1. Introduction
Adequate transport of solutes and water across epithelial barriers is indispensable for maintaining normal physiological homeostasis in all animals [
1,
2]. Fluid is moved either across the plasma membranes of the cells that comprise the epithelial layer (transcellular transport) or between these cells (paracellular transport). The discovery of aquaporin water channels provided a first molecular basis for transcellular water movement [
3].
Paracellular transport involves specialized structures called tight junctions (TJ) that regulate the flow of solutes through paracellular pathways and maintain cell polarity, thereby functioning as a barrier of epithelial and endothelial cellular sheets [
4]. Tricellular tight junctions (tTJs) form at the convergence of bicellular tight junctions (bTJs) where three epithelial cells meet in polarized epithelia [
5,
6]. Claudin family proteins and occludin are main components of bicellular TJs, which are important for the barrier function and permselectivity [
7,
8,
9,
10]. To date, 27 members of the claudin family have been identified in humans. Many of these have sealing functions (claudins 1, 3, 5, 11, 14, 19) while some claudins form channels across TJs, which are permeable either for cations (claudins 2, 10b, 15) or for anions (claudins 10a, 17). For several claudins, their effects on epithelial barriers are inconsistent and the function is unclear (claudins 4, 7, 8, 16) [
11]. The claudin composition of the TJ mainly determines the tightness of an epithelium or an epithelial cell line, respectively.
In addition to their role as channels for small cations, Rosenthal et al. demonstrated that claudin-2 and claudin-15 also transport water [
12,
13,
14].
The mechanisms that determine the use of paracellular versus transcellular routes for water transport are multifaceted. For example, in the kidney, transport of water is achieved in part through the paracellular pathway in the proximal nephron [
15], whereas in the distal nephron and collecting duct, it takes place through transcellular routes [
16]. Mutations in claudins have been identified to cause hereditary diseases, demonstrating that regulation of the paracellular permeability is crucial to the normal functions of various organs [
11].
The tricellular TJ is more complex than bTJ. In the center of tTJ, bTJ elements from each of the three cells come together to form a central tube of about 1 μm in length and 10 nm in diameter [
17]. To date, two types of integral membrane proteins, tricellulin and angulin family proteins, are identified as molecular components of tTJ [
18]. Tricellulin, which was the first tTJ protein to be identified [
5], plays a critical role in sealing the tTJ against the passage of solutes of up to 10 kDa [
19,
20], and it was recently found that it also plays a role in the paracellular water transport in the tight epithelial cell line, MDCK C7 [
21].
As said, angulins contribute to forming the central element of the tTJ. Angulin family proteins, comprising lipolysis-stimulated lipoprotein receptor (LSR), immunoglobulin-like domain containing receptor-1 (ILDR1) and -2 (ILDR2), are type-I transmembrane proteins with an extracellular immunoglobulin-like domain. Due to their common structure and function as tTJs-associated membrane proteins, LSR, ILDR1, and ILDR2 were designated as angulin-1, angulin-2, and angulin-3, respectively [
22].
The original role of LSR/angulin-1 was described in lipid metabolism studies, as LSR was first discovered to be expressed in hepatocytes, where it plays a role in the clearance of triglyceride-rich lipoproteins and low-density lipoproteins, and also acts as an apolipoprotein B/E–containing lipoprotein receptor [
23]. At tricellular contacts, angulin-1 recruits tricellulin, and the interaction between the cytoplasmic domain of angulin-1 and the C-terminal cytoplasmic domain of tricellulin is required for this recruitment [
24,
25]. Angulin-1 together with tricellulin are required for full barrier function of epithelial cells with high transepithelial electrical resistance [
25]. Loss or downregulation of angulin-1 disrupts the barriers with relocalization of tTJ molecule tricellulin in various cell types [
24,
26]. In addition, loss of angulin-1 enhances cancer cell motility [
27,
28] and the LSR knockout mice die before embryonic day 15.5 (E15.5), but the cause of death remains unclear [
29].
The LSR-related proteins immunoglobulin-like domain containing receptors angulin-2 and angulin-3 are also expressed complementarily in many epithelial cell types at tricellular contacts of many epithelial cells and recruit tricellulin [
22], for instance, it is known that angulin-1 and angulin-2 are co-expressed in the large intestine and the kidney [
22].
As for the tTJ, Gong et al. provided evidence that water transport is increased in isolated kidney tubules in the absence of angulin-2, but is normally inhibited in the presence of angulin-2 [
30]. In contrast to this, Hempstock et al. found in the colon and kidney of angulin-2 KO mice no detectable abnormalities in water transport and maintained barrier function of the epithelia. They concluded that angulin-1 changes its expression pattern and location, which compensates for the loss of angulin-2 [
31].
These findings led us to hypothesize that angulin-1 is a direct negative actuator of tTJ water permeability depending on the tightness of the epithelium, in this respect similar to tricellulin. Therefore, we aimed to characterize the effect of angulin-1 expression on water transport in a tight and an intermediate-tight epithelial cell line, MDCK C7 and HT-29/B6, respectively. In order to define the tightness of these cell lines, we previously performed a two-path impedance spectroscopy study where we analyzed paracellular, transcellular, and transepithelial resistance in MDCK C7, HT-29/B6, and MDCK C11 cells. As a result, MDCK C7 cells were defined as a tight and HT-29/B6 cells as a moderate tight epithelial cell line, as defined by the ratio R
para/R
trans and the absolute resistance values of the two pathways [
32]. In our present study, we found that the water passage through the tTJ was changed in the absence of angulin-1 only in MDCK C7 cells, thus in a tight epithelium.
3. Discussion
Tricellulin and angulin family proteins have been identified as molecular constituents of tTJs [
5,
22,
25]. Tricellulin and angulins localize along the central elements of tTJs [
5,
25]. As angulins recruit tricellulin to TJs, the angulin–tricellulin axis is proposed to be of importance in tTJ formation [
22,
25,
36]. Therefore, a dysregulation of angulins may critically affect the permeability for molecules up to 10 kDa in different epithelial organs.
In the present study, our focus was placed on elucidating the role of angulin-1 on paracellular water permeability. For this, the effect of angulin-1 knockout was analyzed in two different cell lines, MDCK C7, a tight epithelial cell line, and HT-29/B6, an intermediate-tight epithelial cell line. It turned out that angulin-1 is involved in water permeability only in the tight epithelial cell line (similar to what we previously reported on tricellulin [
21]), while both angulin-1 and tricellulin have no effect on water permeability in an intermediate-tight epithelium. These results indicate that in tight epithelia, the tTJ is also essential for maintaining a barrier for water.
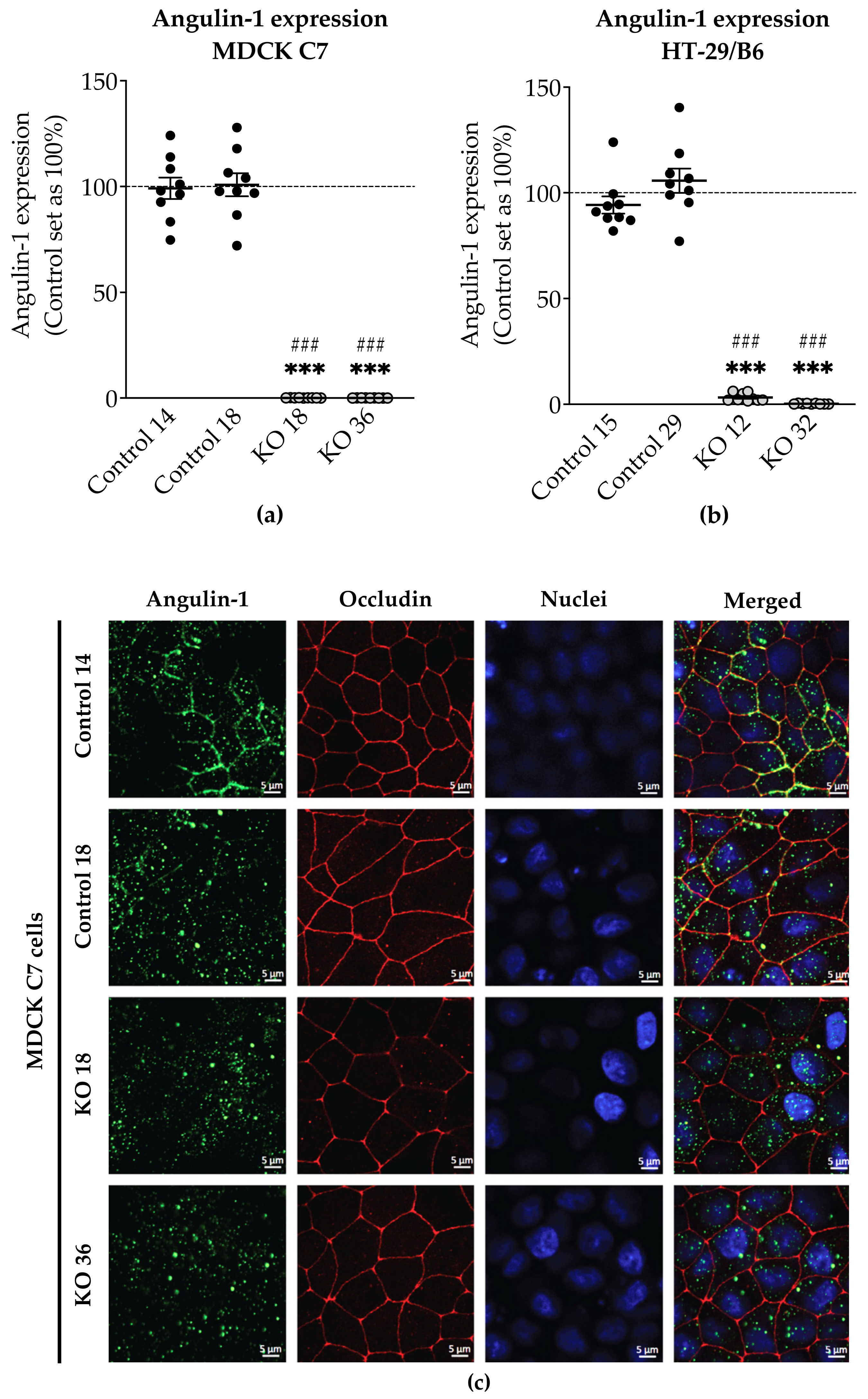
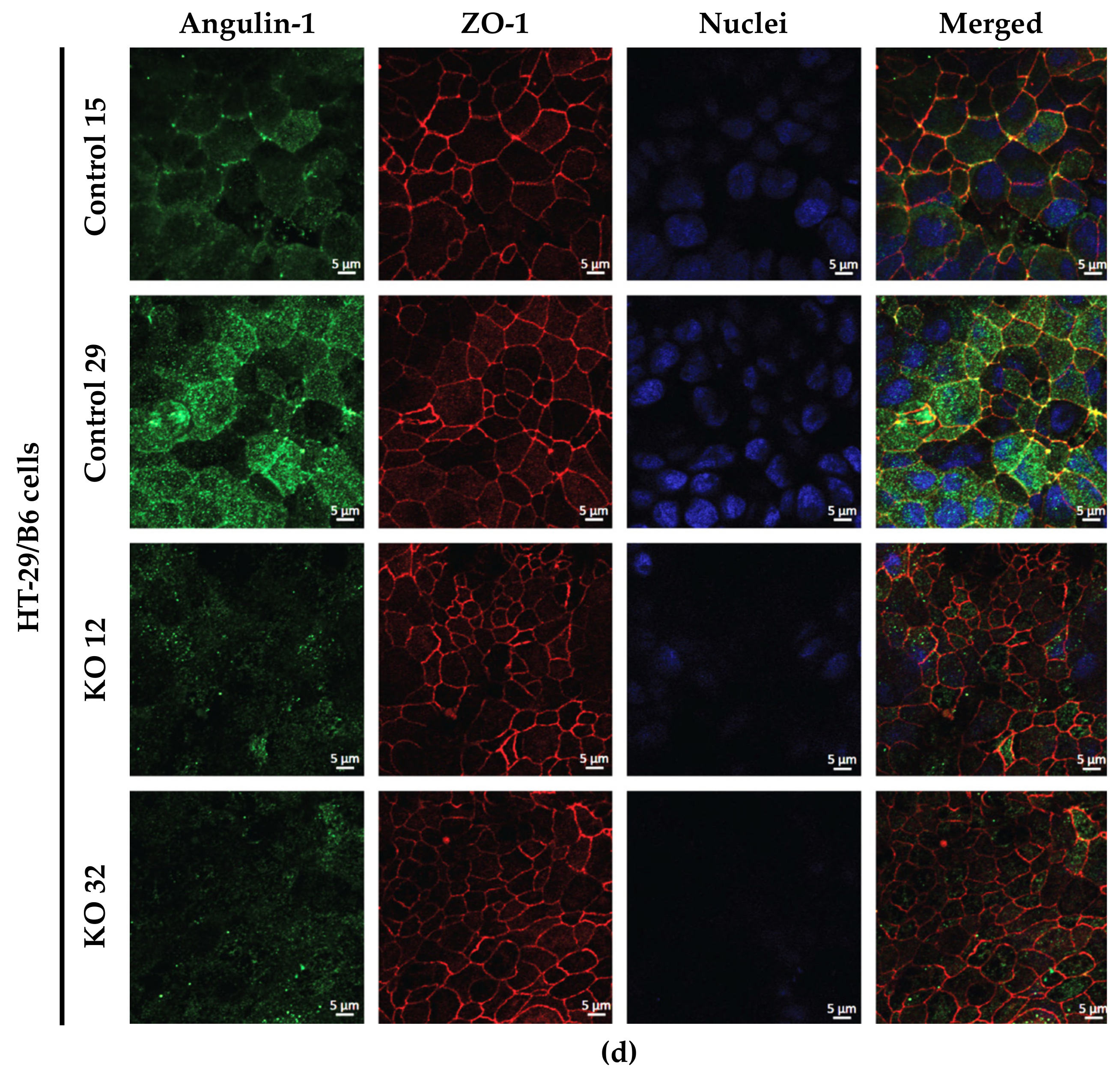
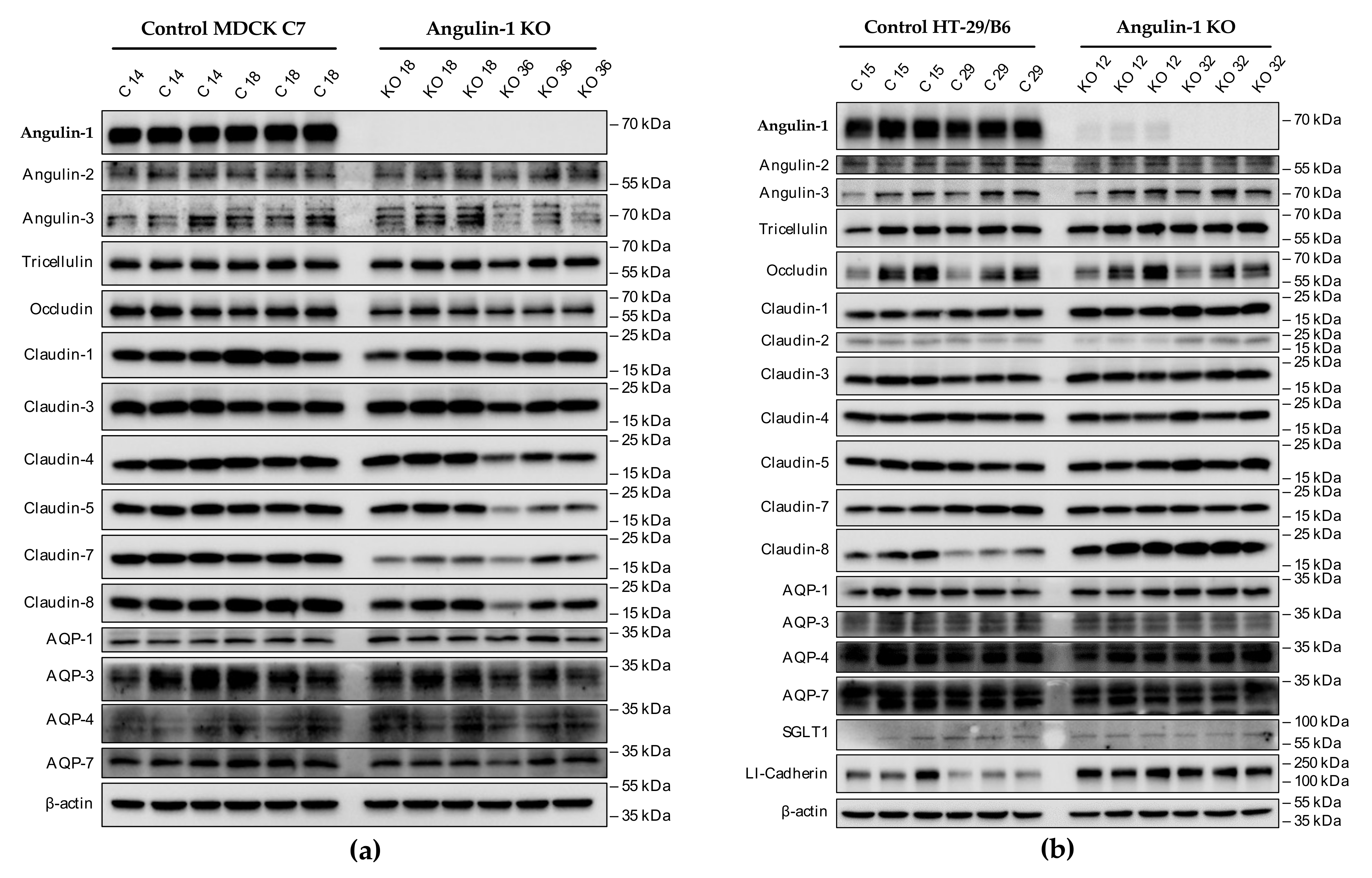
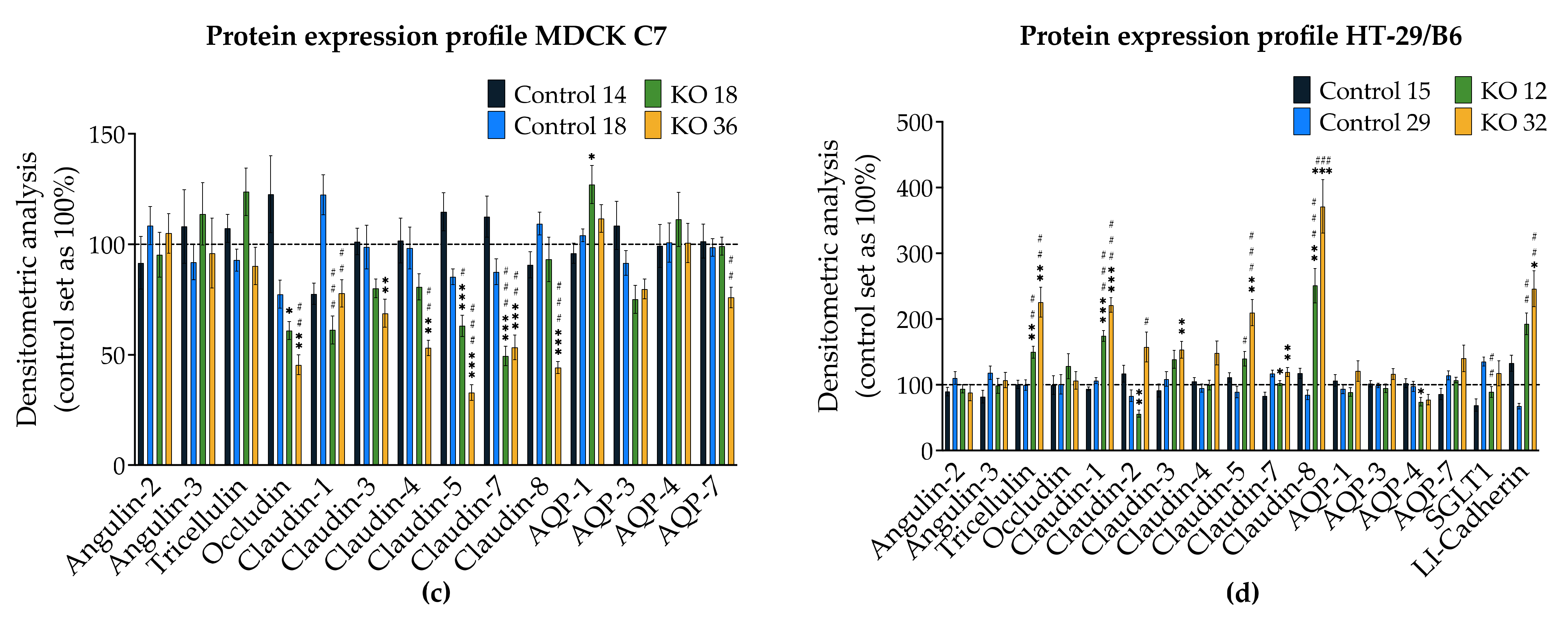
3.1. Angulin-1 Knockout Alters the Expression and Localization of Other Proteins in MDCK C7 and HT-29/B6 Cells
The expression of several tightening claudins in MDCK C7 cells was reduced in angulin-1 knockout cells, suggesting these claudins were partly removed from the TJ strands in the absence of angulin-1, in contrast, claudins and tricellulin expression were increased after angulin-1 knockout in HT-29/B6 cells, suggesting that these claudins were additionally incorporated into TJ strands in the absence of angulin-1. Most important, in both angulin-1 KO cell lines, tricellulin was partly removed from the tTJ and was also detectable in the bTJ [
25].
On one hand, angulin-1 knockout in MDCK C7 cells induced a decrease in the expression level of occludin, claudin-1 and -4 (
Figure 2c). This might affect the barrier function and formation as evidenced by Kirschner et al. through different knockdowns of claudin-1 and -4, occludin, and ZO-1, causing increased paracellular permeability for ions and larger molecules [
37]. Furthermore, the absence of occludin and claudin-1 in both KO clones prevents the specific localization of tricellulin at tricellular contacts and promotes its localization at the bTJ [
38,
39]. These observations suggested that the proteins functionally influence each other.
On the other hand, angulin-1 knockout in HT-29/B6 cells increased the expression of barrier-forming claudins [
39] and tricellulin (
Figure 2d). Therefore, it can be concluded that the observed clonal variation of the claudins and tricellulin in sum is balanced and provides a constant barrier function of the bTJ and tTJ. These results suggested that the effects of angulin-1 and tricellulin relocalization from tTJs to bTJs on epithelial barrier function are controversial [
39]. Interestingly, claudin-2 was downregulated in the KO 12 clone and upregulated in the KO 32 clone and appeared not to affect the water permeability. In relation to transcellular water transport, the major aquaporins of human colon cell line were found unchanged or downregulated, this suggests that changes in transepithelial water permeability could be attributed to the removal of angulin-1 and unlikely due to aquaporins. Lastly, the expression of LI-cadherin was increased in both KO clones, this could affect the water permeability under our experimental conditions. It was shown that water transport was impaired under hypertonic conditions when LI-cadherin is reduced [
40,
41]. Therefore, LI-cadherin might be important for water reabsorption of the gut at intraluminal hypertonic conditions [
40,
41].
Interestingly, the expression of angulin-2 and -3 did not change after the knockout of angulin-1 as a form of compensation in both cell lines. It was shown that compensatory functions exist between the angulin family members, e.g., in the organ of Corti [
42], the large intestine and kidney [
31], but this seems to be not relevant in the cell lines used for this study.
3.2. Angulin-1 Knockout Did Not Alter the Ultrastructure of the Bicellular Tight Junction in MDCK C7 and HT-29/B6 Cells
In previous RNAi experiments, the loss of tricellulin led to an unstable ultrastructure of tight junction, which ultimately caused the junctional complex to collapse completely [
5]. In both cell lines, the loss of angulin-1 did not modify or alter the bTJ compared to the vector control clones, the TJ ultrastructure remains unchanged after angulin-1 knockout in MDCK C7 and HT-29/B6 cells (
Figure 4).
It is established that, depending on the presence of claudin-2, strand discontinuities appear or disappear [
43]. In our study, no discontinuities appear even if claudin-2 is increased in the HT-29/B6 angulin-1 KO 36 clone, possibly due to the upregulation of tricellulin.
Thus, an increase in ion permeability in both cell lines and an increase in macromolecule permeability in HT-29/B6 cells could possibly be caused by the opening of the tTJ because the only difference between the controls and the angulin-1 KO clones was the width of the central tube. However, the morphometric parameters could not be analyzed because of the low abundance of the same structures in both cell lines. After considering all possibilities, it is concluded that the knockout of angulin-1 did not induce resolvable changes in the ultrastructure of bTJs and tTJs that could affect water transport measurements.
3.3. Angulin-1 Knockout Increases the Ion Permeability in MDCK C7 and HT-29/B6 Cells
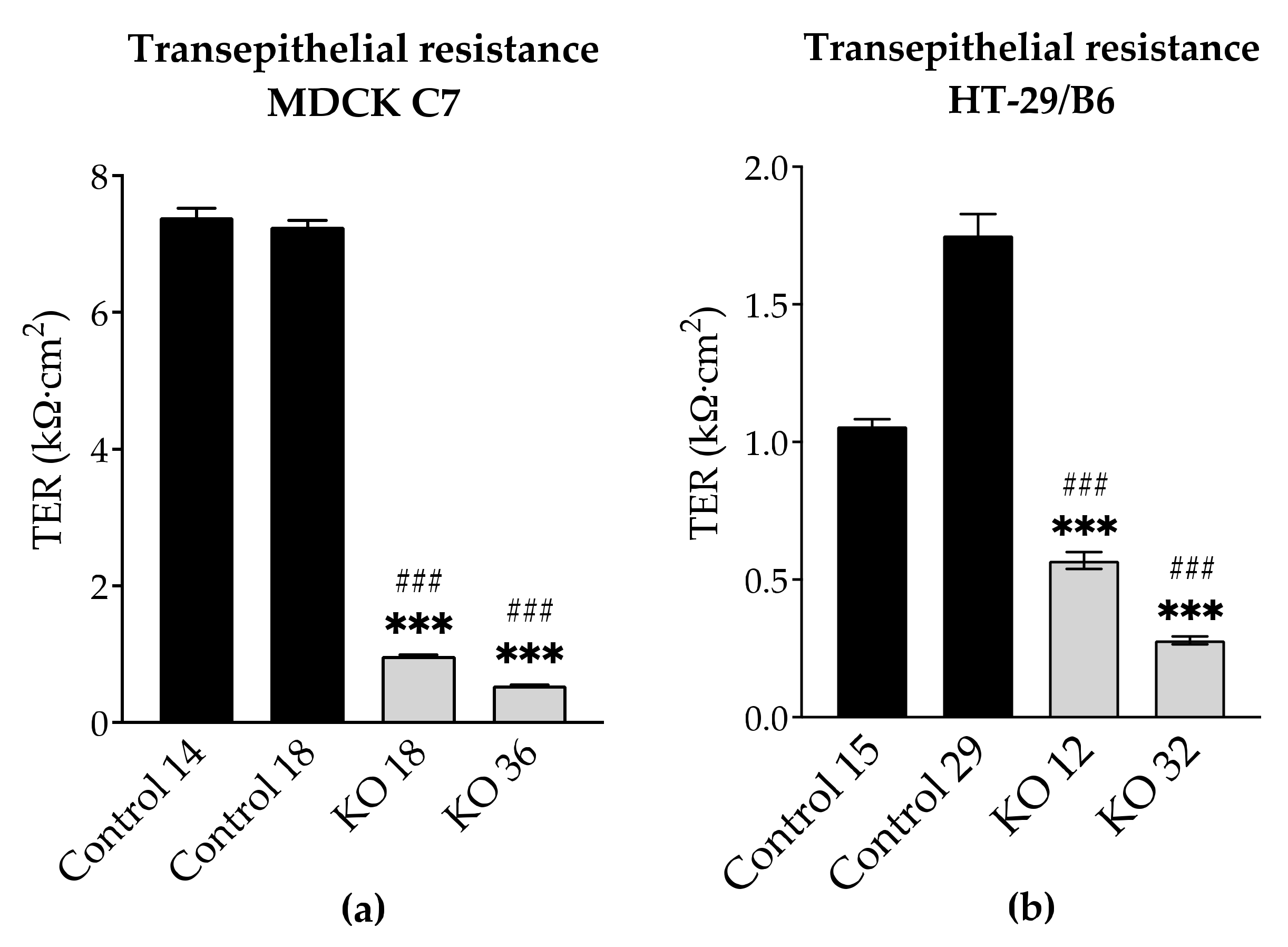
By the knockout of angulin-1, also the pore pathway was addressed (
Figure 5). Similar to what was observed in tricellulin knockdown cells, the reduction in the expression of angulin-1 resulted in a lowered transepithelial resistance (TER). It is important to note that in MDCK C7 cells the removal of angulin-1 resulted in the reduction of the expression of most claudins while in HT-29/B6 the effect was the opposite. Therefore, it could be said that the reduction in TER on HT-29/B6 cells is due only to the reduction in angulin-1; however, in MDCK C7 cells the effect could be intensified due to the downregulation of other tightening TJ proteins. In conclusion, the decrease of angulin-1 within the tTJ also leads in both cell lines to a drop in paracellular resistance, which reciprocally reflects the permeability for ions.
3.4. Angulin-1 Knockout Increases the Macromolecule Permeability Only in HT-29/B6 Cells
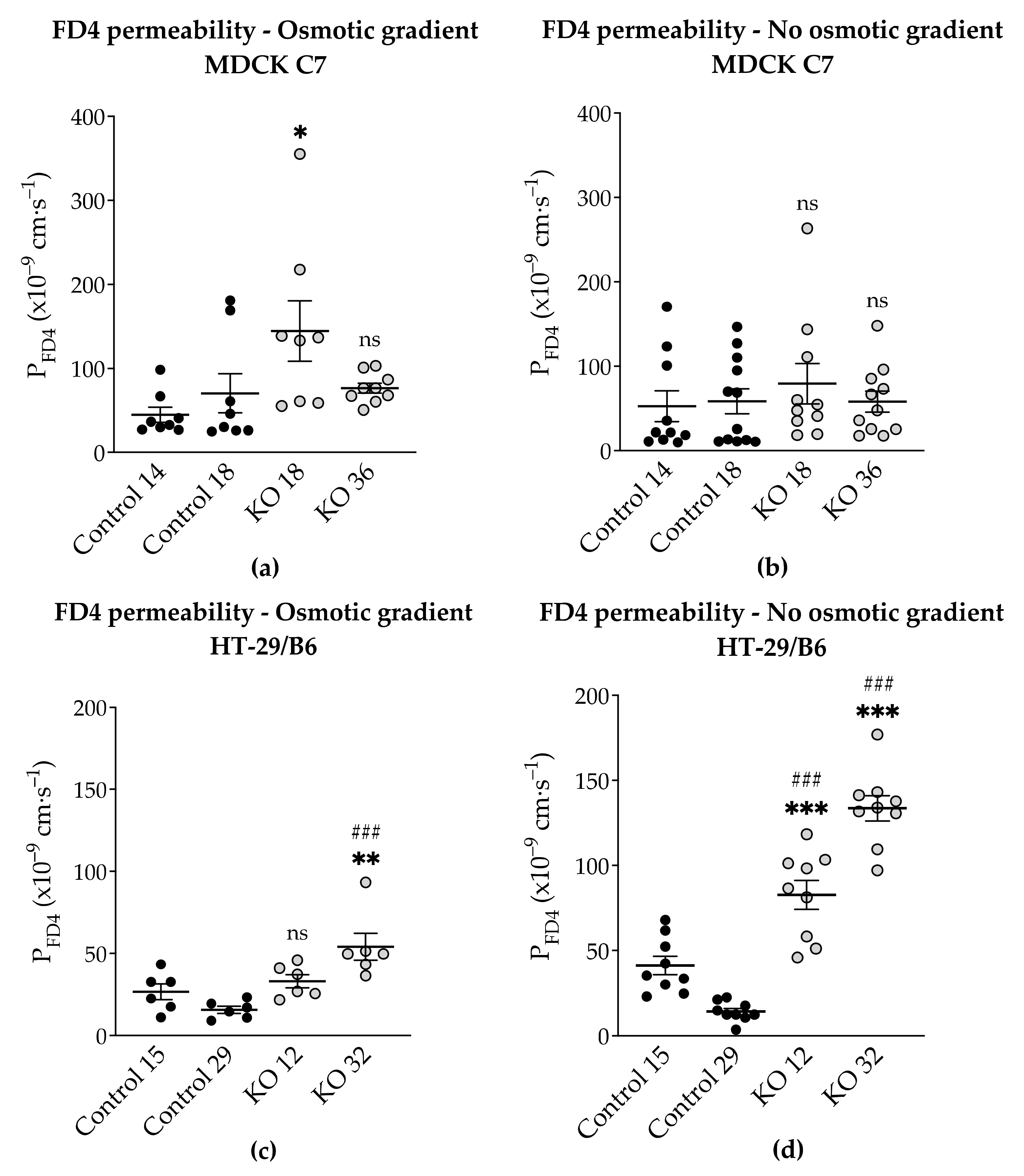
As it is known, knockdown of angulin-1 in the mouse mammary gland Eph4 epithelial cells increases the permeability to macromolecules between 332 Da and 40 kDa [
22]. Considering the above, the leak pathway was assessed (
Figure 6). The present data showed that in HT-29/B6, a removal of angulin-1 strongly regulates the permeability for 4-kDa macromolecules. Nevertheless, it might be surprising that the same phenotype, in MDCK C7 cells, only slightly increased the permeability for macromolecules in one of the KO clones (KO 18), probably because in this KO clone, the tricellulin delocalization from the tTJ was larger than in the KO 36 clone.
Considering that HT-29/B6 is an intermediate-tight epithelial cell line and that its tTJ contributes at least 38% to the total paracellular conductance, it can be said that, not only did it affect the paracellular passage of the small solutes, including inorganic ions, but also of the macromolecules [
20]. However, the contribution of tTJ in MDCK C7 cells is not yet known, but it would be expected to be higher than in HT-29/B6 cells because this cell line is highly tight [
20], and therefore the permeability to FD4 would be higher after angulin-1 knockout, but this was not the case. This is perhaps due to tricellulin which has an affinity for claudin-based TJ strands within the plasma membrane if not directed to tricellular contacts by angulins [
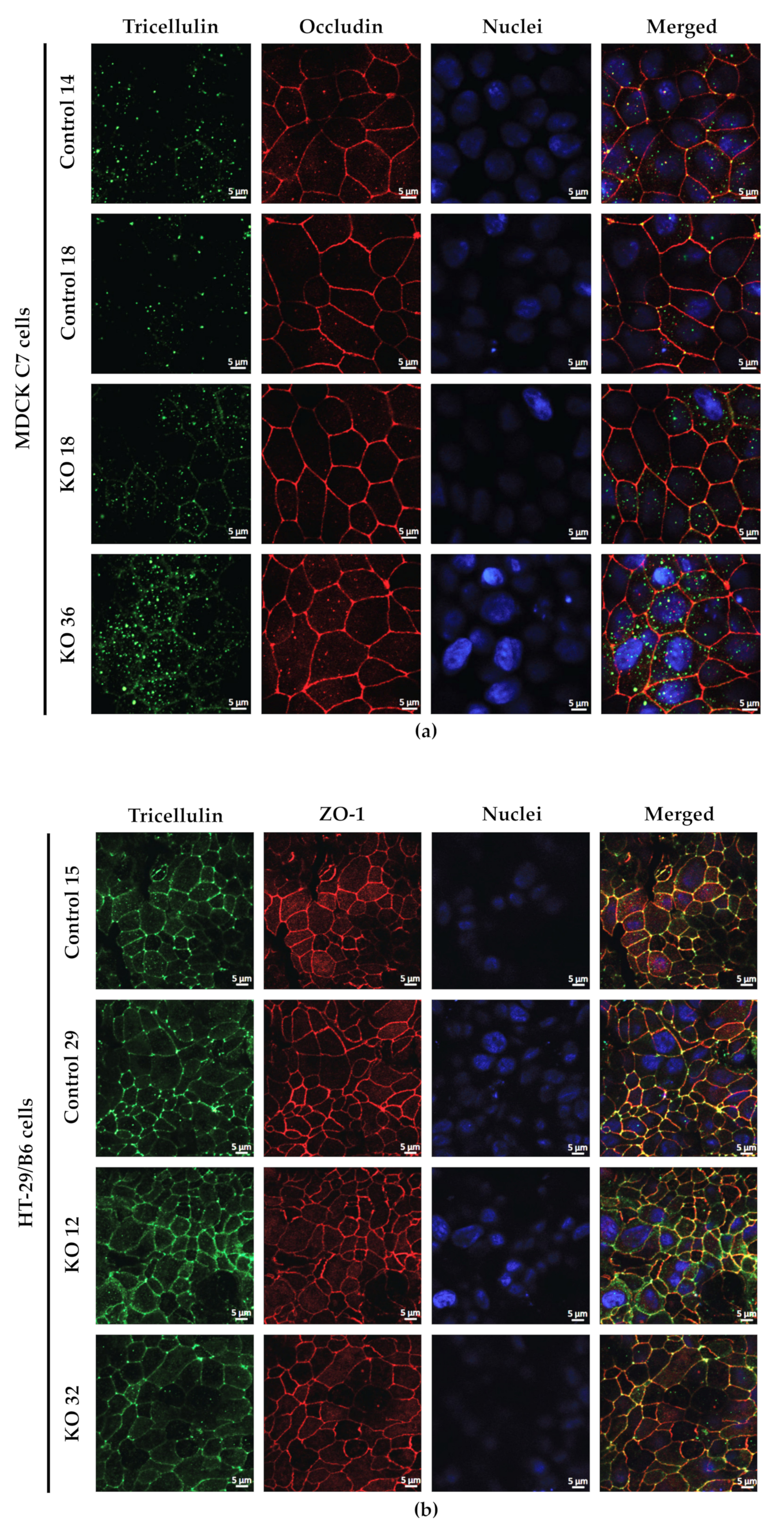
44] and also because occludin was concentrated in the corners of tTJ after angulin-1 KO (
Figure 1c) probably stabilizing this structure. One could assume that due to redistribution of tricellulin into the bTJ a slight enhancement of bTJ barrier properties could occur [
44].
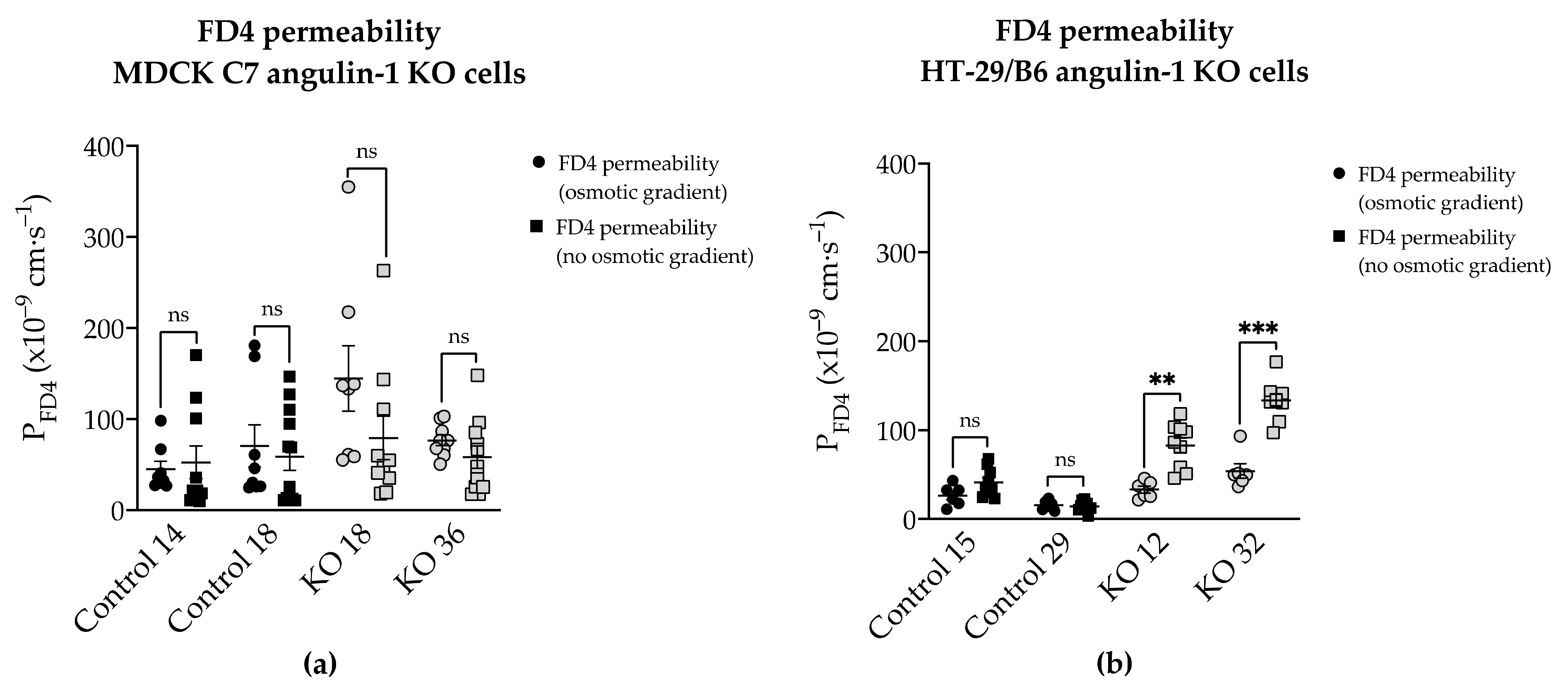
Additionally, the permeability to FD4 was measured in two different conditions after angulin-1 KO and, as can be seen, in MDCK C7 cells there were no differences between isosmotic and osmotic conditions (
Figure 9a). Conversely, in HT-29/B6 cells it was observed that the permeability to FD4 is higher under isosmotic conditions than under an osmotic gradient. A possible explanation for this might be that under osmotic conditions water moves in the opposite direction and this inhibits the movement of 4-kDa FITC-dextran, therefore a lower FD4 permeability was measured (
Figure 9b). This water movement through the tTJ is too small to be detected in the overall transepithelial water flux measured in HT-29/B6 cells.
3.5. Angulin-1 Knockout Increases Transepithelial Water Transport Only in MDCK C7 Cells
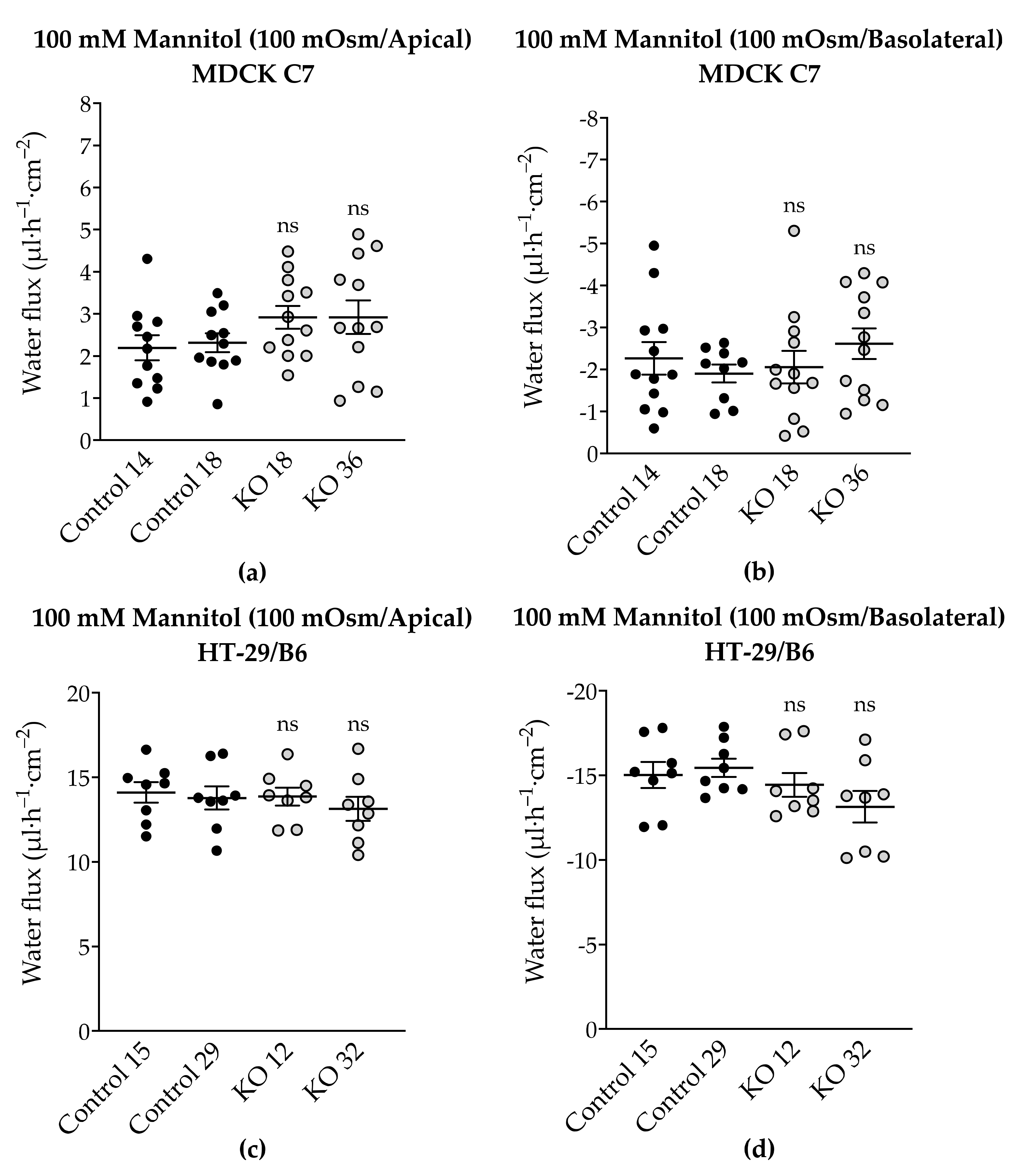
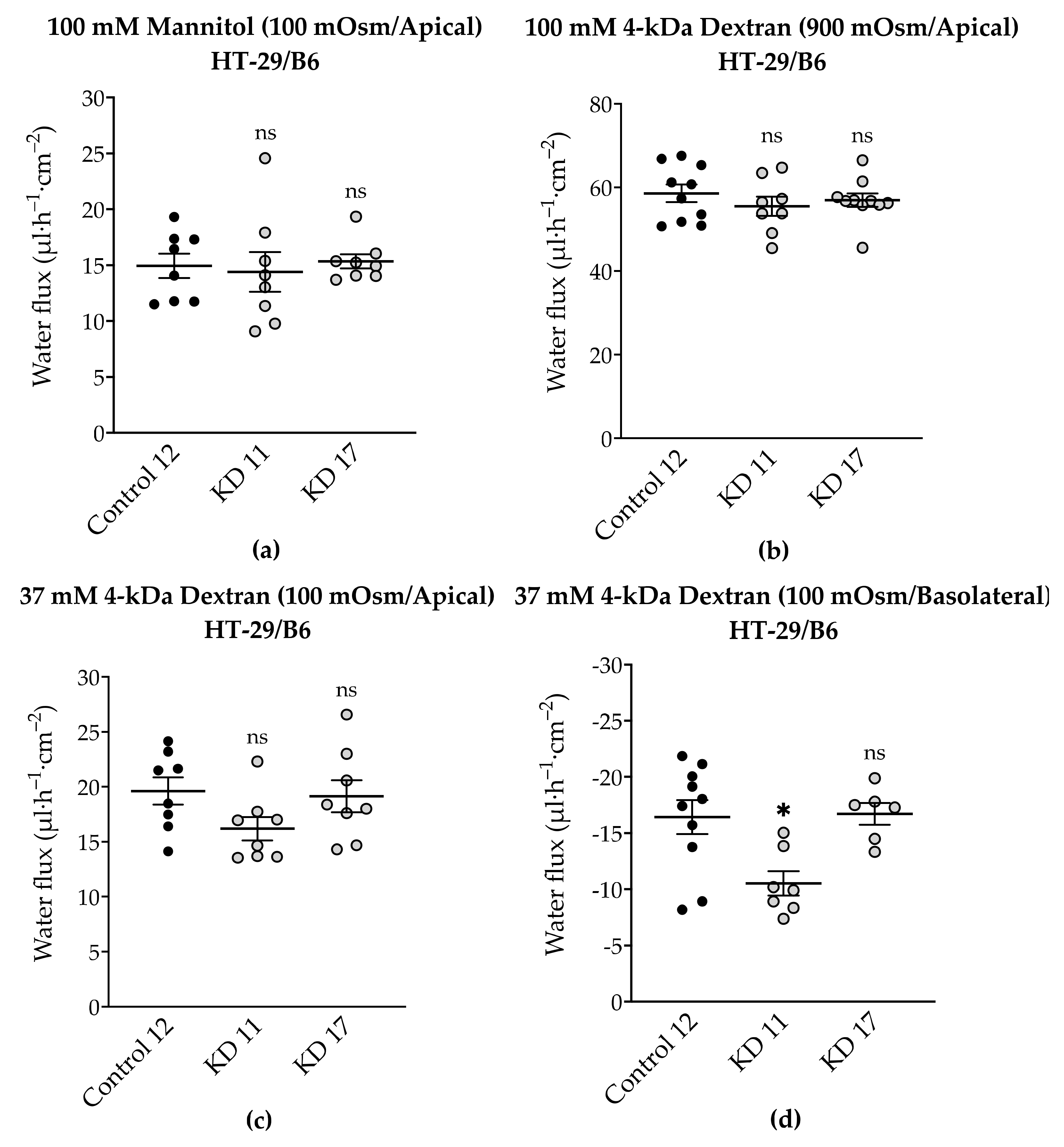
To complete the tTJ permeability pattern, the transepithelial water transport was measured in the angulin-1 KO clones of MDCK C7 and HT-29/B6 cells. As described earlier, while the KO clones caused a decrease in TER in both cell lines, only in HT-29/B6 cells did angulin-1 KO increase the permeability to a 4-kDa macromolecule. Interestingly, the angulin-1 KO did not increase the water flux driven by 100 mM mannitol in both cell lines (
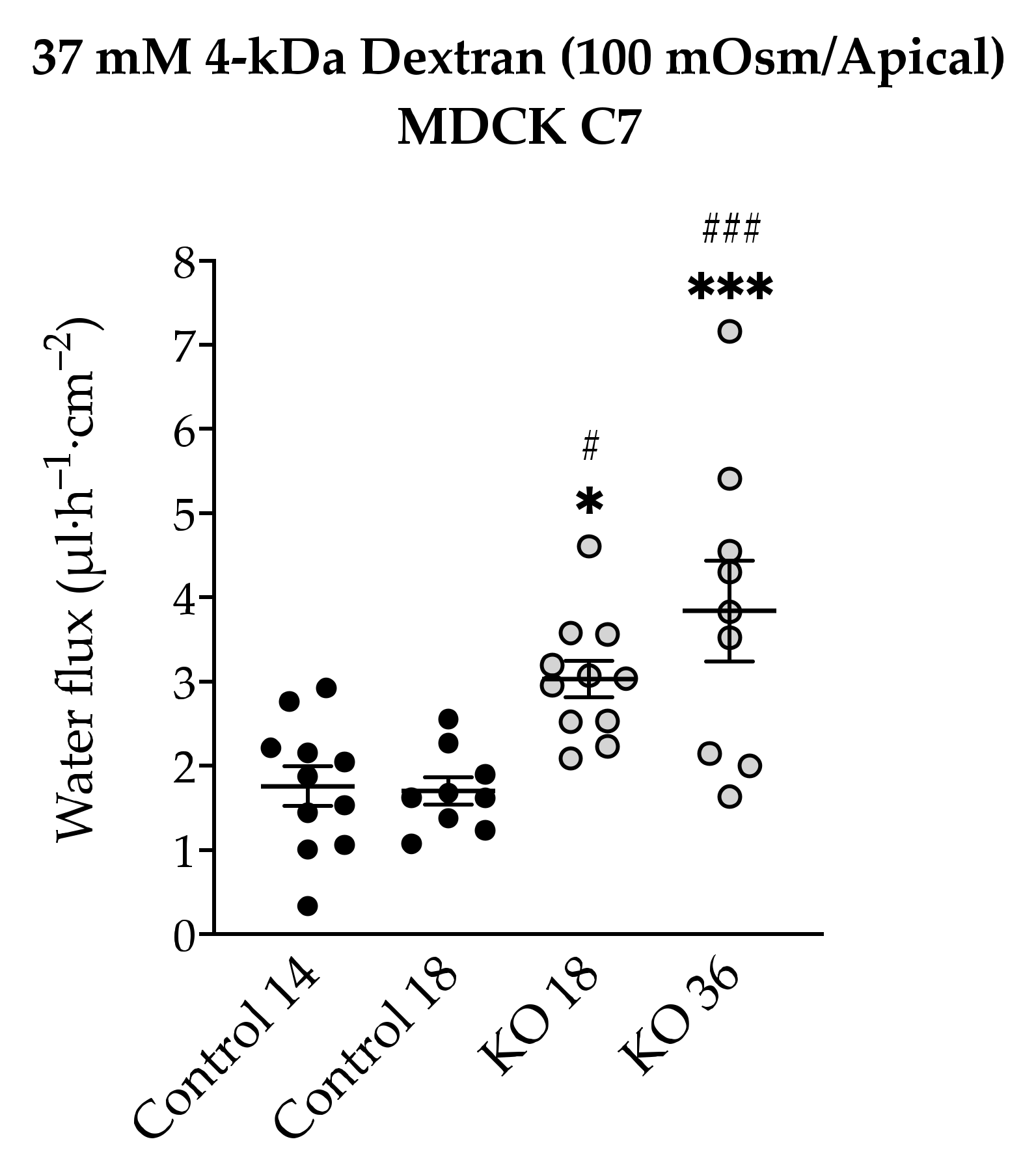
Figure 7) and the total amount of water was independent of the direction of the osmotic gradient. The decision to use 4-kDa dextran was taken due to the dispersion of the values found in MDCK C7 cells when mannitol was used as an osmotic gradient; however, the values obtained with HT-29/B6 cells were more reproducible and therefore their study with 4-kDa dextran was obviated. As result, the water flux increased in the angulin-1 KO clones compared with their controls in MDCK C7 cells when 37 mM 4-kDa dextran was used as osmotic gradient (
Figure 8).
Remarkably, in the case of MDCK C7 cells, an increase in water transport was expected like that found after reducing the expression of tricellulin in tTJ [
21]; however, this was not the case when mannitol was used possibly due to the small diameter of this molecule with respect to the 4-kDa dextran which would facilitate its movement through tTJ thereby inhibiting the water flux in the converse direction. It is important to keep in mind that the reduction in the expression of most barrier-forming claudins could have an influence on the water transport after angulin-1 KO in MDCK C7 cells. As demonstrated by immunofluorescence confocal laser-scanning microscopy, tricellulin was redistributed from tTJ to bTJ, which could maintain and strengthen the epithelial barrier after the downregulation of several claudins. It is also important to note that the water fluxes measured in angulin-1 KO cells were lower than those measured in our previous work on tricellulin KD clones [
21], demonstrating once again the importance of tricellulin in tTJ (for instance, angulin-1 KO 36 showed higher tricellulin displacement leading to higher water flux). In addition, it can be concluded that the reduction of tightening claudins did not increase the water flux in the KO clones with respect to their controls.
This phenomenon was also observed in HT-29/B6 cells; however, here the regulation of TJ expression was the opposite to what we observe in MDCK C7 cells, several claudins were upregulated, nevertheless, and no change in water transport could be measured. As shown in
Figure 9, FD4 permeability in HT-29/B6 angulin-1 KO cells (not in control cells) measured under osmotic conditions is lower than under isosmotic conditions, potentially due to an opposite water flow across the tTJ. This means that the water flow across the tTJ, which was induced by a gradient using mannitol or dextran, could also be inhibited by the opposite passage of the osmotically active substances across the tTJ in these cells. This could also be the case in MDCK C7 angulin-1 KO cells when using mannitol for producing the osmotic gradient. In addition, the strong upregulation of tightening claudins in the bTJ in HT-29/B6 angulin-KO cells could diminish the water flux across the bTJ and thus mask a slight increase in water flux across the tTJ. Thus, we cannot completely exclude an increased water flow across the tTJ after downregulation of tricellulin or knockout of angulin-1, but this does not significantly contribute to the overall large water flow across an intermediate tight epithelial cell line like the HT-29/B6 cells.
In summary, the present study suggests a functional difference between angulin-2 and angulin-1 on the paracellular water transport via tTJs, which is an interesting issue to be pursued for the understanding of variations of tTJs. The effects of angulin-1 knockout on the epithelial barrier function vary by cell type and according to the interdependence between different TJ species affecting cell expression, localization and renewal. The reason could be the differences in angulin-1 interaction partners in bTJs and tTJ [
22,
39]. Therefore, combinations of proteins influenced by angulin-1 knockout in bTJs and tTJ vary by cell type and tissue, and the resulting epithelial barrier function of the mammalian epithelial cell sheet causes cell specific changes.
3.6. Tricellulin Knockdown Does Not Significantly Affect Paracellular Water Transport in the HT-29/B6 Cell Line
As was previously done for tricellulin knockdown clones in MDCK C7 cells [
21], two tricellulin clones were selected in HT-29/B6 cells that differ in the grade of tricellulin reduction (
Supplementary Figure S1). While both KD clones caused a decrease in TER and an increase in permeability to a 4-kDa macromolecule, none of them increased the water flux driven by different osmotic gradients. This may be due, as already described for the HT-29/B6 angulin knockout cells, to an inhibition of the water flux across the tTJ by a backflow of the osmotically active substances like mannitol or dextran. In addition, a large component of water transport may have traveled through the bTJ and a possible contribution from the tTJ may not have reached significant levels (
Figure 10). On the other hand, there may be a threshold in the expression of tricellulin, that must be overcome to find significant differences in water transport, that were not reached with the knockdowns generated in this research. In addition, there is a possibility that in general the paracellular water flow is lower than the transcellular water flux or that there is a claudin-2-independent paracellular pathway for water.
In conclusion, in HT-29/B6 cells (intermediate-tight) the contribution of tricellulin to transepithelial water permeability is negligible, in contrast to MDCK C7 cells (tight) where tricellulin was able to regulate osmotically induced transepithelial water flux.
4. Materials and Methods
4.1. Cell Culture, Transfection and TER Measurement
The kidney cell line MDCK C7 (RRID: CVCL_0423) exhibits a high transepithelial resistance and other basic properties (no claudin-2 expression) making it an excellent model of a tight epithelium, and the intestinal cell line HT-29/B6 (RRID: CVCL_LJ30) exhibits a basic properties (moderate transepithelial resistance, low claudin-2 expression) making it an excellent model of an intermediate-tight epithelium. For stable angulin-1 knockout, MDCK C7 and HT-29/B6 cells were transfected with three sgRNA-CRISPR/ Cas9 vectors containing sequences targeting different exons of angulin-1 and corresponding HDR plasmids for direct homologous repair or sgRNA-CRISPR/Cas9 negative control (Santa Cruz, Heidelberg, Germany). For stable tricellulin knockdown, HT-29/B6 cells were transfected with pLKO.1-puro vector containing a sequence for shRNA targeting tricellulin (tricellulin shRNA; Sigma-Aldrich, Schnelldorf, Germany) or pLKO.1-puro empty vector as a negative control (Sigma-Aldrich, Schnelldorf, Germany).
The transfected cells were incubated at 37 °C and 5% CO2 in sterilization incubators held. For the cultivation of the cells sterile culture vessels made of plastic were used. For MDCK C7 knockout cells, a nutrient medium Earl’s salts MEM (minimal essential medium) supplemented with 10% FBS as well as 100 U/mL penicillin/100 μg/mL streptomycin and 1.5 µg/mL of puromycin was used. For HT-29/B6 knockdown and knockout cells, RPMI-1640 with stable L-glutamine medium supplemented with 10% FBS as well as 100 U/mL penicillin/100 μg/mL streptomycin and 1.5 µg/mL of puromycin was used. Every second to third day the medium was changed. Consumption of nutrients was also visible due to a color change of the medium contained a detecting pH indicator.
For protein quantification, water flux measurements and electrophysiological studies, cell monolayers were cultured on porous culture plate inserts (Millicell PCF filters, pore size 0.4 µm, effective area 0.6 cm2, Millipore GmbH, Schwalbach, Germany) for 7–10 days before they were used for experiments.
Transepithelial resistance (TER) was measured at 37 °C using chopstick electrodes (STX2, World Precision Instruments, Friedberg, Germany). Electrodes were reproducibly positioned by a semi-automatic motor-driven device and signals were processed by a low-frequency clamp (both own design). The resistances of the bathing solution and the blank filter support were subtracted from measured values, which were finally converted to Ω·cm
2 [
45].
4.2. Western Blot Analysis
Western Blot analysis was carried out as described before [
21]. Cells grown on culture-plate inserts were scraped and homogenized in total lysis buffer containing 10 mM of Tris, 150 mM of NaCl, 0.5% of Triton X-100, and 0.1% of sodium dodecyl sulphate (SDS), and protease inhibitors (cOmplete
TM EDTA free; Roche, Basel, Switzerland). The samples were incubated for 2 h at 4 °C (vortexed every 20 min), and then centrifuged at 11,000×
g during 20 min. The pellet was discarded and the protein concentration in the supernatant was determined by the BCA (bicinchoninic acid) method (reagents were purchased from Pierce (Perbio Science, Bonn, Germany)) and quantified with a plate reader (Tecan Deutschland, Crailsheim, Germany). After this, samples were prepared with a loading buffer containing 100 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 100 mM DTT and 0.001% bromophenol blue. Each sample was then boiled at 95 °C for 10 min before loading onto the gel.
Aliquots between 10 and 15 µg protein samples were separated by 12% SDS-polyacrylamide gel electrophoresis and then transferred to a PVDF membrane (Perkin Elmer, Rodgau, Germany) for detection of angulins, TAMPs, claudins, AQPs, SGLT-1 and LI-cadherin. After blocking for 2 h in 1% PVP-40 and 0.05% Tween-20, membranes were incubated overnight with primary antibodies specific for angulin-1 (Sigma-Aldrich, Taufkirchen, Germany), angulin-2 (Aviva Systems Biology, San Diego, CA, USA), and angulin-3 (Sigma-Aldrich, Taufkirchen, Germany), tricellulin (Thermo Fisher Scientific, Invitrogen, Darmstadt, Germany), occludin (Thermo Fisher Scientific, Invitrogen, Darmstadt, Germany), claudin-1, -2, -3, -4, -5, -7, and -8 (Thermo Fisher Scientific, Invitrogen, Darmstadt, Germany), AQP-1 (OriGene Technologies, Herford, Germany), AQP-3, -4 and -7 (Santa Cruz, Heidelberg, Germany), SGLT1 (LSBio, Corston, UK) and LI-cadherin (Santa Cruz, Heidelberg, Germany).
After removing the first antibody and three washing steps, the membranes were incubated for 2 h with the second peroxidase-conjugated antibody (anti-mouse or anti-rabbit) in 1.5% of milk powder prepared in TBST 1X. The primary antibodies were used in dilutions of 1:1000 with the exception of AQP antibodies (1:1500), tricellulin (1:2000), and angulin-1 (1:3000). The secondary antibodies were used in dilutions of 1:1000. For detection of the chemiluminescence signal induced by addition of Lumi-LightPLUS Western blotting kit (Roche, Mannheim, Germany) a Fusion FX7 (Vilber Lourmat, Eberhardzell, Germany) was used. Densitometric analysis was performed with a quantification software (Image Studio™ Lite, LI-COR Biosciences, Lincoln, NE, USA). Equal protein loading in each lane was verified by comparison with signals for β-actin (Sigma-Aldrich, Taufkirchen, Germany). For Western blot analysis, lysates of at least three individual cell cultures were used and one representative experiment is shown.
4.3. Immunofluorescent Staining
Immunofluorescence analysis was carried out as described before [
21]. Immunofluorescence studies were performed on culture-plate inserts. Confluent monolayers were rinsed with PBS, fixed with 4% paraformaldehyde for 20 min, washed three times with PBS, and permeabilized for 10 min with PBS containing 0.5% (
v/
v) Triton X-100. To block non-specific binding sites, cells were then incubated in PBS containing 1% (
w/
v) BSA and 5% (
v/
v) goat serum (blocking solution; Biochrom) for 60 min. All subsequent washing procedures were performed with this blocking solution.
After blocking, cells were incubated overnight at 4 °C with primary antibodies for angulin-1 (1:1000; Sigma-Aldrich), tricellulin (1:500; Thermo Fisher Scientific, Invitrogen), occludin (1:250; Thermo Fisher Scientific, Invitrogen) and ZO-1 (1:250; Thermo Fisher Scientific, Invitrogen), followed by washing steps and incubation during 60 min at room temperature with the respective secondary antibodies (Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 594 goat anti-mouse, each 1:500; Thermo Fisher Scientific, Waltham MA, USA) and 4′,6-diamidino-2-phenylindole (DAPI, 1:1000; Roche, Mannheim, Germany). Cell culture inserts were mounted on microscope slides using ProTag MountFluor (Biocyc, Luckenwalde, Germany). Images were obtained with a confocal laser-scanning microscope (LSM 780, Zeiss, Jena, Germany) and processed using ZEN software (Carl Zeiss, Oberkochen, Germany, ZEN black edition 2012 SP1, ver. 8.1).
4.4. Freeze Fracture Electron Microscopy
At a confluency of 100%, cells were washed with PBS (containing Ca2+/Mg2+) and fixed with 2.5% glutaraldehyde at RT for 2 h. After washing twice with PBS (containing Ca2+/Mg2+), the cells were stored at 4 °C in 0.25% glutaraldehyde. Small rectangles of the bottom of the cell culture filters were cut out, the attached cells were cryoprotected in 30% glycerol for 30 min, placed between two gold specimen holders and shock frozen in R422D (TEGA GmbH, Würzburg, Germany) cooled by liquid nitrogen (−210 °C). The samples were fractured using the freeze-fracture device Denton DV-502 (Denton Vacuum, Moorestown, NJ, USA) at −100 °C and 2 × 10−10 Torr. The samples were vaporized at −150 °C (2 × 10−10 Torr) with a layer of platinum and then a layer of carbon. This results in a thin metal film on the broken sample. The replicas were cleaned with 12% sodium hypochlorite, washed several times in ddH2O and mounted on a copper mesh grid. The Zeiss 902A electron microscope was used to examine the replicas at 80 kV. Magnifications between 20,000 and 50,000 was used.
4.5. Measurement of 4-kDa FITC-Dextran Flux
Flux studies were performed in conventional Ussing chambers for cell-culture inserts [
45] under voltage clamp conditions. Dextran flux was measured in 5 mL circulating 111-Ringer’s ((in mM) 119.7 NaCl, 21.4 NaHCO
3, 2.5 Na
2HPO
4, 0.6 NaH
2PO
4, 5.7 KCl, 1.3 MgCl
2, 1.2 CaCl
2, and 10.0 D(+)-glucose) containing also 37 mM unlabeled 4-kDa dextran (SERVA, Heidelberg, Germany) on both sides of the cells for isosmotic conditions and only on the apical side for osmotic conditions. After addition of 0.2 mM 4-kDa FITC-labeled dialyzed dextran (Sigma-Aldrich) to the apical chamber (final concentration), basolateral samples (200 μL) were collected at 0-, 20-, 40-, 60-, 80-, 100-, 120- and 140-min. Tracer fluxes were determined from FITC-dextran samples, which were measured with a fluorometer at 520 nm (Spectramax Gemini, Molecular Devices, Ismaning, Germany). Dextran permeability was calculated from
p = J/Δc with
p = permeability (cm·s
−1), J = flux (mol·h
−1·cm
−2) and c = concentration (mol/L).
4.6. Dilution Potential Measurements
Dilution potential measurements for the determination of ion permeabilities were performed in Ussing chambers for cell-culture inserts [
45]. Water-jacketed gas lifts kept at 37 °C were filled with 10 mL circulating fluid on each side. The bathing solution contained (in mM) 119.7 NaCl, 21.4 NaHCO
3, 5.7 KCl, 1.3 MgCl
2, 1.2 CaCl
2, 3.0 HEPES, and 10.0 D(+)-glucose, and was gassed with 95% O
2 and 5% CO
2 to ensure a pH value of 7.4. All experimental data were corrected for the resistance of the empty filter and the bathing solution. Dilution potentials were measured with modified bathing solution on the apical or basolateral side of the epithelial monolayer. In the modified bathing solution, NaCl was iso-osmotically replaced by mannitol. The ratio of P
Na and P
Cl and the absolute permeabilities for Na
+ and Cl
− were calculated as described before [
46].
4.7. Measurement of Transepithelial Water Transport
Water flux measurements were performed using a modified Ussing chamber developed by our group [
12,
13,
14,
21]. The gas lifts of the common Ussing chamber were replaced by two glass tubules with a small diameter, where the menisci of the perfusion solution are clearly visible. From changes in the menisci, the water flux is calculated. Throughout these experiments, transepithelial voltage (mV) was clamped to 0 mV and transepithelial resistance (TER, Ω·cm
2) and short-circuit current (I
SC, μA·cm
−2) were recorded. Resistances of bathing solution and blank filter support were measured prior to each experiment and subtracted. The stability of TER served as an indicator of cell viability.
Cell filters were mounted in Ussing chambers and perfused with HEPES-buffered solution with the following composition (in mM): 144.8 NaCl, 2.4 Na
2HPO
4, 0.6 NaH
2PO
4, 5.4 KCl, 1.2 MgCl
2, 1.2 CaCl
2, 10.6 HEPES, and 10.0 D(+)-glucose. The pH value of the perfusion solution was pH 7.4. A rotary pump ensured constant circulation of the perfusion solution (4.0 mL·min
−1) and thus a fast fluid exchange in both hemichambers (volume 500 μL) to avoid effects of unstirred layers on water permeability. Water flux was induced by a transepithelial osmotic gradient: (i) 100 mM mannitol, (ii), 37 mM 4 kDa-dextran or (iii) 100 mM 4 kDa-dextran. The solution was added in the apical or basolateral compartment of the Ussing chamber. The osmolality of the perfusion solutions (mosmol/kg, abbreviated mOsm) was determined using a freezing point depression osmometer (Osmomat 3000, Gonotec, Berlin, Germany). The osmolality of the HEPES-buffered solutions is annotated in
Table 3.
The fluid level in both glass tubes was monitored by a visual system ColorView XS (Olympus Soft Imaging Solutions GmbH, Munster, Germany) at time 0 min and with intervals of 10 min over a period of 120 min. Transepithelial water flux, given as flux per square centimeter and hour, was calculated after special calibration from the difference between the menisci at the registration times. Fluxes directed from the basolateral to the apical compartment were defined as positive flux.
4.8. Statistical Analysis
Data are expressed as mean values ± SEM, indicating n as the number of single measurements, and N is the number of independent experiments, which means independent seeding of cells. Statistical analysis was performed using Student’s t-test between the tricellulin KD clones and their corresponding control (KD 11 and KD 17 versus control 12). In case of angulin-1 KO clones, statistical significance of the difference between means was evaluated using one-way ANOVA and the Tukey and Dunnett adjustment for multiple testing.
MDCK C7 cells: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 with regard to control 14 and # p ≤ 0.05, ## p ≤ 0.01, ### p ≤ 0.001 with regard to control 18. HT-29/B6 cells: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001 with regard to control 15 and # p ≤ 0.05, ## p ≤ 0.01, ### p ≤ 0.001 with regard to control 29. Statistical analysis was performed using GraphPad Prism 8 software (GraphPad; San Diego, CA, USA). A probability value of p ≤ 0.05 was considered statistically significant.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}