
Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
2.2. Detection and Validation of Hepatitis B Virus (HBV) Integration Breakpoints
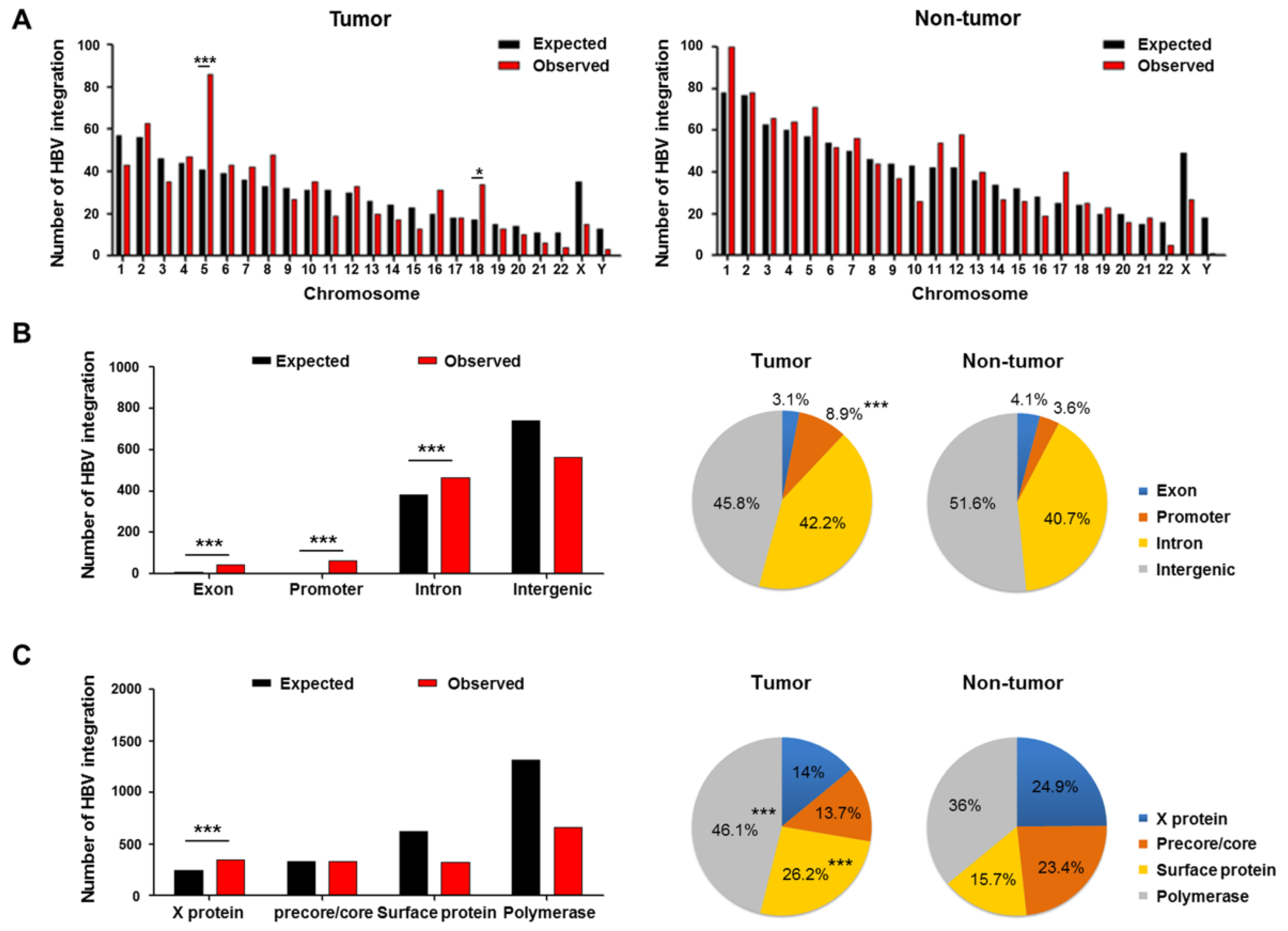
2.3. Genomic Locations of HBV Integration Breakpoints
2.4. Patterns of HBV Integration Breakpoints in Tumors and Non-Tumors
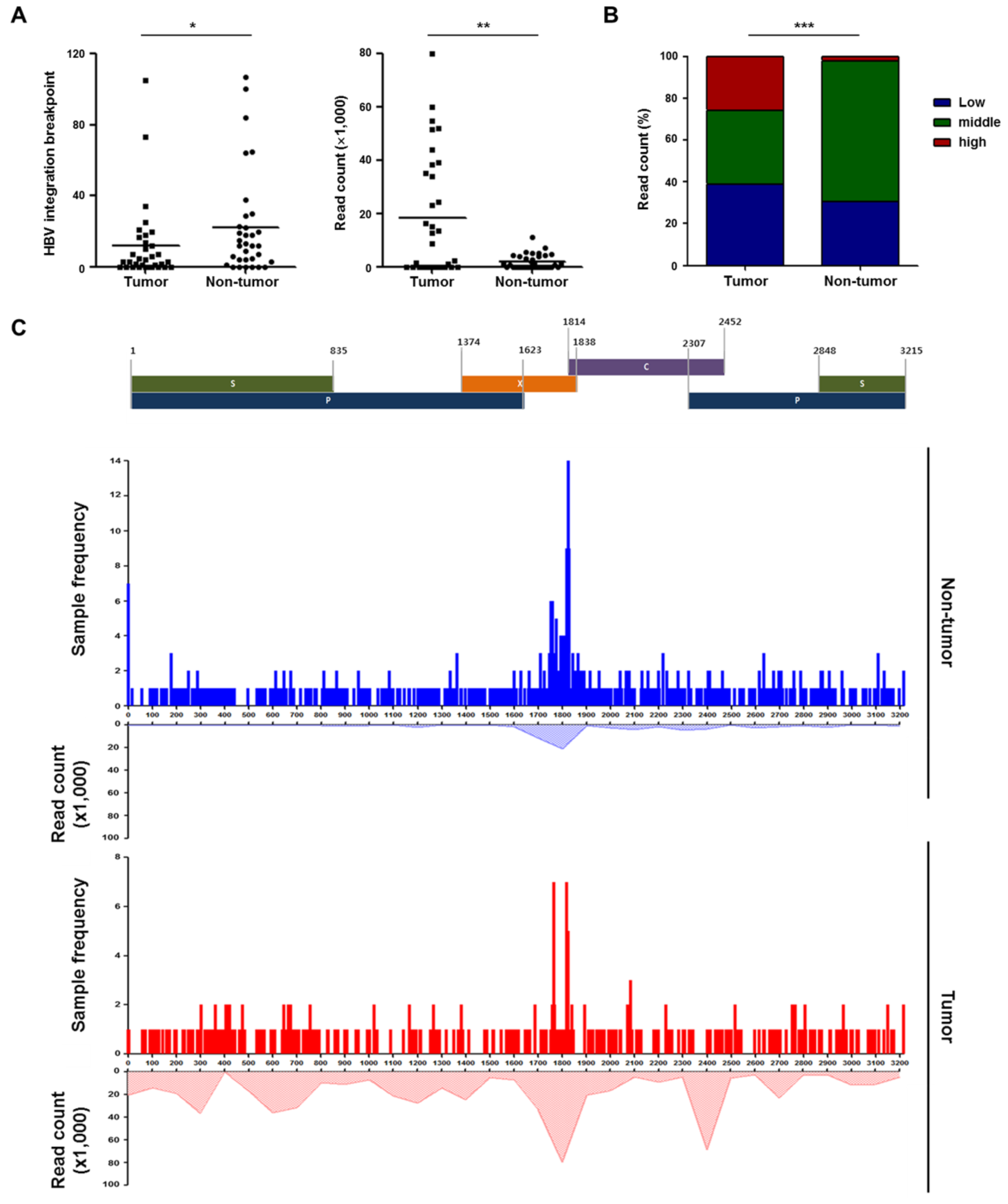
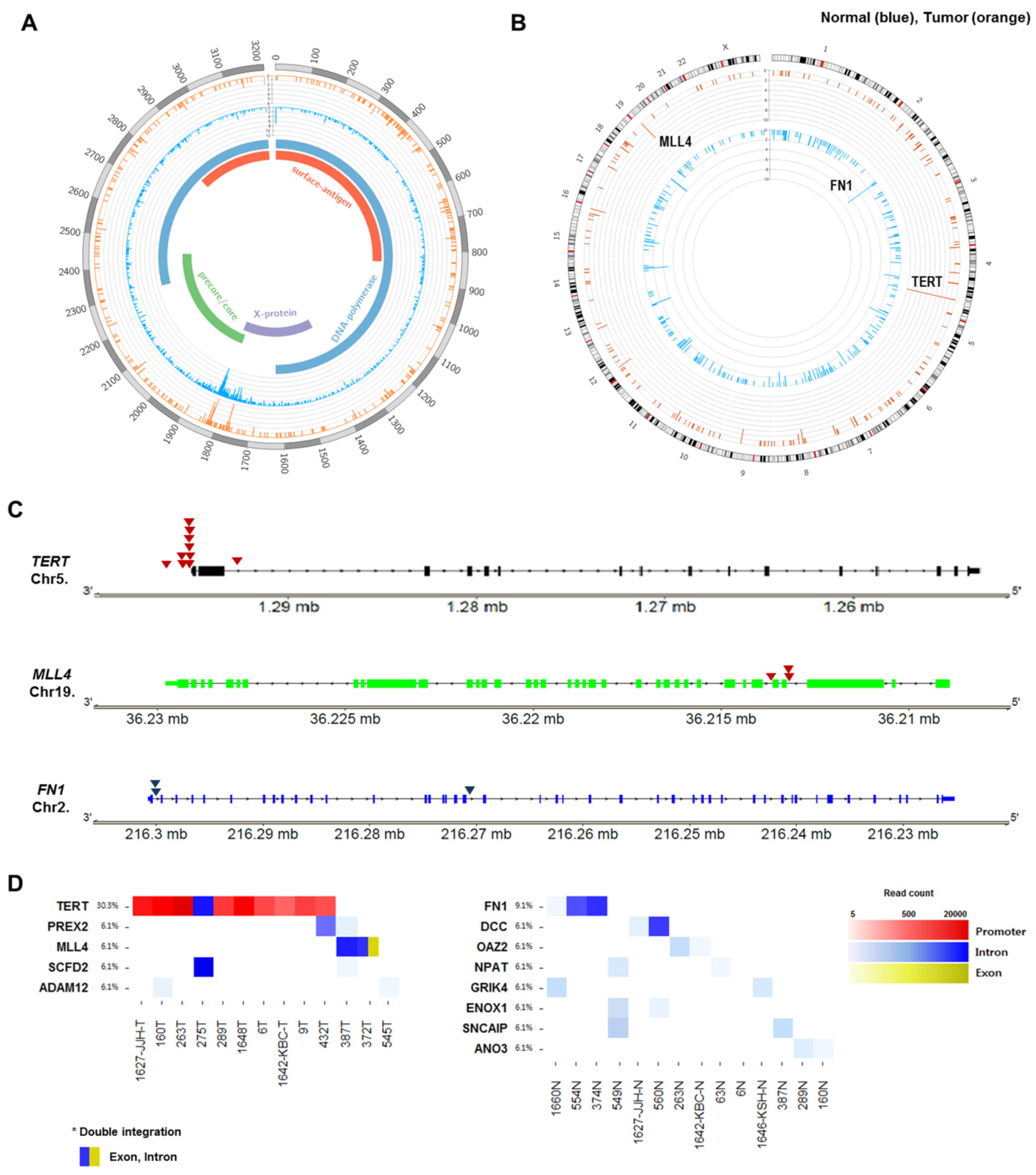
2.5. Recurrent HBV Integration Sites
2.6. Telomerase Reverse Transcriptase (TERT) Promoter Hot Spot Mutation
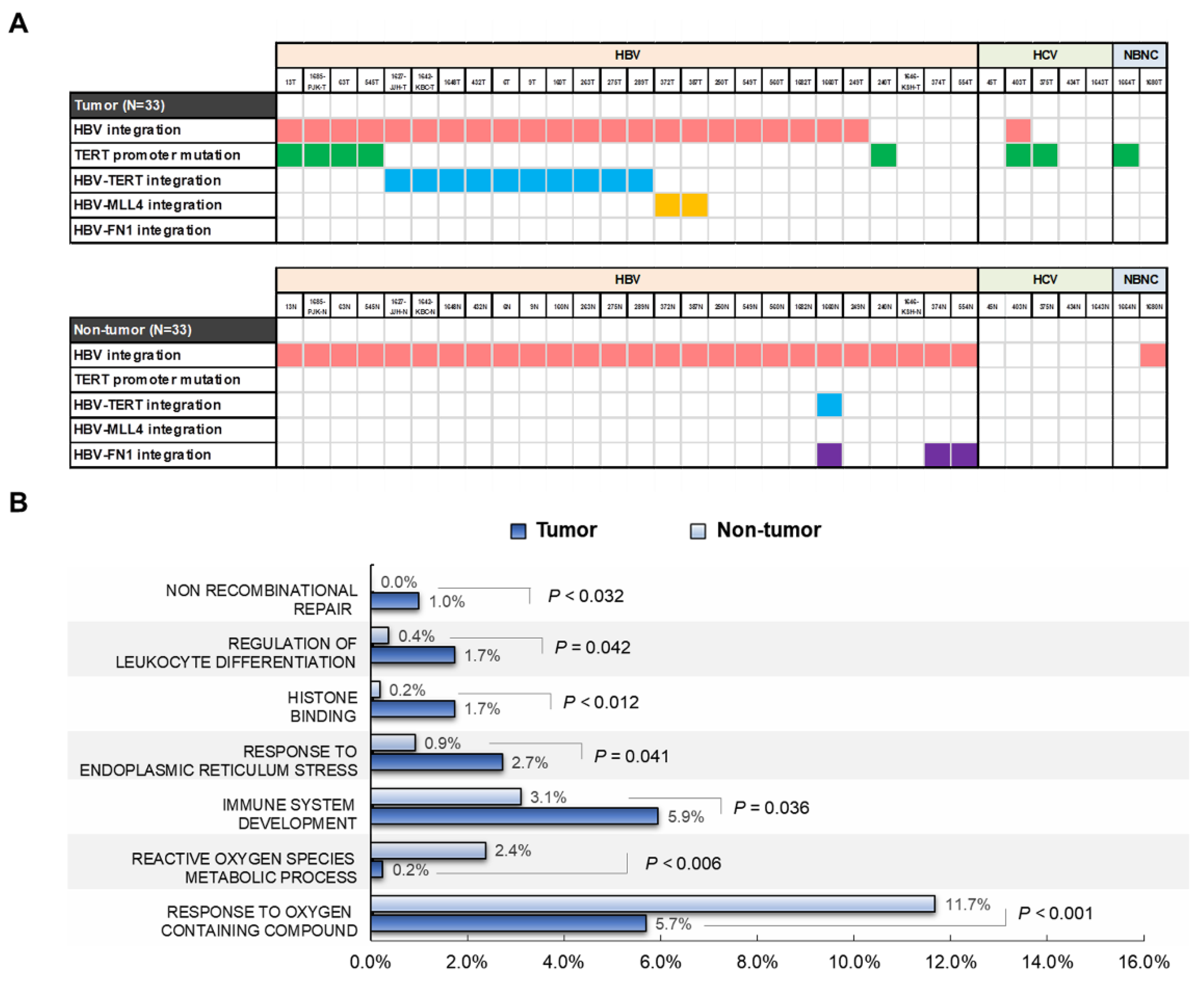
2.7. Functional Enrichment Analysis of HBV-Integrated Genes
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Preparation of HBV Probes
4.3. Enrichment of HBV-Integrated Fragments and Capture Sequencing
4.4. Detection of the HBV-Human Chimeric Reads
4.5. PCR-Based Sanger Sequencing for Validation
4.6. Sequencing of the TERT Promoter Region
4.7. Functional Enrichment Analysis of HBV-Integrated Genes
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM12 | ADAM metallopeptidase domain 12 |
| AFP | alpha-fetoprotein |
| ALT | alanine aminotransferase |
| ANO3 | anoctamin 3 |
| BCLC | Barcelona Clinic Liver Cancer |
| BWA-MEM | Burrows-Wheeler Aligner |
| CH | chronic hepatitis |
| CHB | chronic hepatitis B |
| Chr | chromosome |
| DCC | DCC netrin 1 receptor |
| ENOX1 | Ecto-NOX disulfide-thiol exchanger 1 |
| GRIK4 | glutamate ionotropic receptor kainate type subunit 4 |
| HBsAg | hepatitis B surface antigen |
| HBV | hepatitis B virus |
| HCC | hepatocellular carcinoma |
| HCV | hepatitis C virus |
| INR | international normalized ratio |
| LC | liver cirrhosis |
| MLL4 | mixed lineage leukemia 4 |
| NBNC | non-HBV non-HCV |
| NCBI | National Center for Biotechnology Information |
| NPAT | nuclear protein, coactivator of histone transcription |
| OAZ2 | ornithine decarboxylase antizyme 2 |
| PREX2 | phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor 2 |
| PT | prothrombin time |
| SCFD2 | sec1 family domain containing 2 |
| SNCAIP | synuclein alpha interacting protein |
| TERT | telomerase reverse transcriptase) |
References
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Edman, J.C.; Gray, P.; Valenzuela, P.; Rall, L.B.; Rutter, W.J. Integration of hepatitis B virus sequences and their expression in a human hepatoma cell. Nature 1980, 286, 535–538. [Google Scholar] [CrossRef]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Ningarhari, M.; Rebouissou, S.; Zucman-Rossi, J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 544–558. [Google Scholar] [CrossRef]
- Okamoto, K.; Seimiya, H. Revisiting Telomere Shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- In der Stroth, L.; Tharehalli, U.; Gunes, C.; Lechel, A. Telomeres and Telomerase in the Development of Liver Cancer. Cancers 2020, 12, 2048. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Zucman-Rossi, J. Genetics of hepatocellular carcinoma: The next generation. J. Hepatol. 2014, 60, 224–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinyol, R.; Tovar, V.; Llovet, J.M. TERT promoter mutations: Gatekeeper and driver of hepatocellular carcinoma. J. Hepatol. 2014, 61, 685–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brechot, C.; Gozuacik, D.; Murakami, Y.; Paterlini-Brechot, P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2000, 10, 211–231. [Google Scholar] [CrossRef]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Cellular Genomic Sites of Hepatitis B Virus DNA Integration. Genes 2018, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.W.; Kim, J.S.; Kim, H.S.; Tak, K.Y.; Lee, S.K.; Nam, H.C.; Sung, P.S.; Kim, C.M.; Park, J.Y.; Bae, S.H.; et al. Significance of TERT Genetic Alterations and Telomere Length in Hepatocellular Carcinoma. Cancers 2021, 13, 2160. [Google Scholar] [CrossRef] [PubMed]
- Paterlini-Brechot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Brechot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Montoya, D.P.; Yu, C.; Aderca, I.; McGee, A.; Thorland, E.C.; Nagorney, D.M.; Gostout, B.S.; Burgart, L.J.; Boix, L.; et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003, 22, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Sze, K.M.; Ho, D.W.; Chiu, Y.T.; Tsui, Y.M.; Chan, L.K.; Lee, J.M.; Chok, K.S.; Chan, A.C.; Tang, C.N.; Tang, V.W.; et al. Hepatitis B Virus-Telomerase Reverse Transcriptase Promoter Integration Harnesses Host ELF4, Resulting in Telomerase Reverse Transcriptase Gene Transcription in Hepatocellular Carcinoma. Hepatology 2021, 73, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.; Chung, A.Y.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Wong, N. Frequent Integration of HBV DNA in Genomes of Hepatocellular Carcinoma Cells From Patients With Occult Infections. Clin. Gastroenterol. Hepatol. 2020, 18, 302–303. [Google Scholar] [CrossRef] [Green Version]
- Doolittle-Hall, J.M.; Cunningham Glasspoole, D.L.; Seaman, W.T.; Webster-Cyriaque, J. Meta-Analysis of DNA Tumor-Viral Integration Site Selection Indicates a Role for Repeats, Gene Expression and Epigenetics. Cancers 2015, 7, 2217–2235. [Google Scholar] [CrossRef] [PubMed]
- Fourel, G.; Couturier, J.; Wei, Y.; Apiou, F.; Tiollais, P.; Buendia, M.A. Evidence for long-range oncogene activation by hepadnavirus insertion. EMBO J. 1994, 13, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Kim, J.S.; Kim, H.S.; Tak, K.Y.; Nam, H.; Sung, P.S.; Bae, S.H.; Choi, J.Y.; Yoon, S.K.; Roberts, L.R. Persistence of intrahepatic hepatitis B virus DNA integration in patients developing hepatocellular carcinoma after hepatitis B surface antigen seroclearance. Clin. Mol. Hepatol. 2021, 27, 207–218. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | HCC (n = 33) |
|---|---|
| Sex (male/female, %) | 29 (87.9)/4 (12.1) |
| Age (years) | 52.6 ± 10.2 (29–72) |
| Etiology (HBV/HCV/NBNC, %) | 26 (78.8)/5 (15.2)/2 (6.1) |
| AST (U/L) | 54 (16−97) |
| ALT (U/L) | 36 (14−73) |
| Total bilirubin (mg/dL) | 0.8 (0.2–29.9) |
| Albumin (g/dL) | 3.8 ± 0.5 |
| PT (INR) | 1.2 ± 0.2 |
| Child-Pugh class (A/B/C, %) | 29 (87.9)/3 (9.1)/1 (3.0) |
| Tumor size (cm) | 4.1 ± 2.0 |
| Tumor number (single/multiple, %) | 27 (81.8)/6 (28.2) |
| α-fetoprotein (ng/mL) | 71.3 (1.7–59403) |
| BCLC stage (A/B/C, %) | 20 (60.6)/10 (30.3)/3 (9.1) |
| Treatment (resection/LT) | 25 (75.8)/8 (24.2) |
| Gene Name | Chr | Location | HBV Proteins | Samples |
|---|---|---|---|---|
| TERT | 5 | Promoter | X protein | 9T, 1648T |
| 5 | Promoter | X protein, Precore/core protein | 1627-JJH-T, 263T | |
| 5 | Promoter | Precore/core protein | 289T, 432T, 1648T | |
| 5 | Promoter | Polymerase, Surface antigen | 1642-KBC-T, 6T, 160T | |
| 5 | Promoter | Polymerase | 6T, 9T, 289T, 432T | |
| 5 | Intron | Polymerase, Surface antigen | 275T | |
| MLL4 | 19 | Intron | Polymerase, X protein | 372T, 387T |
| 19 | Exon | Polymerase, surface antigen | 372T | |
| 19 | Intron | Polymerase | 387T | |
| PREX2 | 8 | Intron | Precore/core protein | 387T |
| 8 | Intron | Polymerase, X protein | 432T | |
| SCFD2 | 4 | Intron | Polymerase | 275T |
| 4 | Intron | X protein | 387T | |
| ADAM12 | 10 | Intron | Polymerase, Surface antigen | 160T |
| 10 | Intron | Polymerase | 545T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, J.-W.; Kim, H.-S.; Kim, J.-S.; Lee, S.-K.; Han, J.-W.; Sung, P.-S.; Bae, S.-H.; Choi, J.-Y.; Yoon, S.-K.; Han, D.-J.; et al. Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 7056. https://doi.org/10.3390/ijms22137056
Jang J-W, Kim H-S, Kim J-S, Lee S-K, Han J-W, Sung P-S, Bae S-H, Choi J-Y, Yoon S-K, Han D-J, et al. Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(13):7056. https://doi.org/10.3390/ijms22137056
Chicago/Turabian StyleJang, Jeong-Won, Hye-Seon Kim, Jin-Seoub Kim, Soon-Kyu Lee, Ji-Won Han, Pil-Soo Sung, Si-Hyun Bae, Jong-Young Choi, Seung-Kew Yoon, Dong-Jin Han, and et al. 2021. "Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 13: 7056. https://doi.org/10.3390/ijms22137056
APA StyleJang, J.-W., Kim, H.-S., Kim, J.-S., Lee, S.-K., Han, J.-W., Sung, P.-S., Bae, S.-H., Choi, J.-Y., Yoon, S.-K., Han, D.-J., Kim, T.-M., & Roberts, L. R. (2021). Distinct Patterns of HBV Integration and TERT Alterations between in Tumor and Non-Tumor Tissue in Patients with Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(13), 7056. https://doi.org/10.3390/ijms22137056