
Brain Symptoms of Tuberous Sclerosis Complex: Pathogenesis and Treatment


Abstract
:1. Introduction
2. Pathology and Clinical Picture of TSC
2.1. Systemic Findings
2.2. Brain Symptoms
2.2.1. Epilepsy
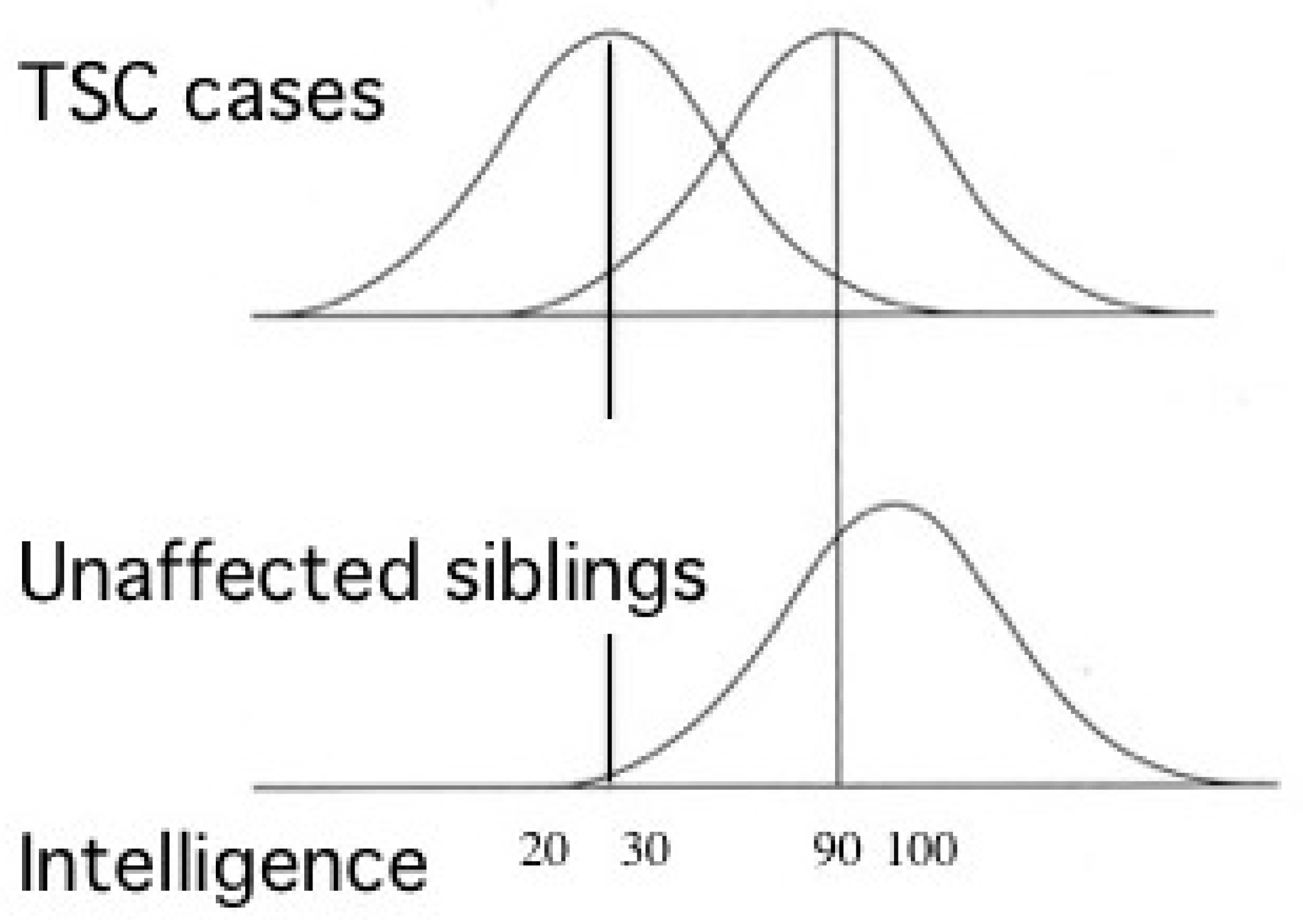
2.2.2. ID and ASD
2.2.3. Brain Tumor
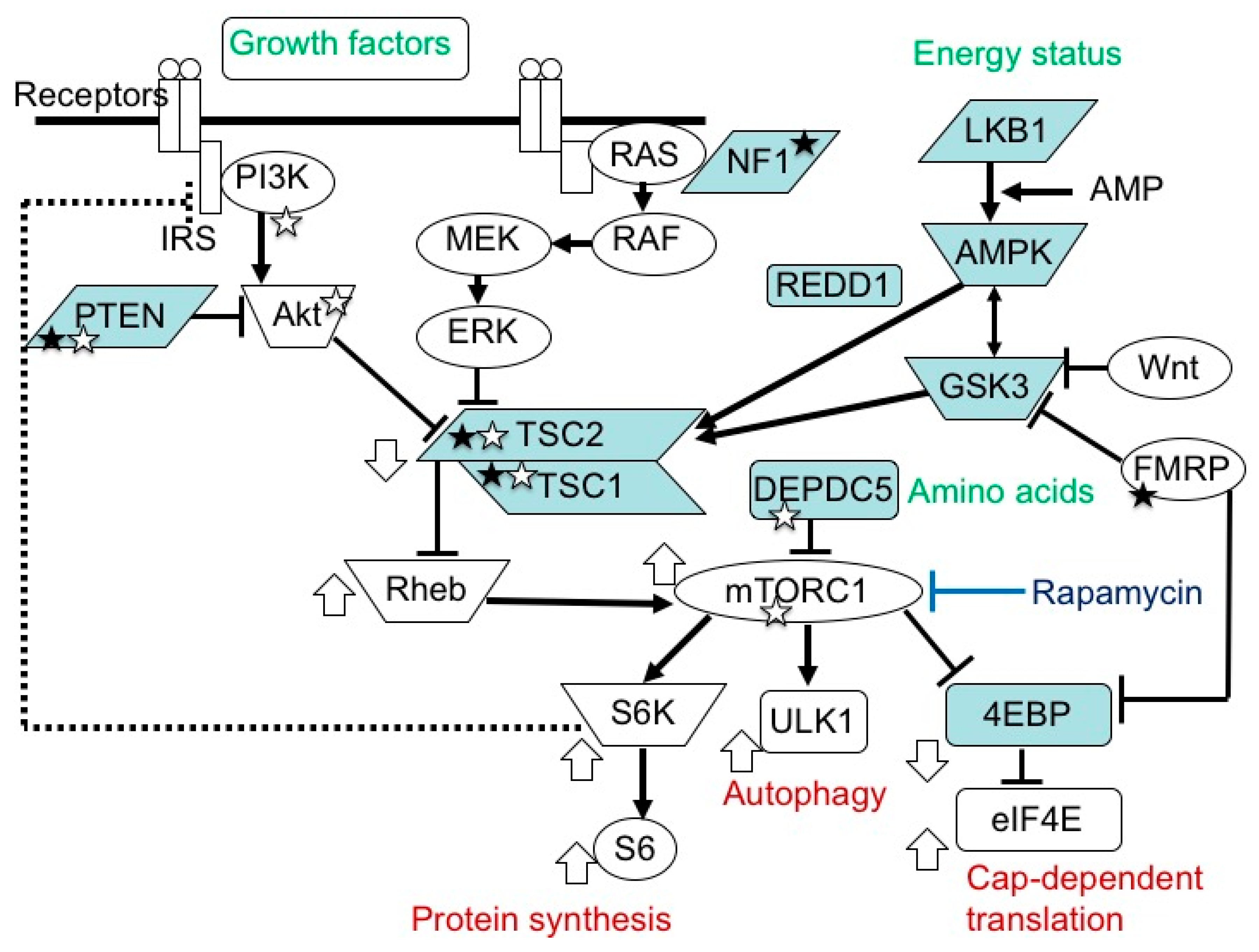
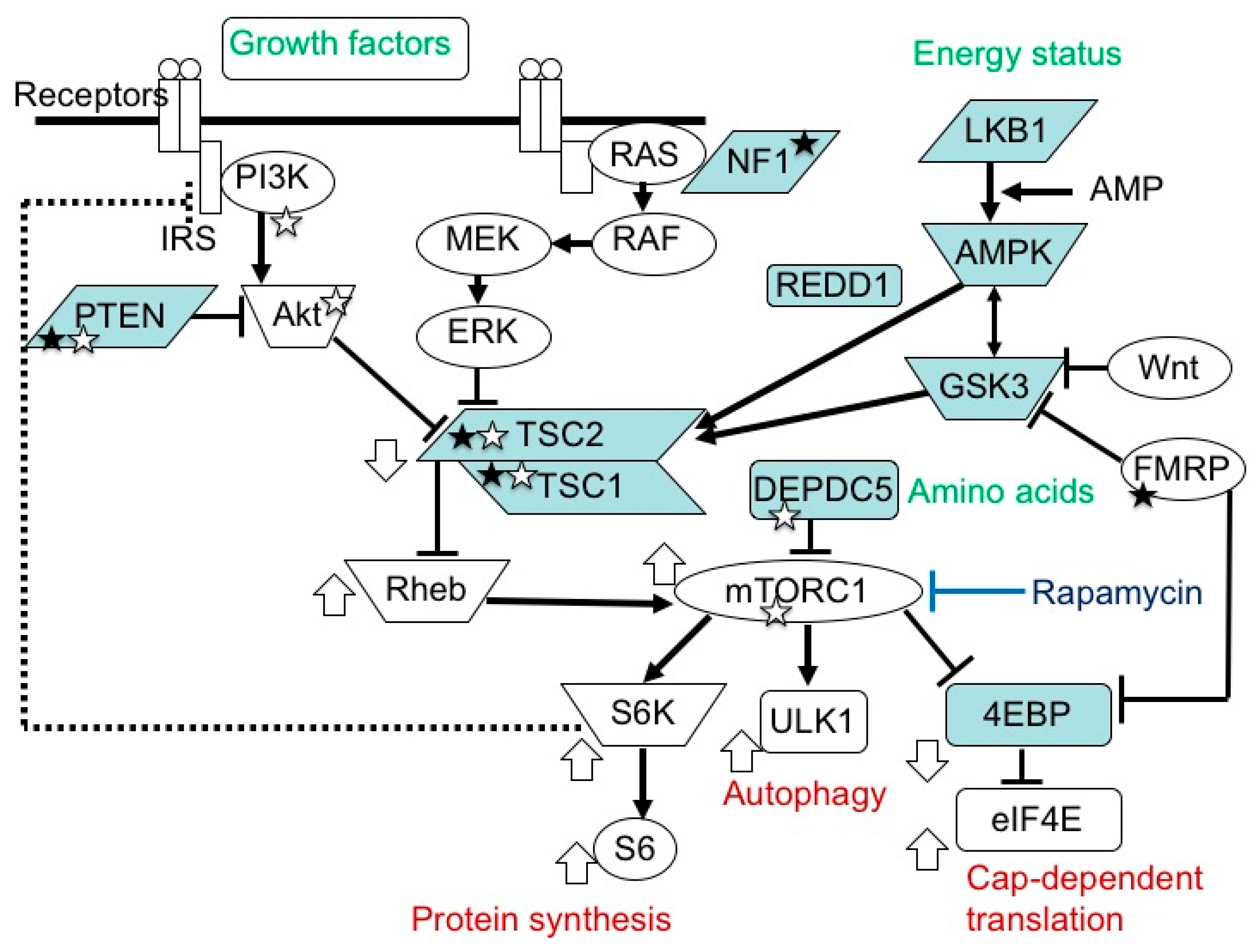
3. Etiology and Pathogenesis of TSC
3.1. TSC Gene Mutations and Their Consequences
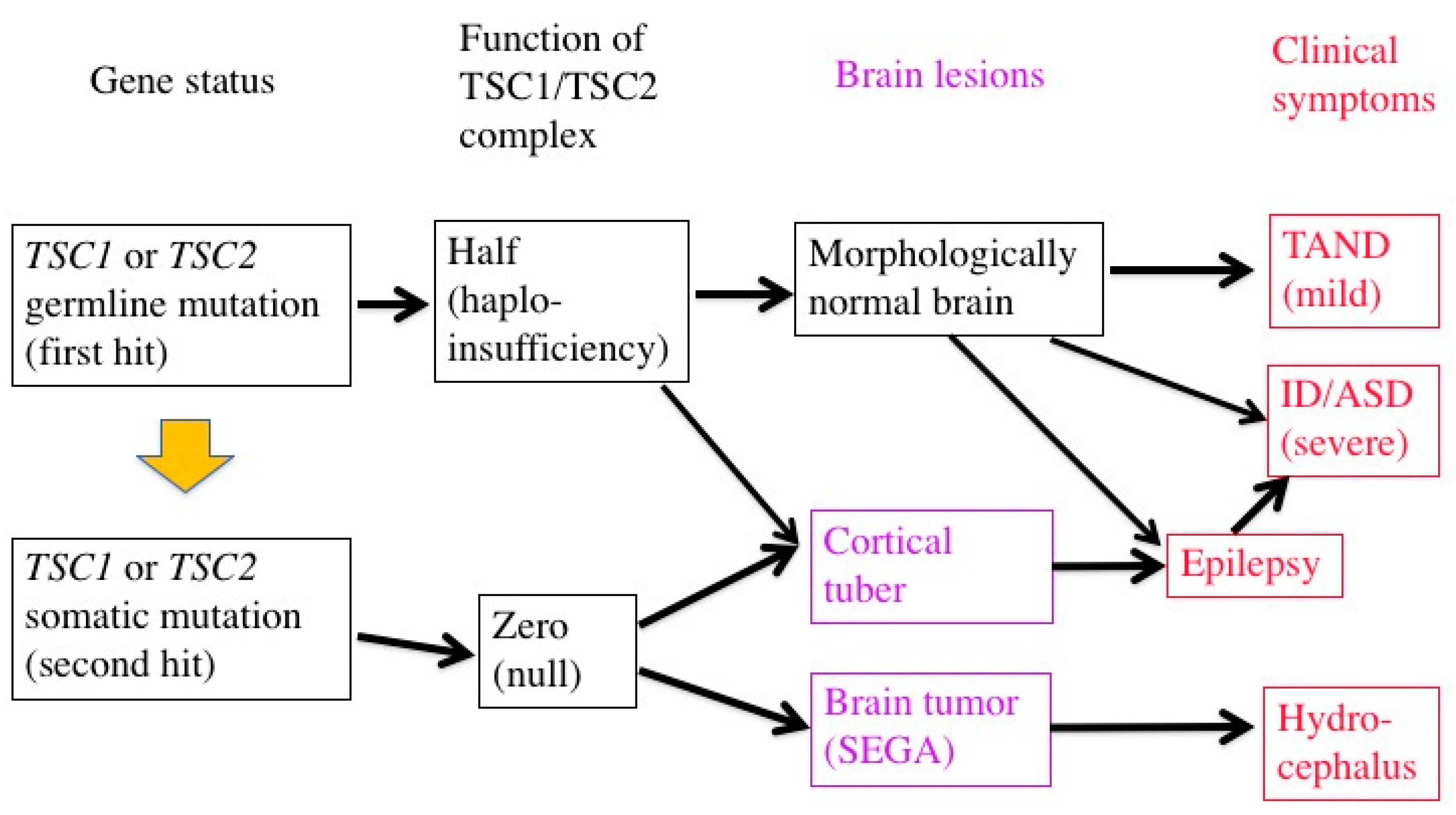
3.2. Germline and Somatic Mutations
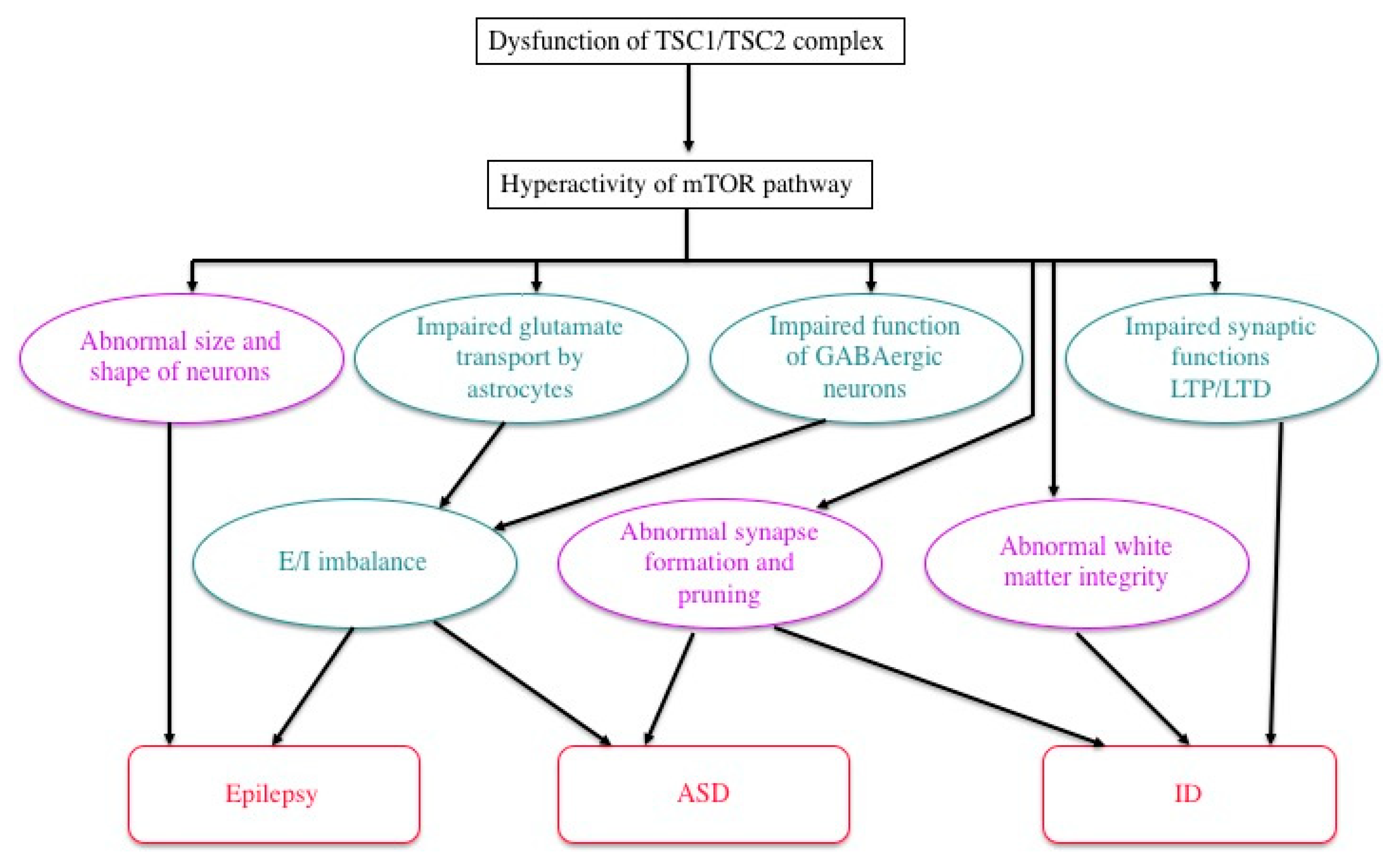
4. Brain Dysfunction in TSC
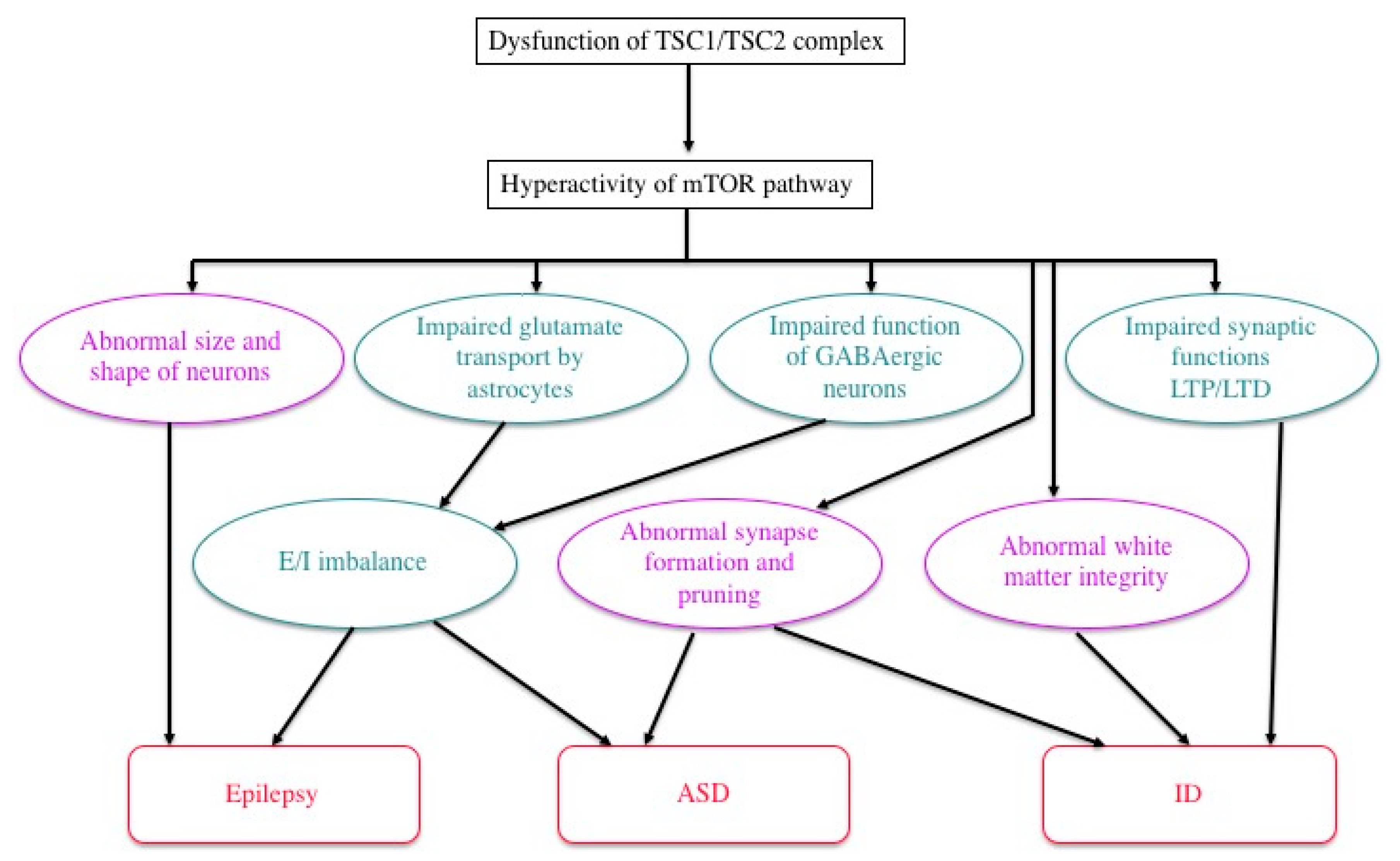
4.1. ID, ASD, and Epilepsy
4.2. Pathophysiology: Neural and Glial Dysfunction
4.3. Efficacy of mTOR Inhibitors: Animal Experiment
4.4. Efficacy of mTOR Inhibitors: Clinical Trials
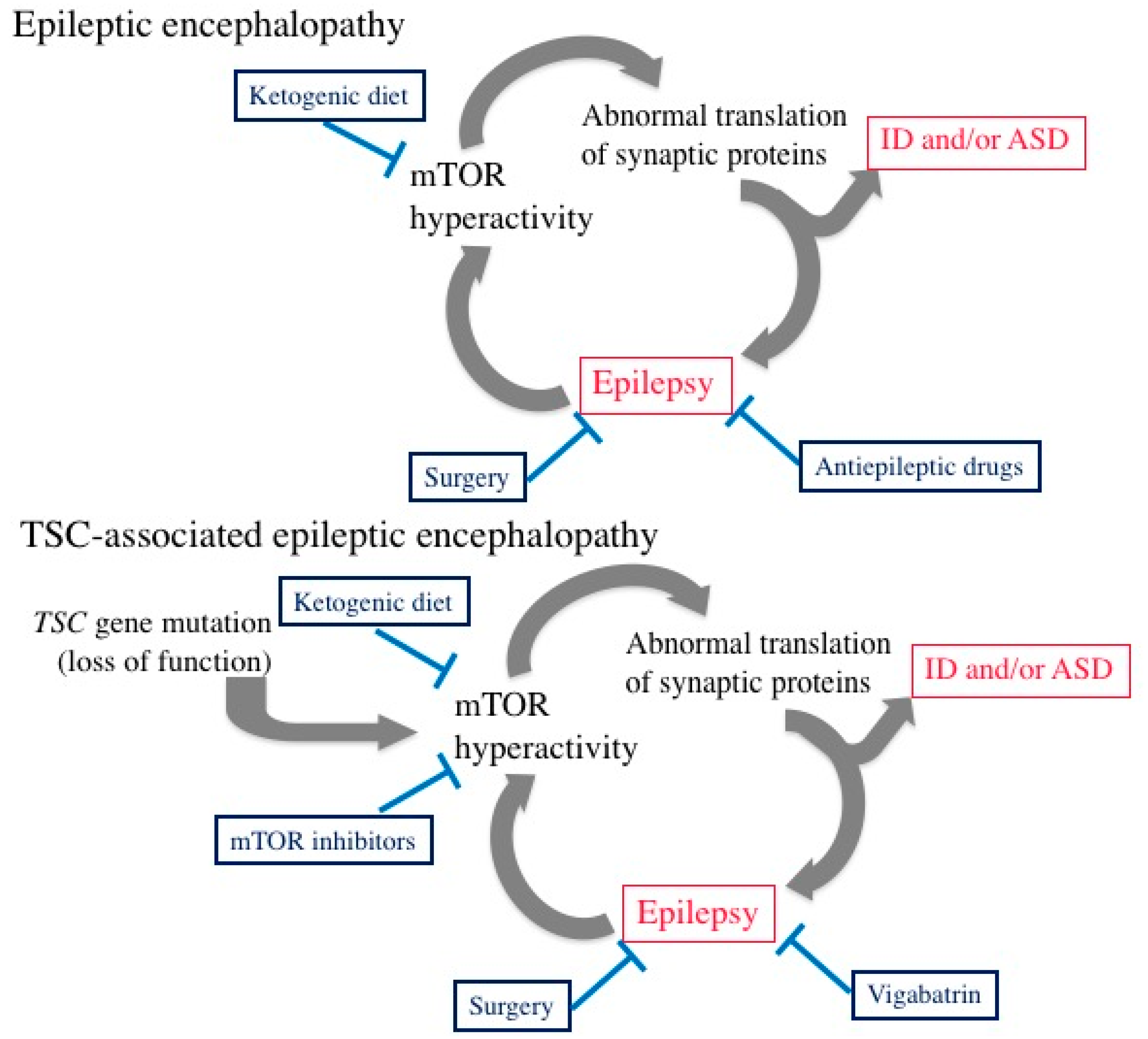
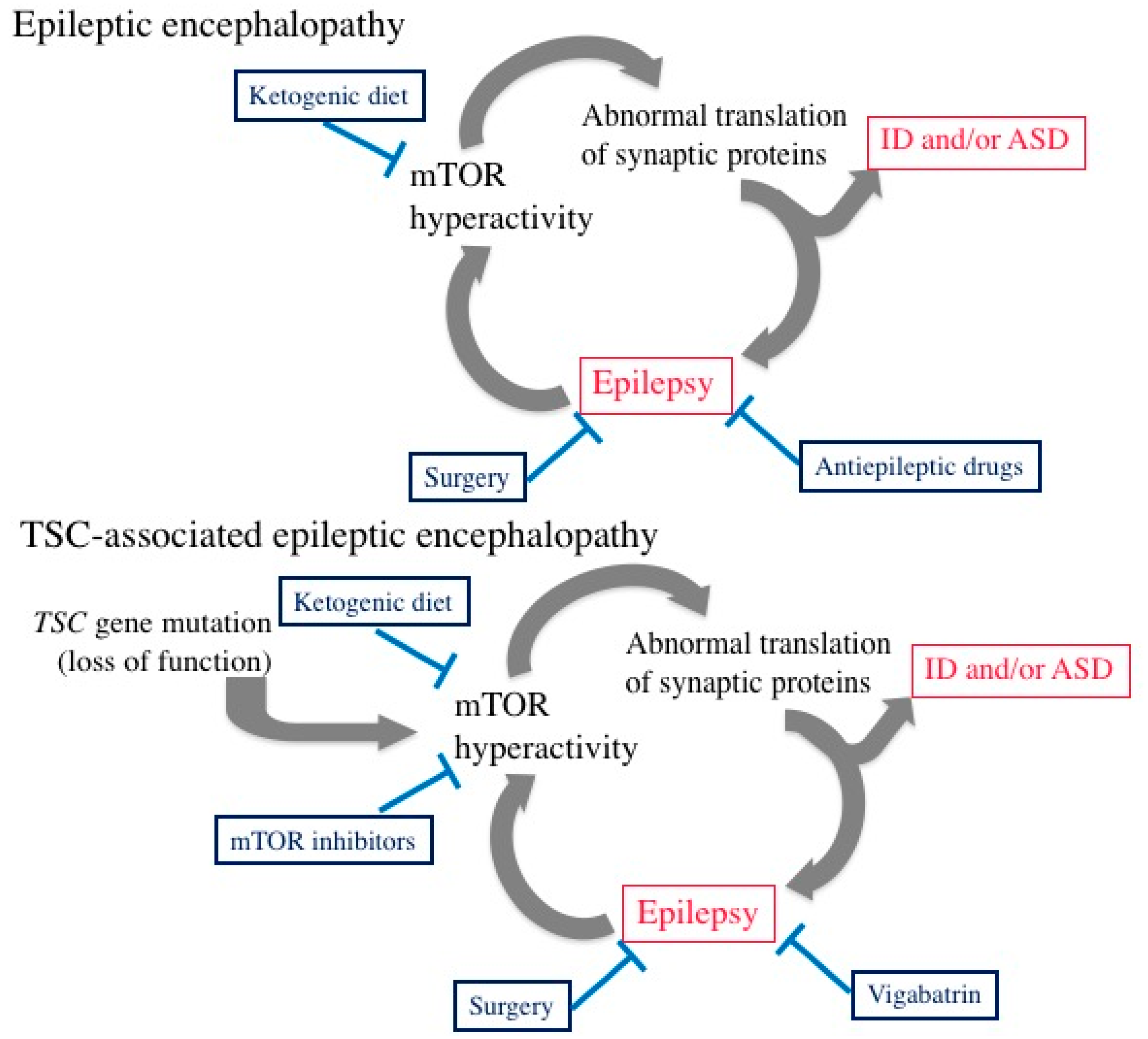
5. Epileptic Encephalopathy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| AKT | Protein kinase B |
| AML | Angiomyolipoma |
| ASD | Autism spectrum disorder |
| 4EBP | Eukaryotic translation initiation factor-4E-binding proteins |
| eIF4E | Eukaryotic translation initiation factor-4E |
| ERK | Extracellular signal-related kinase |
| GABA | Gamma aminobutyric acid |
| ID | Intellectual disability |
| IGF | Insulin-like growth factor |
| IQ | Intelligence quotient |
| LAM | Lymphangioleiomyomatosis |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MEK | Mitogen-activated protein kinase kinase |
| MMPH | Multifocal micronodular pneumocyte hyperplasia |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| PARS | Pervasive Developmental Disorders Autism Society Japan Rating Scale |
| PI3K | Phosphatidylinositol 3-kinase |
| Rheb | Ras homolog enriched in brain |
| S6 | Ribosomal protein S6 |
| SEGA | Subependymal giant cell astrocytoma |
| S6K | Ribosomal protein S6 kinase |
| TAND | Tuberous sclerosis complex-associated neuropsychiatric disorders |
| TSC | Tuberous sclerosis complex |
| ULK1 | Unk-5l-like kinase 1 |
References
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef]
- Wienecke, R.; König, A.; DeClue, J.E. Identification of tuberin, the tuberous sclerosis-2 product. Tuberin possesses specific Rap1GAP activity. J. Biol. Chem. 1995, 270, 16409–16414. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.H.; Shoarinejad, F.; Jin, F.; Golemis, E.A.; Yeung, R.S. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J. Biol. Chem. 1997, 272, 6097–6100. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, G.; Li, S.; Brown, S.J.; Braverman, R.; Vass, W.C.; Cheadle, J.P.; Halley, D.J.; Sampson, J.R.; Wienecke, R.; DeClue, J.E. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000, 19, 6306–6316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [Green Version]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donzé, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Di Nardo, A.; Wertz, M.H.; Kwiatkowski, E.; Tsai, P.T.; Leech, J.D.; Greene-Colozzi, E.; Goto, J.; Dilsiz, P.; Talos, D.M.; Clish, C.B.; et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 2014, 23, 3865–3874. [Google Scholar] [CrossRef] [Green Version]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Fabrizi, C.; Frati, A.; Fornai, F. mTOR-related cell-clearing systems in epileptic seizures, an update. Int. J. Mol. Sci. 2020, 21, 1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LiCausi, F.; Hartman, N.W. Role of mTOR complexes in neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [Green Version]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef]
- Crino, P.B. The mTOR signaling cascade: Paving new roads to cure neurological disease. Nat. Rev. 2016, 12, 379–392. [Google Scholar]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993, 75, 1305–1315. [Google Scholar] [CrossRef]
- Green, A.J.; Smith, M.; Yates, J.R. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat. Genet. 1994, 6, 193–196. [Google Scholar] [CrossRef]
- Henske, E.P.; Scheithauer, B.W.; Short, M.P.; Wollmann, R.; Nahmias, J.; Hornigold, N.; Slegtenhorst, M.; Welsh, C.T.; Kwiatkowski, D.J. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet. 1996, 59, 400–406. [Google Scholar] [PubMed]
- Crino, P.B.; Aronica, E.; Baltuch, G.; Nathanson, K.L. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010, 74, 1716–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Frost, M.; Belousova, E.; Sauter, M.; Nonomura, N.; Brakemeier, S.; de Vries, P.J.; et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 817–824. [Google Scholar] [CrossRef]
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N.; et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- Northrup, H.; Krueger, D.A. International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.R. Definition and criteria for diagnosis. In Tuberous Sclerosis Complex, 3rd ed.; Gomez, M.R., Sampson, J.R., Whittemore, V.H., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 10–23. [Google Scholar]
- Gomez, M.R. Natural history of cerebral tuberous sclerosis. In Tuberous Sclerosis Complex, 3rd ed.; Gomez, M.R., Sampson, J.R., Whittemore, V.H., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 29–46. [Google Scholar]
- Wataya-Kaneda, M.; Tanaka, M.; Hamasaki, T.; Katayama, I. Trends in the prevalence of tuberous sclerosis complex manifestations: An epidemiological study of 166 Japanese patients. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef] [Green Version]
- Niida, Y.; Wakisaka, A.; Tsuji, T.; Yamada, H.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J. Hum. Genet. 2013, 58, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Kato, M.; Yamanouchi, H.; Ikeda, K.; Takashima, S. Tuberin immunohistochemistry in brain, kidneys and heart with or without tuberous sclerosis. Acta Neuropathol. 1997, 94, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Ikeda, K.; Takashima, S. Simultaneous loss of hamartin and tuberin from the cerebrum, kidney and heart with tuberous sclerosis. Acta Neuropathol. 2000, 99, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Hengstschläger, M.; Rodman, D.M.; Miloloza, A.; Hengstschläger-Ottnad, E.; Rosner, M.; Kubista, M. Tuberous sclerosis gene products in proliferation control. Mutat. Res. 2001, 488, 233–239. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Yamanouchi, H.; Becker, L.E.; Itoh, M.; Takashima, S. Doublecortin immunoreactivity in giant cells of tuberous sclerosis and focal cortical dysplasia. Acta Neuropathol. 2002, 104, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, M.; Kobayashi, T.; Okura, H.; Igarashi, T.; Mizuguchi, M.; Hino, O. TSC1 controls distribution of actin fibers through its effect on function of Rho family of small GTPases and regulates cell migration and polarity. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Jiang, X.; Kenerson, H.; Aicher, L.; Miyaoka, R.; Eary, J.; Bissler, J.; Yeung, R.S. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am. J. Pathol. 2008, 172, 1748–1756. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Kaelin, W.G., Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell 2004, 6, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Niida, Y.; Stemmer-Rachamimov, A.O.; Logrip, M.; Tapon, D.; Perez, R.; Kwiatkowski, D.J.; Sims, K.; MacCollin, M.; Louis, D.N.; Ramesh, V. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet. 2001, 69, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Wolf, H.K.; Normann, S.; Green, A.J.; von Bakel, I.; Blümcke, I.; Pietsch, T.; Wiestler, O.D.; von Deimling, A. Tuberous sclerosis-like lesions in epileptogenic human neocortex lack allelic loss at the TSC1 and TSC2 regions. Acta Neuropathol. 1997, 93, 93–96. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Mori, M.; Nozaki, Y.; Momoi, M.Y.; Itoh, M.; Takashima, S.; Hino, O. Absence of allelic loss in cytomegalic neurons of cortical tuber in the Eker rat model of tuberous sclerosis. Acta Neuropathol. 2004, 107, 47–52. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Takashima, S. Neuropathology of tuberous sclerosis. Brain Dev. 2001, 23, 508–515. [Google Scholar] [CrossRef]
- Aw, F.; Goyer, I.; Raboisson, M.J.; Boutin, C.; Major, P.; Dahdah, N. Accelerated cardiac rhabdomyoma regression with everolimus in infants with tuberous sclerosis complex. Pediatr. Cardiol. 2017, 38, 394–400. [Google Scholar] [CrossRef]
- Wataya-Kaneda, M.; Ohno, Y.; Fujita, Y.; Yokozeki, H.; Niizeki, H.; Ogai, M.; Fukai, K.; Nagai, H.; Yoshida, Y.; Hamada, I.; et al. Sirolimus gel treatment vs. placebo for facial angiofibromas in patients with tuberous sclerosis complex: A randomized clinical trial. JAMA Dermatol. 2018, 154, 781–788. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joinson, C.; O’Callaghan, F.J.; Osborne, J.; Martyn, C.; Harris, T.; Bolton, P.F. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol. Med. 2003, 33, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chu-Shore, C.J.; Major, P.; Camposano, S.; Muzykewicz, D.; Thiele, E.A. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010, 51. [Google Scholar] [CrossRef] [Green Version]
- van Eeghen, A.M.; Pulsifer, M.B.; Merker, V.L.; Neumeyer, A.M.; van Eeghen, E.E.; Thibert, R.L.; Cole, A.J.; Leigh, F.A.; Plotkin, S.R.; Thiele, E.A. Understanding relationships between autism, intelligence, and epilepsy: A cross-disorder approach. Dev. Med. Child Neurol. 2013, 55. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C.; Thomas, H.V.; Murphy, K.C.; Sampson, J.R. Genotype and psychological phenotype in tuberous sclerosis. J. Med. Genet. 2004, 41, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, P.J.; Hunt, A.; Bolton, P.F. The psychopathologies of children and adolescents with tuberous sclerosis complex (TSC): A postal survey of UK families. Eur. Child Adolesc. Psychiatry 2007, 16, 16–24. [Google Scholar] [CrossRef]
- de Vries, P.J.; Whittemore, V.H.; Leclezio, L.; Byars, A.W.; Dunn, D.; Ess, K.C.; Hook, D.; King, B.H.; Sahin, M.; Jansen, A. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr. Neurol. 2015, 52, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, M. Abnormal giant cells in the cerebral lesions of tuberous sclerosis complex. Cong. Anom. 2007, 47, 2–8. [Google Scholar] [CrossRef]
- Scheithauer, B.W.; Reagan, T.J. Neuropathology. In Tuberous Sclerosis Complex, 3rd ed.; Gomez, M.R., Sampson, J.R., Whittemore, V.H., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 29–46. [Google Scholar]
- McMahon, J.; Huang, X.; Yang, J.; Komatsu, M.; Yue, Z.; Qian, J.; Zhu, X.; Huang, Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 2012, 32, 15704–15714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasin, S.A.; Ali, A.M.; Tata, M.; Picker, S.R.; Anderson, G.W.; Latimer-Bowman, E.; Nicholson, S.L.; Harkness, W.; Cross, J.H.; Paine, S.M.; et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: Evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. 2013, 126, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Ess, K.C.; Uhlmann, E.J.; Jansen, L.A.; Li, W.; Crino, P.B.; Mennerick, S.; Yamada, K.A.; Gutmann, D.H. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann. Neurol. 2003, 54, 251–256. [Google Scholar] [CrossRef]
- White, R.; Hua, Y.; Scheithauer, B.; Lynch, D.R.; Henske, E.P.; Crino, P.B. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann. Neurol. 2001, 49, 67–78. [Google Scholar] [CrossRef]
- Talos, D.M.; Sun, H.; Kosaras, B.; Joseph, A.; Folkerth, R.D.; Poduri, A.; Madsen, J.R.; Black, P.M.; Jensen, F.E. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 2012, 71, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Valencia, I.; Legido, A.; Yelin, K.; Khurana, D.; Kothare, S.V.; Katsetos, C.D. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: Aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J. Child Neurol. 2006, 21, 1058–1063. [Google Scholar] [CrossRef]
- Wang, Y.; Greenwood, J.S.; Calcagnotto, M.E.; Kirsch, H.E.; Marmaro, N.M.; Baraban, S.C. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann. Neurol. 2007, 61, 139–152. [Google Scholar] [CrossRef]
- Zhao, J.-P.; Yoshii, A. Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol. Brain 2019, 12, 6. [Google Scholar]
- Lippa, C.F.; Pearson, D.; Smith, T.W. Cortical tubers demonstrate reduced immunoreactivity for synapsin I. Acta Neuropathol. 1993, 85, 449–451. [Google Scholar] [CrossRef]
- Sugiura, H.; Yasuda, S.; Katsurabayashi, S.; Kawano, H.; Endo, K.; Takasaki, K.; Iwasaki, K.; Ichikawa, M.; Kobayashi, T.; Hino, O.; et al. Rheb activation disrupts spine synapse formation through accumulation of syntenin in tuberous sclerosis complex. Nat. Commun. 2015, 6, 6842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J. Neurosci. 2011, 31, 8862–8869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Napolioni, V.; Moavero, R.; Curatolo, P. Recent advances in neurobiology of Tuberous Sclerosis Complex. Brain Dev. 2009, 31, 104–113. [Google Scholar] [CrossRef]
- Lewis, W.W.; Sahin, M.; Scherrer, B.; Peters, J.M.; Suarez, R.O.; Vogel-Farley, V.K.; Jeste, S.S.; Gregas, M.C.; Prabhu, S.P.; Nelson, C.A., 3rd; et al. Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb. Cortex 2013, 23, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Tillema, J.-M.; Leach, J.L.; Krueger, D.A.; Franz, D.N. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 2012, 78, 526–531. [Google Scholar] [CrossRef]
- Hino, O.; Klein-Szanto, A.J.; Freed, J.J.; Testa, J.R.; Brown, D.Q.; Vilensky, M.; Yeung, R.S.; Tartof, K.D.; Knudson, A.G. Spontaneous and radiation-induced renal tumors in the Eker rat model of dominantly inherited cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Everitt, J.I.; Goldsworthy, T.L.; Wolf, D.C.; Walker, C.L. Hereditary renal cell carcinoma in the Eker rat: A rodent familial cancer syndrome. J. Urol. 1992, 148, 1932–1936. [Google Scholar] [CrossRef]
- Kobayashi, T.; Hirayama, Y.; Kobayashi, E.; Kubo, Y.; Hino, O. A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat. Genet. 1995, 9, 70–74. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Takashima, S.; Yamanouchi, H.; Nakazato, Y.; Mitani, H.; Hino, O. Novel cerebral lesions in the Eker rat model of tuberous sclerosis: Cortical tuber and anaplastic gangliogioma. J. Neuropathol. Exp. Neurol. 2000, 59, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, H.J.; Patel, L.S.; Robbins, C.A.; Emmi, A.; Yeung, R.S.; Schwartzkroin, P.A. Morphology of cerebral lesions in the Eker rat model of tuberous sclerosis. Acta Neuropathol. 2004, 108, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.J.; Dinday, M.T.; Barbaro, N.M.; Baraban, S.C. Abnormal cortical cells and astrocytomas in the Eker rat model of tuberous sclerosis complex. Epilepsia 2004, 45, 1525–1530. [Google Scholar] [CrossRef]
- Schneider, M.; DeVries, P.J.; Schönig, K.; Rößner, V.; Waltereit, R. mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and developmental status epilepticus. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, D.M.; Su, T.; Lopez, J.C.; Bordey, A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Investig. 2011, 121, 1596–1607. [Google Scholar] [CrossRef]
- Gataullina, S.; Lemaire, E.; Wendling, F.; Kaminska, A.; Watrin, F.; Riquet, A.; Ville, D.; Moutard, M.L.; de Saint Martin, A.; Napuri, S.; et al. Epilepsy in young Tsc1(+/−) mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex. Epilepsia 2016, 57, 648–659. [Google Scholar] [CrossRef] [Green Version]
- Goorden, S.M.; van Woerden, G.M.; van der Weerd, L.; Cheadle, J.P.; Elgersma, Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann. Neurol. 2007, 62, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Atsuta, M.; Inaba, H.; Endo, K.; Egawa, S. Effect of everolimus treatment for renal angiomyolipoma associated with tuberous sclerosis complex: An evaluation based on tumor density. Int. J. Clin. Oncol. 2018, 23, 547–552. [Google Scholar] [CrossRef]
- Malissen, N.; Vergely, L.; Simon, M.; Roubertie, A.; Malinge, M.C.; Bessis, D. Long-term treatment of cutaneous manifestations of tuberous sclerosis complex with topical 1% sirolimus cream: A prospective study of 25 patients. J. Am. Acad. Dermatol. 2017, 77, 464–472. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Ikeda, H.; Kagitani-Shimono, K.; Yoshinaga, H.; Suzuki, Y.; Aoki, M.; Endo, M.; Yonemura, M.; Kubota, M. Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3substudy in Japan. Brain Dev. 2019, 41, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, I.E.; Liao, J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy”. Eur. J. Paediatr. Neurol. 2020, 24, 11–14. [Google Scholar] [CrossRef]
- Talos, D.M.; Sun, H.; Zhou, X.; Fitzgerald, E.C.; Jackson, M.C.; Klein, P.M.; Lan, V.J.; Joseph, A.; Jensen, F.E. The interaction between early life epilepsy and autistic-like behavioral consequences: A role for the mammalian target of rapamycin (mTOR) pathway. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Vigevano, F.; Arzimanoglou, A.; Plouin, P.; Specchio, N. Therapeutic approach to epileptic encephalopathies. Epilepsia 2013, 54 (Suppl. S8), 45–50. [Google Scholar] [CrossRef]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [Green Version]
- Wong, M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia 2010, 51, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samueli, S.; Dressler, A.; Gröppel, G.; Scholl, T.; Feucht, M. Everolimus in infants with tuberous sclerosis complex-related West syndrome: First results from a single-center prospective observational study. Epilepsia 2018, 59, e142–e146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffari, A.; Brösse, I.; Wiemer-Kruel, A.; Wilken, B.; Kreuzaler, P.; Hahn, A.; Bernhard, M.K.; van Tilburg, C.M.; Hoffmann, G.F.; Gorenflo, M.; et al. Safety and efficacy of mTOR inhibitor treatment in patients with tuberous sclerosis complex under 2 years of age-a multicenter retrospective study. Orphanet J. Rare Dis. 2019, 14, 96. [Google Scholar] [CrossRef]
- Jóźwiak, S.; Kotulska, K.; Domańska-Pakieła, D.; Lojszczyk, B.; Syczewska, M.; Chmielewski, D.; Dunin-Wąsowicz, D.; Kmieć, T.; Szymkiewicz-Dangel, J.; Kornacka, M.; et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2011, 15, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Jozwiak, S.; Słowinska, M.; Borkowska, J.; Sadowski, K.; Łojszczyk, B.; Domańska-Pakieła, D.; Chmielewski, D.; Kaczorowska-Frontczak, M.; Głowacka, J.; Sijko, K.; et al. Preventive antiepileptic treatment in tuberous sclerosis complex: Long-term, prospective trial. Pediatr. Neurol. 2019, 101, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Karenfort, M.; Kruse, B.; Freitag, H.; Pannek, H.; Tuxhorn, I. Epilepsy surgery outcome in children with focal epilepsy due to tuberous sclerosis complex. Neuropediatrics 2002, 33, 255–261. [Google Scholar] [CrossRef]
- Fohlen, M.; Taussig, D.; Ferrand-Sorbets, S.; Chipaux, M.; Dorison, N.; Delalande, O.; Dorfmüller, G. Refractory epilepsy in preschool children with tuberous sclerosis complex: Early surgical treatment and outcome. Seizure 2018, 60, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Grayson, L.E.; Peters, J.M.; McPherson, T.; Krueger, D.A.; Sahin, M.; Wu, J.Y.; Northrup, H.A.; Porter, B.; Cutter, G.R.; O’Kelley, S.E.; et al. Pilot study of neurodevelopmental impact of early epilepsy surgery in tuberous sclerosis complex. Pediatr. Neurol. 2020, 109, 39–46. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Hamartias | Hamartomas |
|---|---|---|
| Skin | Hypomelanotic macules Confetti skin lesions | Facial angiofibromas Fibrous cephalic plaque Ungual fibromas Shagreen patch |
| Brain | Cortical tuber | SEGA Subependymal nodules |
| Eye | Retinal achromic patch | Retinal hamartoma |
| Mouth | Gingival fibromas | Dental enamel pits |
| Lung | Pulmonary LAM MMPH | |
| Heart | Cardiac rhabdomyoma | |
| Arteries | Wall defects/Aneurysm | Renal AML |
| Kidney | Renal cysts | |
| Bone | Bone cysts |
| Lesion/Symptom | Pathology | Age of Occurrence or Worsening | Department in Charge | Note |
|---|---|---|---|---|
| Cardiac rhabdomyoma | Tumor | Fetal–neonatal period | Pediatric cardiology | Spontaneous regression in infancy |
| Cortical tuber | Dysplasia | Fetal–neonatal period/Infancy | Pediatric neurology | Focus of epileptic seizures |
| Hypopigmented macule | Dysplasia | Fetal–neonatal period/Infancy | Dermatology | |
| Epilepsy | Brain dysfunction | Infancy/Childhood | Pediatric neurology | Intractable in many cases |
| ID/ASD | Brain dysfunction | Infancy/Childhood | Pediatric neurology/Psychiatry | |
| Retinal hamartoma | Tumor | Infancy/Childhood | Ophthalmology | |
| Renal cyst | Dysplasia | Infancy/Childhood | Pediatrics | |
| SEGA | Tumor | Childhood/Adolescence | Neurosurgery | Hydrocephalus, potentially fatal |
| Facial angiofibroma | Tumor | Childhood/Adolescence | Dermatology | |
| TAND | Brain dysfunction | Childhood/Adolescence | Pediatrics/Psychiatry | |
| Renal AML | Tumor | Childhood/Adolescence/Adulthood | Pediatrics/Urology | Hemorrhage, potentially fatal |
| Pulmonary LAM | Tumor | Adolescence/Adulthood | Pulmonology | Predominantly affecting women, potentially fatal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizuguchi, M.; Ohsawa, M.; Kashii, H.; Sato, A. Brain Symptoms of Tuberous Sclerosis Complex: Pathogenesis and Treatment. Int. J. Mol. Sci. 2021, 22, 6677. https://doi.org/10.3390/ijms22136677
Mizuguchi M, Ohsawa M, Kashii H, Sato A. Brain Symptoms of Tuberous Sclerosis Complex: Pathogenesis and Treatment. International Journal of Molecular Sciences. 2021; 22(13):6677. https://doi.org/10.3390/ijms22136677
Chicago/Turabian StyleMizuguchi, Masashi, Maki Ohsawa, Hirofumi Kashii, and Atsushi Sato. 2021. "Brain Symptoms of Tuberous Sclerosis Complex: Pathogenesis and Treatment" International Journal of Molecular Sciences 22, no. 13: 6677. https://doi.org/10.3390/ijms22136677
APA StyleMizuguchi, M., Ohsawa, M., Kashii, H., & Sato, A. (2021). Brain Symptoms of Tuberous Sclerosis Complex: Pathogenesis and Treatment. International Journal of Molecular Sciences, 22(13), 6677. https://doi.org/10.3390/ijms22136677