sequence relative to an MTS (and thus the target protein) is MTS- and/or cargo-protein-dependent. Therefore, the position of TAT and the selection of MTS have to be optimized for each individual therapeutic protein desired for targeting to mitochondria by TAT-mediated protein transduction.
Although accumulative evidence has proven that TAT-mediated delivery of proteins into cells is applicable, the mechanism through which proteins are transduced to mitochondria by incorporation with TAT still remains elusive. Some reports considered that the TAT-conjugated protein enters mitochondria via the canonical mitochondrial import pathway [
30], but others suggested that the TAT delivery system may be achieved through a different mechanism that does not involve the canonical mitochondrial import pathway [
29]. To clarify this unresolved mystery, we applied the transient transfection approach using TAT-derived NDUFS8 plasmids as the classic mode of the mitochondrial import pathway and compared the results to those of the TAT-mediated NDUFS8 transduction system under similar experimental conditions. Unlike the finding from the transient transfection experiment where only NDUFS8-TAT was successfully imported and processed in mitochondria, both the exogenously produced TAT-NDUFS8 and NDUFS8-TAT entered mitochondria and were then processed. In addition, when NDUFS8-TAT fusion was used for comparison, the efficiency of processing NDUFS8-TAT into its corresponding mature protein was more prominent in the transient transfection experiment than that in the protein transduction experiment. The above findings all indicate that the pathway for the mitochondrial import of TAT-conjugated NDUFS8 by protein transduction is different from the canonical mitochondrial import pathway as demonstrated by the transient transfection experiment.
Interestingly, we presented here that the translocation of exogenously produced TAT-NDUFS8 and NDUFS8-TAT across the inner mitochondrial membrane through protein transduction is independent of ΔΨm. This unexpected finding was also reported by Del Gaizo et al. when the aforementioned TAT-MTS
mMDH-eGFP was applied in a mitochondrial targeting study [
29]. The nuclear-encoded complex I protein NDUFS8 is predicated by the Mitoprot II program [
34] to contain the first 34 amino acids in the N-terminus as an MTS, and the presence of two peptides corresponding to the precursor and mature forms of NDUFS8 in the results of our studies proves this prediction. For a cleavable, presequence-type mitochondrial matrix protein, NDUFS8 is expected to be imported into mitochondria through the outer mitochondrial membrane (TOM) to a classical presequence import pathway, which is involved with the presequence translocase of the inner membrane (TIM23) before being excised by MPPs. It is well-accepted that the positively charged MTS initiates electrophoretic translocation through the inner membrane, and ΔΨm is critical for translocase activation and presequence transport [
25,
26,
27]. Therefore, the feature of ΔΨm independency of our TAT-mediated NDUFS8 transduction system suggests that it is unlike the well-recognized presequence import pathway and may be a yet-to-be-discovered route for mitochondrial matrix protein import. One example of a TIM23-dependent process of mitochondrial protein import but without ΔΨm dependency was reported in the literature [
35]. The subunit e of the F
1F
o ATP-synthase is a nuclear-encoded, single-spanning mitochondrial protein that is anchored to the inner membrane and lacks a recognizable MTS. It was shown that the assembly of subunit e into the inner mitochondrial membrane does not require ΔΨm. It was assumed that this ΔΨm-independence is due to the absence of positively charged amino acid residues at the N-terminus of the subunit. However, how TIM23 function is involved in the mitochondrial import of subunit e is still poorly understood. NDUFS8 possesses a cleavable N-terminal MTS with four positively charged residues that are unlike the features shown in the subunit e of the F
1F
o ATP synthase. Therefore, the reasons contributing to the ΔΨm-independence of subunit e mitochondrial import may not be applicable to TAT-conjugated NDUFS8. The exact mechanism through which exogenous TAT fusion proteins are delivered into mitochondria remains unknown at this stage and further investigation is needed.
After the binding of TAT-conjugated proteins to cell membranes, the fusion proteins have to penetrate cell membranes to enter cells. Based on the available data, it appears that multiple cellular entry routes exist for TAT-derived cargoes, which include direct membrane translocation and various types of endocytotic processes [
24]. Among them, macropinocytosis is the most recognized cell entry route for TAT-mediated protein transduction [
36,
37]. Following internalization by macropinocytosis, several reports indicated that TAT-conjugated cargoes have to escape from endosomes so that they can be released into the cytoplasm to execute the desired function [
36,
38]. On the basis of our current data of TAT-NDUFS8 from the transient transfection experiment and their comparison with the corresponding TAT-NDUFS8 transduction experiment (
Figure 1 and
Figure 3, respectively), the exogenously produced TAT-NDUFS8 from protein transduction appears to not be released from endosomes to the cytosol, as the fusion protein would otherwise not correctly enter mitochondria, just like the result shown in the transfection study. Therefore, we propose that the exogenously produced TAT-fused NDUFS8 may enter mitochondria via an as yet not well-known endosome-mitochondria juxtaposition route (contact or even fusion) exemplified by the entry of
Helicobacter pylori vacuolating cytotoxin A (VacA) into mitochondria [
39,
40]. VacA is a pore-forming toxin of
H. pylori and can enter host gastric cells via endocytosis. After internalization, this toxin is enclosed by endosomes. In one of the downstream pathways, the VacA-enriched endosomes can move close to mitochondria to form endosomes-mitochondria juxtaposition and further transport VacA into mitochondria to trigger apoptosis in host cells. Based on the discovered VacA traffic pathway, it would be interesting in the future to explore the possible involvement of endosomes-mitochondria juxtaposition in the import of TAT-conjugated NDUFS8 into mitochondria through protein transduction.
4. Materials and Methods
4.1. Molecular Cloning of NDUFS8 and Its TAT-Containing Constructs
A DNA fragment of
NDUFS8 with
NdeI and
XhoI restriction enzyme cutting sites was amplified by pfu polymerase with a primer pair,
NdeI-NDUFS8-F and NDUFS8-
XhoI-R, using the pcDNA4-
NDUFS8 construct as the template [
41]. The pET28b plasmid with
TAT sequence insert (provided by Dr. Steven F. Dowdy from the University of California, San Diego School of Medicine, CA, USA) was digested with
NdeI and
XhoI restriction enzymes and then ligated to the processed
NDUFS8 PCR product to generate the pET28b
-TAT-NDUFS8 construct. For the generation of the pET28a
-NDUFS8-TAT construct, a DNA fragment of
NDUFS8 with
EcoRI and
XhoI restriction enzyme cutting sites was amplified by pfu polymerase with a primer pair
EcoRI-NDUFS8-F and NDUFS8-
XhoI-R using the pcDNA4-
NDUFS8 construct as the template. The processed PCR product was then ligated to the pET28a plasmid pre-digested with
EcoRI and
XhoI restriction enzymes to generate the pET28a-
NDUFS8 construct. Using the obtained pET28a-
NDUFS8 construct as the template, a primer with an extending oligonucleotide
TAT-
EcoRV was designed and applied to PCR amplification to generate the
EcoRI-
NDUFS8-
XhoI-
TAT-
EcoRV insert. The sticky end of the
XhoI restriction site generated by pre-digestion of the pET28a plasmid with
EcoRI and
XhoI restriction enzymes was filled to become a blunt end with T4 DNA polymerase. Finally, the
EcoRI-
NDUFS8-
XhoI-
TAT-
EcoRV insert was ligated to the above modified plasmid to generate the pET28a-
NDUFS8-TAT construct. The resulting pET28b
-TAT-NDUFS8 and pET28a
-NDUFS8-TAT, and previously generated pET28a
-NDUFS8, were used in later experiments for TAT-NDUFS8, NDUFS8-TAT, and NDUFS8 overexpression in
Escherichia coli and protein purification, respectively. Furthermore, using the pET28b
-TAT-NDUFS8 construct as the template with primers
HindIII-TAT-NDUFS8-F and NDUFS8-
XhoI-R, a DNA fragment containing
TAT-NDUFS8 with
HindIII and
XhoI cutting sites was generated and ligated with the pcDNA4 vector to form the pcDNA4
-TAT-NDUFS8 construct. Similarly, using the pET28a
-NDUFS8-TAT construct as the template with primers
EcoRI-NDUFS8-2-F and NDUFS8-TAT-
XbaI-R, a DNA fragment containing
NDUFS8-TAT with
EcoRI and
XbaI cutting sites was generated and ligated with the pcDNA4 plasmid to form the pcDNA4
-NDUFS8-TAT construct. The pcDNA4
-TAT-NDUFS8, pcDNA4
-NDUFS8-TAT, and previously generated pcDNA4
-NDUFS8 plasmids were used in later experiments for transient transfection. All oligonucleotide sequences of the primers used in this study are listed in
Table 1.
4.2. Overexpression and Purification of TAT-NDUFS8, NDUFS8-TAT, and NDUFS8 Proteins
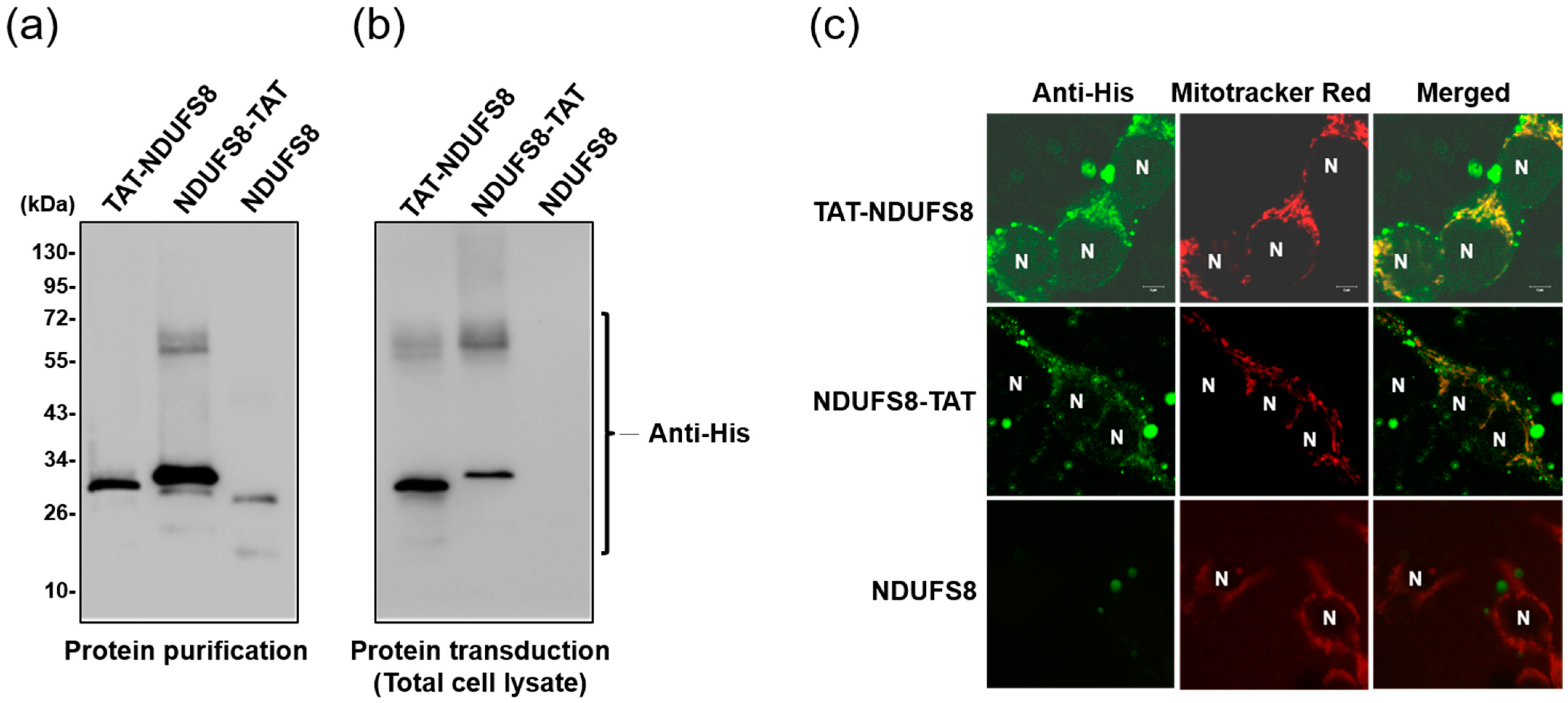
E. coli BL21 (DE3) cells transformed with pET28b-TAT-NDUFS8, pET28a-NDUFS8-TAT, or pET28a-NDUFS8 were cultivated in Luria-Bertani medium overnight at 37 °C. The bacterial culture was then added into Yeast Extract Tryptone (2YT) medium and cultivated at 37 °C until the OD600nm value reached 0.8. Then, 1.0 mM isopropy-β-D-thiogalactoside (IPTG; VWR-Amresco, Radnor, PA, USA) was used to induce protein overexpression for 5 h. Then, bacterial cells were collected by centrifugation with an R10A3 rotor at 5200× g rpm for 15 min (Hitachi, Tokyo, Japan) and resuspended in buffer Z (8 M urea, 100 mM NaCl and 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 8.0). After, sonication was used to rupture cells (conditions: 10 s on/off for 30 min). The supernatant and pellet fractions of cells were collected by centrifugation (Hitachi R15A rotor, 14,000× g rpm for 20 min at 4 °C). The collected TAT-NDUFS8, NDUFS8-TAT, and NDUFS8 supernatant fractions were added into the affinity chromatography column packed with IMAC Sepharose 6 FastFlow resins (GE Healthcare, Chalfont St. Giles, UK) and CoCl2 chelating reagents. The protein bound resins were sequentially washed with the elution buffer containing different concentrations of imidazole (20–1000 mM, pH 5.5). The fractions collected from 250 and 500 mM imidazole elution were concentrated using an Amicon Ultra concentrator (Merck-Millipore, Darmstadt, Germany) and exchanged with PBS. The purified proteins were further analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Finally, 20% glycerol was added to the collected recombinant proteins and kept at −20 °C until later analyses.
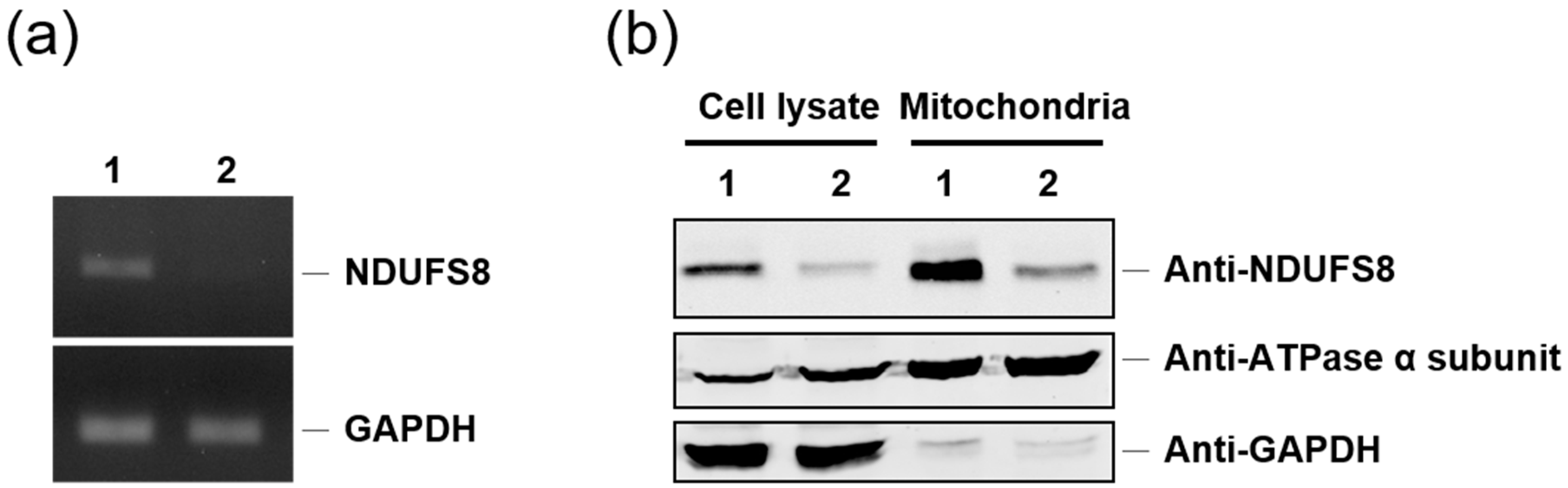
4.3. Generation of an NDUFS8 Knockdown Stable Cell Line
A short-hairpin RNA (shRNA) clone targeting base pairs 243–263 of the NDUFS8 coding region was purchased from National RNAi Core Facility (NRC, Taipei, Taiwan). The short-hairpin oligonucleotide sequence (shRNA-C3) inserted in the plasmid pLKO.1-puro is shown in
Table 1. Cells in 6-well culture plates were transfected with the shRNA-C3-containing plasmid by Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). After 24 h incubation, cells were transferred to 10 cm culture dishes and selected in a medium containing 2.5 μg/mL puromycin (Sigma-Aldrich, St. Louis, MO, USA) for 4 weeks.
4.4. Cell Culture
T-REx-293 cells, human embryonic kidney cells with a tetracycline-regulated expression system (Thermo Fisher Scientific), and their derived NDUFS8 stable knockdown cells (shRNA-C3) were cultivated in Dulbecco’s modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS; Biological Industries, Beit Haemek, Israel), 100 U/mL penicillin, and 100 μg/mL streptomycin (Biological Industries) with 5% CO2 at 37 °C. shRNA-C3 cells were maintained in the above cell culture medium containing 2.5 μg/mL puromycin (Sigma-Aldrich).
4.5. Transient Transfection
T-REx-293 cells were transfected with MaestroFectin transfection reagents (MaestroGen, Hsinchu City, Taiwan) as recommended by the manufacturer. Cells were seeded in 6 cm dishes and grown to 70–80% confluency before the transfection. The transfection procedure was conducted as follows: 3 μL MaestroFectin transfection reagent was first added to 90 μL serum-free medium (without antibiotics) in an Eppendorf tube and mixed gently. After incubating for 5 min at room temperature, 3 μg plasmid DNA was added to the diluted MaestroFectin transfection reagent and mixed gently. The mixture was incubated for 15 min at room temperature to form a MaestroFectin transfection reagent-DNA complex, and then 500 ng/mL tetracycline was added to the cells to induce the overexpression of target proteins for 18 h at 37 °C.
4.6. Transduction of Proteins into Cells
Cells were cultured in 6 or 10 cm dishes to 70–80% confluency. Before protein transduction, cells were washed once with PBS and then incubated with different amounts of TAT-NDUFS8, NDUFS8-TAT, or NDUFS8 in serum-free growth medium for 3–48 h. We added 10% FBS to the cell culture when the incubation time was more than 3 h to prevent cell starvation.
4.7. Subcellular Fractionation
The cytosolic and mitochondrial fractions of cells were separated by differential centrifugation with some modifications to a previously established method [
39]. All the steps of centrifugation were performed at 4 °C. Briefly, cells transfected with expression plasmids or treated with transduction proteins were obtained by trypsinization. The resulting cells were resuspended with 3 times the pellet volume of homogenization buffer (HB; 250 mM sucrose, 3 mM imidazole, and 1mM ethylenediaminetetraacetic acid (EDTA); pH 7.4) on ice and centrifuged at 1300×
g for 10 min. Cells were then resuspended with 4 times the pellet volume of HB and homogenized using 45 strokes with a glass homogenizer. The obtained homogenate was centrifuged at 3000×
g for 10 min. The post-nuclear supernatant (PNS) was transferred to a clean tube for further centrifugation at 12,000×
g for 10 min. The supernatant collected was the cytosolic fraction. The nuclear pellet (NP) was resuspended with 0.4 mL assay buffer (120 mM mannitol, 80 mM KCl, 1 mM EDTA, and 20 mM HEPES; pH 7.4) and centrifuged at 600×
g for 10 min again. The resulting supernatant was layered onto a cushion of 1 M mannitol in the assay buffer (0.6 mL). After centrifugation at 9000×
g for 15 min, the obtained pellet was resuspended in 0.5 mL assay buffer and further centrifuged at 10,000×
g for 10 min. Finally, the pellet was resuspended in 30 μL lysis buffer as the mitochondrial fraction.
4.8. Trypsin Treatment
After subcellular fractionation, the mitochondrial pellet was incubated with trypsin to the concentration of 1 μg trypsin/100 μg mitochondrial pellet at 37 °C for 5 min to obtain the trypsin-treated mitochondrial fraction.
4.9. Carbonyl Cyanid p-(Trifluoromethoxy) Phenylhydrazone Treatment
Cells were first cultivated in the cell culture medium containing 10 μM carbonyl cyanid p-(trifluoromethoxy) phenylhydrazone (FCCP) for 18 h. Later, the serum-free medium containing 0.06 mg/mL TAT-NDUFS8 or NDUFS8-TAT was replaced by the original cell culture medium and incubated for another 3 h. The resulting cell culture was applied for subcellular fractionation.
4.10. Prepartion of Total Cell Lysates
Cells were first cultivated in 6 cm dishes to 90% confluency and then collected by trypsinization, followed by washing once with PBS. Later, the lysis buffer (150 mM NaCl, 5 mM EDTA, 1% triton-x 100, and 10 mM Tris-Cl; pH 7.4) was added to cells for cell disruption to obtain the total cell lysates.
4.11. Protein Electrophoresis and Immunoblotting Analysis
A bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific) with bovine serum albumin (BSA) as the standard was used to determine the protein concentration of samples. We added 4× protein sample dye into the samples and the mixtures were boiled for 10 min before gel loading. Protein samples were separated by 10% SDS-PAGE gels. The resolved gels were then transferred to 0.45 µm nitrocellulose membranes (GE Healthcare, Chicago, IL, USA) at a 400 mA current for 90 min. The transferred membranes were blocked with 5% skim milk in PBS for 1 h at room temperature, and then incubated at 4 °C overnight with one of the following primary antibodies: rabbit anti-His antibody (Bioman, New Taipei city, Taiwan; 1:5000 dilution), rabbit anti-NDUFS8 antibody (GeneTex, Hsinchu City, Taiwan; 1:1000 dilution), mouse anti-ATPase α subunit antibody (Thermo Fisher Scientific; 1:10,000 dilution), mouse anti-β tubulin antibody (Santa Cruz Biotechnology, Dallas, TX, USA; 1:500 dilution), mouse anti-GAPDH antibody (Merck-Millipore; 1:10,000 dilution), mouse anti-Tom20 antibody (Santa Cruz Biotechnology; 1:250 dilution), or mouse anti-NDUFS3 antibody (Thermo Fisher Scientific; 1:500 dilution). After 5 washes with PBST (PBS containing 0.02% Tween-20) for 7 min each, the membranes were incubated with the secondary antibodies of goat anti-mouse IRDye 680LT (LI-COR Biosciences, Lincoln, NE, USA; 1:10,000 dilution) or goat anti-rabbit IRDye 800CW (LI-COR Biosciences; 1:10,000 dilution) for 45 min in a light-proof container. The membranes were then washed 5 times for 7 min each with PBST and scanned using a LI-COR Odyssey® infrared imaging system (LI-COR Biosciences).
4.12. Cell Viability Assay
The 3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma-Aldrich, St. Louis, MO, USA) was used to determine the cell viability. A total of 5 × 104 cells were seeded and cultivated in each well of 96-well plates for 24 h, followed by treatment with different amounts of TAT-NDUFS8 or NDUFS8-TAT recombinant protein for 48 h. Later, we added 250 μg/mL MTT to the cell culture for 2 h. The resulting yellowish solution was reduced to purple formazan in living cells, and 200 μL dimethyl sulfoxide (DMSO; Scharlau, Sentmenat, Spain) was then added to each well to dissolve the insoluble MTT product into a purplish solution. The cell viability index was measured at 565 nm by a Wallac 1420 VICTOR3 V plate reader (PerkinElmer, Waltham, MA, USA).
4.13. High-Resolution Clear Native Gel Electrophoresis (HrCNE) and Complex I In-Gel Activity Assay
T-REx-293 cells and the derived NDUFS8 stable knockdown cells (shRNA-C3) were cultured with or without 0.03 mg/mL TAT-NDUFS8 recombinant protein for different periods of time and then harvested by trypsinization. After PBS washing and centrifugation, cells were resuspended in ice-cold mitochondrial extraction buffer (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 20 mM HEPES/KOH, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail; Roche, Penzberg, Germany) and homogenized by 45 strokes with a glass homogenizer on ice. The homogenized suspensions were first centrifuged at 700× g for 15 min to remove cell debris, followed by centrifugation at 10,000× g for 10 min to obtain crude mitochondrial pellets. The pellets were weighed and solubilized on ice with 35 μL solubilization buffer (50 mM NaCl, 50 mM imidazole/ HCl, 2 mM 6-aminocaproic acid, and 1mM EDTA) per 20 mg of pellets. Later, 6 μL 20% (w/v) n-dodecyl β-D-maltoside (DDM; Sigma-Aldrich) per 20 mg of pellets was added and mixed by flicking the Eppendorf tube. The resulting suspension was incubated on ice for 10 min, followed by centrifugation at 20,000× g for 20 min. The supernatant containing the DDM-solubilized mitochondrial complexes was collected and mixed with the loading buffer containing 10% glycerol and 0.1% Ponceus S. We used 6.5% acrylamide non-gradient native gel with anode buffer (25 mM imidazole/HCl, pH 7.0) and cathode buffer (50 mM Tricine, 7.5 mM imidazole/HCl, 0.05% deoxycholate, and 0.02% DDM, pH 7.0) to separate the mitochondrial protein complexes. The gel was first run at a voltage of 45 V with a fixed current of 20 mA for 30 min, followed by at a voltage of 100 V with a fixed current of 20 mA for 20 min, and then at a voltage of 250 V with a fixed current of 15 mA for 90 min at 4 °C. After the completion of HrCNE, the resulting native gels were applied in the following 3 assays: First, the protein profile in the native gel was observed by gel staining with Coomassie blue solution (0.1% Coomassie brilliant blue R-250 (Sigma-Aldrich), 50% methanol, and 10% glacial acetic acid) for 1 h and destained with the destaining solution (40% methanol and 10% glacial acetic acid) overnight. Secondly, the degree of NDUFS8 assembled in the mitochondrial complex I was detected using an immunoblotting analysis with rabbit anti-NDUFS8 antibody (GeneTex; 1:1000 dilution). The obtained signals were calculated by ImageJ software (NIH, Bethesda, MA, USA). The intensity of NDUFS8 in the samples was normalized by the loading control, complex V (detected by anti-ATPase α subunit antibody), at 100%. Lastly, the activity of complex I was assessed using the in-gel activity assay. The native gel was incubated with 5 mM Tris/HCl buffer (pH 7.4) containing 0.1 mg/mL NADH and 2.5 mg/mL nitroblue tetrazolium (NBT; USB, Cleveland, OH, USA) at room temperature for 30 min and then fixed with the fixing solution (50% methanol and 10% acetic acid) at room temperature for 30 min, followed by washing with ddH2O and scanned to quantify complex I in-gel activity.
4.14. Oxygen Consumption Measurement
Cells were cultured with or without the treatment of TAT-NDUFS8 (0.03 mg/mL) for 48 h and then harvested by trypsinization. After PBS washing and centrifugation, 5 × 106 cells were resuspended in 1 mL of the respiration buffer (RB; 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, and 10 mM K2PO4; pH 7.4) and permeabilized by the addition of non-ionic detergent digitonin (Sigma-Aldrich, 60 μg/mL) under continuous shaking in a 15 mL tube for 2 min at room temperature. Then, 5 mL RB supplemented with 1 mg/mL BSA was added to the cell suspension to stop the membrane permeabilization. The resulting mixtures were then centrifuged at 1000× g for 5 min, and the obtained pellets were resuspended in 500 μL RB buffer supplemented with 1 mg/mL BSA and 0.5 mM ADP. The digitonin-permeabilized cell suspension was ready for oxygen consumption measurements. The assay was conducted by the Mitocell S200 micro respirometry system (Strathkelvin Instruments, North Lanarkshire, U.K.) after the addition of complex I substrates, 8 mM pyruvate, and 0.2 mM malate. The reaction was stopped by the addition of a complex I inhibitor, rotenone (3 μM).
4.15. Immunofluorescence Detection
Cells were seeded onto cover glasses in 24-well plates and grown to 70–80% confluency, followed by treatment with 0.03 mg/mL TAT-NDUFS8 for 3 h. To label mitochondria, 0.1 mM Mitotracker Red (Thermo Fisher Scientific) was were added to cells, which were cultivated for another 30 min. Cells were then fixed by adding the acetone/methanol mixture (acetone: methanol = 3:1 in volume proportion) for 5 min on ice. After that, cells were incubated with the culture medium for another 1 h at room temperature, followed by treatment with rabbit anti-His antibody (Bioman, New Taipei city, Taiwan; 1:100 dilution). After incubating at room temperature for 1 h, the cover glasses were washed with PBS 3 times for 5 min each. Then, cells were incubated with anti-rabbit IgG-FITC antibody (AnaSpec, Fremont, CA, USA; 1:100 dilution) at room temperature for 1 h, and washed with PBS 3 times for 5 min each. Lastly, the cover glasses were mounted with VECTASHIELD Mounting Medium (Vector Laboratories, Burlingame, CA, USA). Fluorescence was visualized using an LSM 5 PASCAL confocal microscope (ZEISS, Welwyn Garden City, Germany).
4.16. Statistical Analysis
Results are presented as mean and standard deviation (±SD) from n ≥ 3 independent experiments. Data were analyzed using the unpaired Student’s t-test with two-tailed distribution, and the results were considered significant when the p value was smaller than 0.05, 0.01, and 0.001.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}