
Lack of Glutamate Receptor Subunit Expression Changes in Hippocampal Dentate Gyrus after Experimental Traumatic Brain Injury in a Rodent Model of Depression

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
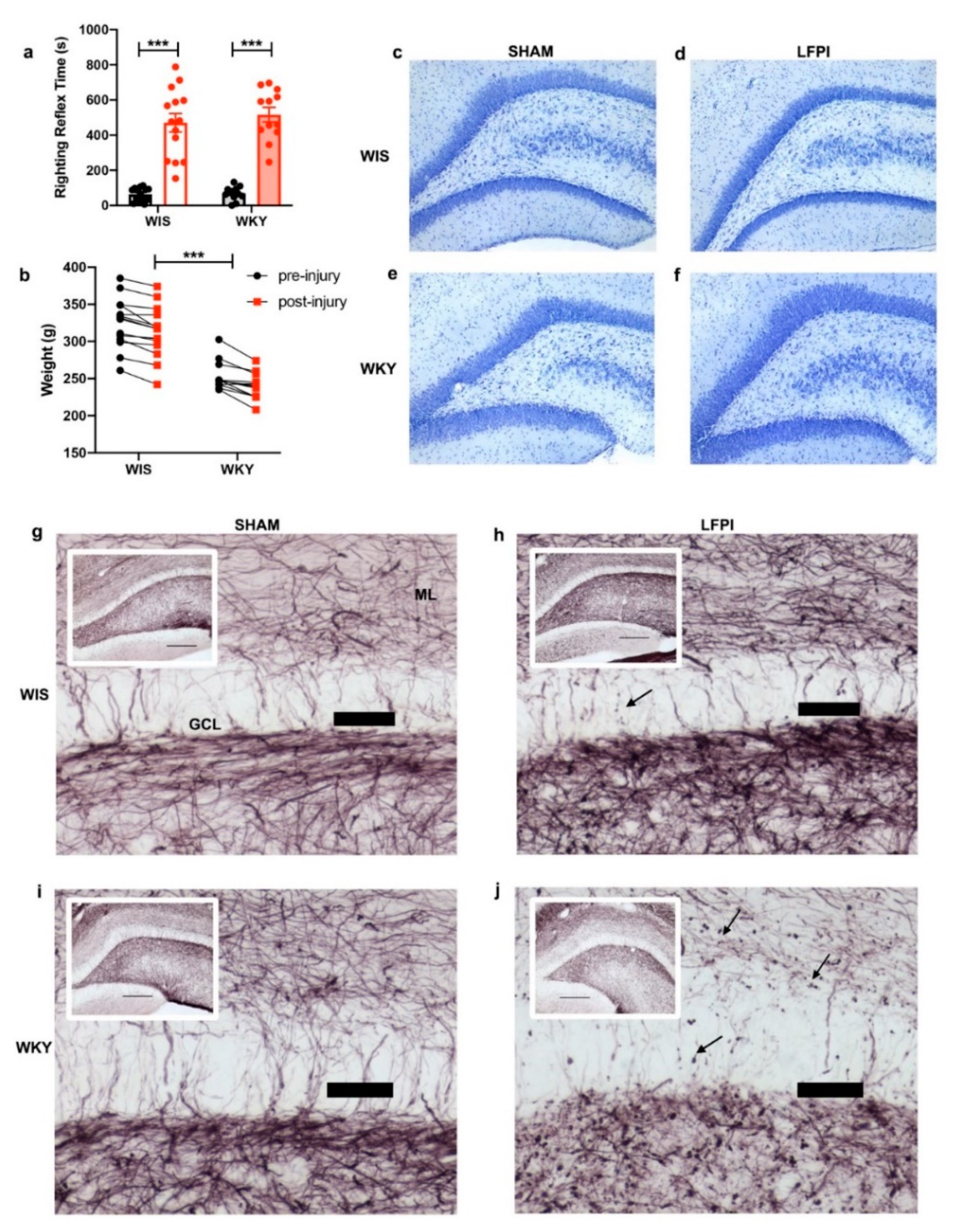
2.1. WKY Animals Show Deficits in Behavior Consistent with a Depression Phenotype
2.2. Similar Injury Phenotypes in WIS and WKY
2.3. AMPA Receptor Subunits Were Not Significantly Altered with Regards to Injury or Strain
2.4. Divergent Changes in NMDA Receptor Subunit Expression with Injury
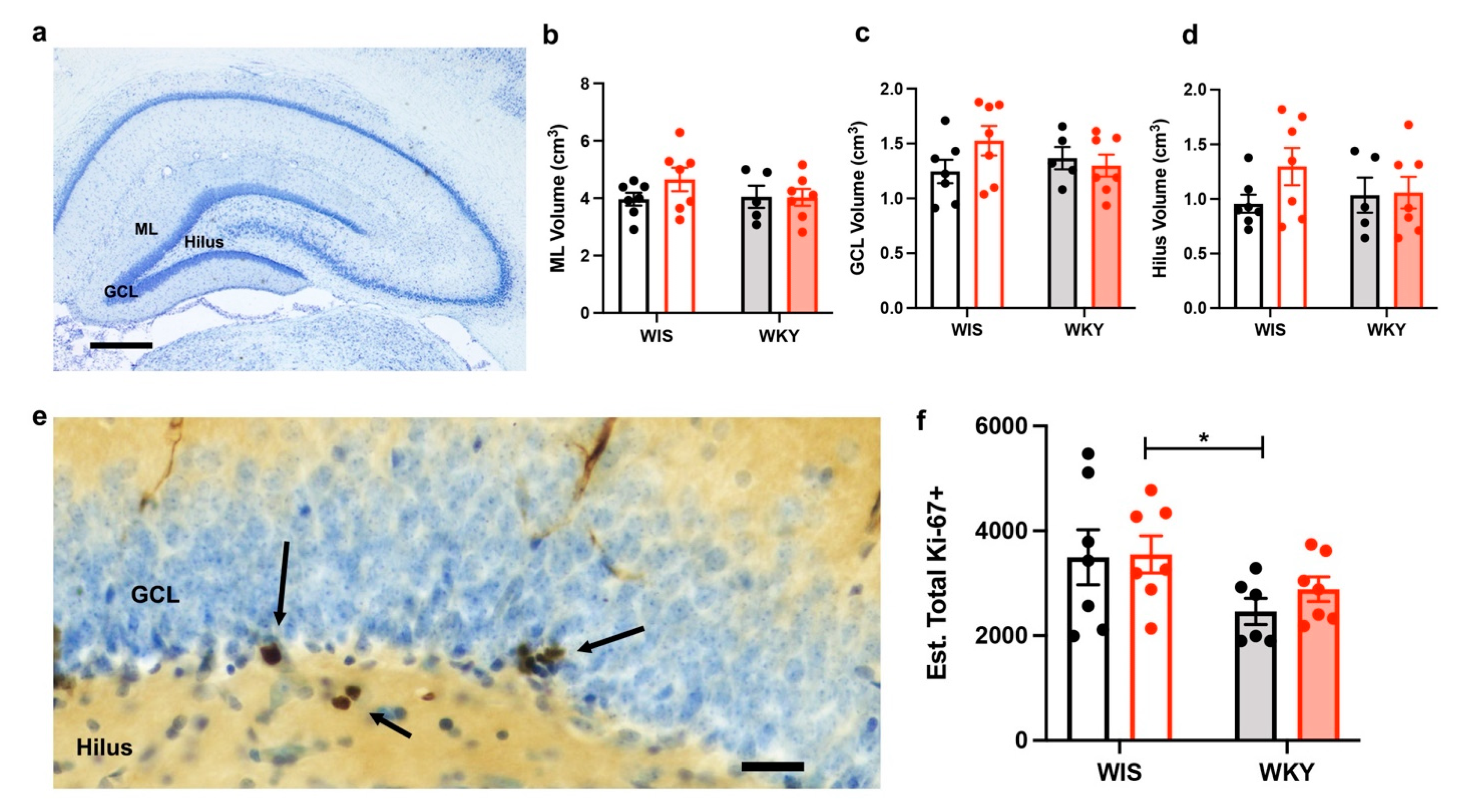
2.5. No Changes in the Dentate Gyrus Volume after Injury
2.6. WKY Animals Have Fewer Ki-67+ Cells but No Change with Injury
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Behavioral Testing
4.2.1. Open Field Activity Task
4.2.2. Radial Arm Maze
4.2.3. Novel Object Recognition
4.3. Lateral Fluid Percussion Injury
4.4. Tissue Acquisition
4.5. Immunohistochemistry and Histological Analysis
4.5.1. Nissl Staining and Analysis of Dentate Gyrus Volume
4.5.2. Gold Chloride Staining
4.5.3. Ki-67 Immunohistochemistry and Analysis
4.6. Immunoblotting
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, C.A.; Bell, J.M.; Breiding, M.J.; Xu, L. Traumatic Brain Injury–Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR Surveill. Summ. 2017, 66, 1–16. [Google Scholar] [CrossRef]
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Fann, J.R. Psychiatric illness and subsequent traumatic brain injury: A case control study. J. Neurol. Neurosurg. Psychiatry 2002, 72, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassallo, J.L.; Proctor-Weber, Z.; Lebowitz, B.K.; Curtiss, G.; Vanderploeg, R.D. Psychiatric risk factors for traumatic brain injury. Brain Inj. 2007, 21, 567–573. [Google Scholar] [CrossRef]
- Preece, M.H.; Geffen, G.M. The contribution of pre-existing depression to the acute cognitive sequelae of mild traumatic brain injury. Brain Inj. 2007, 21, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.G.; Bracken, M.B.; Clark, A.N.; Nick, T.G.; Melguizo, M.S.; Sander, A.M. Relationship of preinjury depressive symptoms to outcomes 3 mos after complicated and uncomplicated mild traumatic brain injury. Am. J. Phys. Med. Rehabil. 2014, 93, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef]
- Lang, U.E.; Borgwardt, S. Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef]
- Campbell, S.; MacQueen, G. An update on regional brain volume differences associated with mood disorders. Curr. Opin. Psychiatry 2006, 19, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Videbech, P. Hippocampal Volume and Depression: A Meta-Analysis of MRI Studies. Am. J. Psychiatry 2004, 161, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin. Neurosci. 2018, 72, 3–12. [Google Scholar] [CrossRef]
- Banasr, M.; Valentine, G.W.; Li, X.-Y.; Gourley, S.L.; Taylor, J.R.; Duman, R.S. Chronic Unpredictable Stress Decreases Cell Proliferation in the Cerebral Cortex of the Adult Rat. Biol. Psychiatry 2007, 62, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Aan Het Rot, M.; Mathew, S.J.; Charney, D.S. Neurobiological mechanisms in major depressive disorder. Can. Med. Assoc. J. 2009, 180, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Duman, R.S.; Sanacora, G.; Krystal, J.H. Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 2019, 102, 75–90. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aan Het Rot, M.; Zarate, C.A.; Charney, D.S.; Mathew, S.J. Ketamine for Depression: Where Do We Go from Here? Biol. Psychiatry 2012, 72, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- O’Neil, D.A.; Nicholas, M.A.; Lajud, N.; Kline, A.E.; Bondi, C.O. Preclinical Models of Traumatic Brain Injury: Emerging Role of Glutamate in the Pathophysiology of Depression. Front. Pharmacol. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuire, J.L.; Ngwenya, L.B.; McCullumsmith, R.E. Neurotransmitter changes after traumatic brain injury: An update for new treatment strategies. Mol. Psychiatry 2019, 24, 995–1012. [Google Scholar] [CrossRef]
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; DePasquale, E.A.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsett, C.R.; McGuire, J.L.; DePasquale, E.A.; Gardner, A.E.; Floyd, C.L.; McCullumsmith, R.E. Glutamate Neurotransmission in Rodent Models of Traumatic Brain Injury. J. Neurotrauma 2016, 34, 263–272. [Google Scholar] [CrossRef]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Aiba, I.; Shuttleworth, C.W. Sustained NMDA receptor activation by spreading depolarizations can initiate excitotoxic injury in metabolically compromised neurons. J. Physiol. 2012, 590, 5877–5893. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Rungta, R.L.; Malik, A.; Han, H.; Wu, D.C.; MacVicar, B.A. Regenerative glutamate release by presynaptic NMDA receptors contributes to spreading depression. J. Cereb. Blood Flow Metab. 2013, 33, 1582–1594. [Google Scholar] [CrossRef] [Green Version]
- Leao, A. Spreading depression of activity in the cerebral cortex. Physiology 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Barhorst, K.A.; Alfawares, Y.; McGuire, J.L.; Danzer, S.C.; Hartings, J.A.; Ngwenya, L.B. Remote and Persistent Alterations in Glutamate Receptor Subunit Composition Induced by Spreading Depolarizations in Rat Brain. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Andaluz, N.; Bullock, M.R.; Hinzman, J.M.; Mathern, B.; Pahl, C.; Puccio, A.; Shutter, L.A.; Strong, A.J.; Vagal, A.; et al. Prognostic Value of Spreading Depolarizations in Patients With Severe Traumatic Brain Injury. JAMA Neurol. 2020, 77, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Hartings, J.A.; Watanabe, T.; Bullock, M.R.; Okonkwo, D.O.; Fabricius, M.; Woitzik, J.; Dreier, J.P.; Puccio, A.; Shutter, L.A.; Pahl, C.; et al. Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain 2011, 134, 1529–1540. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Bullock, M.R.; Okonkwo, D.O.; Murray, L.S.; Murray, G.D.; Fabricius, M.; Maas, A.I.R.; Woitzik, J.; Sakowitz, O.; Mathern, B.; et al. Spreading depolarisations and outcome after traumatic brain injury: A prospective observational study. Lancet Neurol. 2011, 10, 1058–1064. [Google Scholar] [CrossRef]
- Hartings, J.; Ngwenya, L.B.; Carroll, C.P.; Foreman, B. Letter to the Editor: Ketamine sedation for the suppression of spreading depolarizations. J. Neurosurg. 2018, 130, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Hertle, D.N.; Dreier, J.P.; Woitzik, J.; Hartings, J.A.; Bullock, R.; Okonkwo, D.O.; Shutter, L.A.; Vidgeon, S.; Strong, A.J.; Kowoll, C.; et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain 2012, 135, 2390–2398. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.P.; Abbas, M.; Alunday, R.L.; Qeadan, F.; Shuttleworth, C.W. Spreading depolarization in acute brain injury inhibited by ketamine: A prospective, randomized, multiple crossover trial. J. Neurosurg. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakowitz, O.W.; Kiening, K.L.; Krajewski, K.L.; Sarrafzadeh, A.S.; Fabricius, M.; Strong, A.J.; Unterberg, A.W.; Dreier, J.P. Preliminary evidence that ketamine inhibits spreading depolarizations in acute human brain injury. Stroke 2009, 40, e519–e522. [Google Scholar] [CrossRef]
- Greene, J.; Greenamyre, J. Bioenergetics and glutamate excitotoxicity. Prog. Neurobiol. 1996, 48, 613–634. [Google Scholar] [CrossRef]
- Overstreet, D.H. Modeling depression in animal models. Methods Mol. Biol. 2012, 829, 125–144. [Google Scholar] [CrossRef]
- Aleksandrova, L.R.; Wang, Y.T.; Phillips, A.G. Evaluation of the Wistar-Kyoto rat model of depression and the role of synaptic plasticity in depression and antidepressant response. Neurosci. Biobehav. Rev. 2019, 105, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Malkesman, O.; Braw, Y.; Weller, A. Assessment of antidepressant and anxiolytic properties of NK1 antagonists and Substance P in Wistar Kyoto rats. Physiol. Behav. 2007, 90, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Carr, G.V.; Bangasser, D.A.; Bethea, T.; Young, M.; Valentino, R.J.; Lucki, I. Antidepressant-Like Effects of κ-Opioid Receptor Antagonists in Wistar Kyoto Rats. Neuropsychopharmacology 2010, 35, 752–763. [Google Scholar] [CrossRef]
- Paré, W.P. The performance of WKY rats on three tests of emotional behavior. Physiol. Behav. 1992, 51, 1051–1056. [Google Scholar] [CrossRef]
- Stanford, S.C. The Open Field Test: Reinventing the wheel. J. Psychopharmacol. 2007, 21, 134–135. [Google Scholar] [CrossRef]
- Belovicova, K.; Bogi, E.; Csatlosova, K.; Dubovicky, M. Animal tests for anxiety-like and depression-like behavior in rats. Interdiscip. Toxicol. 2017, 10, 40–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denenberg, V.H. Open-Field Behavior in the Rat: What Does It Mean? Ann. N. Y. Acad. Sci. 1969, 159, 852–859. [Google Scholar] [CrossRef]
- Hall, C.; Ballachey, E.L. A study of the rat’s behavior in a field. A contribution to method in comparative psychology. Univ. Calif. Publ. Psychol. 1932, 6, 1–12. [Google Scholar]
- Gould, N.F.; Holmes, M.K.; Fantie, B.D.; Luckenbaugh, D.A.; Pine, D.S.; Gould, T.D.; Burgess, N.; Manji, H.K.; Zarate, C.A. Performance on a Virtual Reality Spatial Memory Navigation Task in Depressed Patients. Am. J. Psychiatry 2007, 164, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Grauer, E.; Kapon, Y. Wistar-Kyoto rats in the Morris water maze: Impaired working memory and hyper-reactivity to stress. Behav. Brain Res. 1993, 59, 147–151. [Google Scholar] [CrossRef]
- Clements, K.; Wainwright, P. Spontaneously hypertensive, Wistar-Kyoto and Sprague-Dawley rats differ in performance on a win-shift task in the water radial arm maze. Behav. Brain Res. 2006, 167, 295–304. [Google Scholar] [CrossRef]
- Shoval, G.; Shbiro, L.; Hershkovitz, L.; Hazut, N.; Zalsman, G.; Mechoulam, R.; Weller, A. Prohedonic Effect of Cannabidiol in a Rat Model of Depression. Neuropsychobiology 2016, 73, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Slattery, D.A.; Cryan, J.F. Using the rat forced swim test to assess antidepressant-like activity in rodents. Nat. Protoc. 2012, 7, 1009–1014. [Google Scholar] [CrossRef]
- Castagné, V.; Porsolt, R.D.; Moser, P. Use of latency to immobility improves detection of antidepressant-like activity in the behavioral despair test in the mouse. Eur. J. Pharmacol. 2009, 616, 128–133. [Google Scholar] [CrossRef]
- McNally, K.J.; Peters, A. A New Method for Intense Staining of Myelin. J. Histochem. Cytochem. 1998, 46, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Witherspoon, J.; Smirnova, I.; McIff, T. Improved gold chloride staining method for anatomical analysis of sensory nerve endings in the shoulder capsule and labrum as examples of loose and dense fibrous tissues. Biotech. Histochem. 2014, 89, 355–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchet, M.D.; Gotlib, I.H. Myelination of the brain in Major Depressive Disorder: An in vivo quantitative magnetic resonance imaging study. Sci. Rep. 2017, 7, 2200. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.-W.; Williams, R.A.; Lescher, J.D.; Jikaria, N.; Turtzo, L.C.; Frank, J.A. Radiological-pathological correlation of diffusion tensor and magnetization transfer imaging in a closed head traumatic brain injury model. Ann. Neurol. 2016, 79, 907–920. [Google Scholar] [CrossRef] [Green Version]
- Hylin, M.J.; Orsi, S.A.; Zhao, J.; Bockhorst, K.; Perez, A.; Moore, A.N.; Dash, P.K. Behavioral and Histopathological Alterations Resulting from Mild Fluid Percussion Injury. J. Neurotrauma 2013, 30, 702–715. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.C.; Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M. White matter involvement after TBI: Clues to axon and myelin repair capacity. Exp. Neurol. 2016, 275, 328–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA Imbalance Following Traumatic Brain Injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M.; Kallarackal, A.J.; Kvarta, M.D.; Van Dyke, A.M.; Legates, T.A.; Cai, X. An excitatory synapse hypothesis of depression. Trends Neurosci. 2015, 38, 279–294. [Google Scholar] [CrossRef] [Green Version]
- Kallarackal, A.J.; Kvarta, M.D.; Cammarata, E.; Jaberi, L.; Cai, X.; Bailey, A.M.; Thompson, S.M. Chronic Stress Induces a Selective Decrease in AMPA Receptor-Mediated Synaptic Excitation at Hippocampal Temporoammonic-CA1 Synapses. J. Neurosci. 2013, 33, 15669–15674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, C.M.; Chen, S.; Alonso, O.F.; Dietrich, W.D.; Hu, B.-R. Activation of Calcium/Calmodulin-Dependent Protein Kinases after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2006, 26, 1507–1518. [Google Scholar] [CrossRef] [Green Version]
- Kharlamov, E.A.; Lepsveridze, E.; Meparishvili, M.; Solomonia, R.O.; Lu, B.; Miller, E.R.; Kelly, K.M.; Mtchedlishvili, Z. Alterations of GABAA and glutamate receptor subunits and heat shock protein in rat hippocampus following traumatic brain injury and in posttraumatic epilepsy. Epilepsy Res. 2011, 95, 20–34. [Google Scholar] [CrossRef]
- Bell, J.D.; Ai, J.; Chen, Y.; Baker, A.J. Mild in vitro trauma induces rapid Glur2 endocytosis, robustly augments calcium permeability and enhances susceptibility to secondary excitotoxic insult in cultured Purkinje cells. Brain 2007, 130, 2528–2542. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.D.; Park, E.; Ai, J.; Baker, A.J. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009, 16, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.J.; Zukin, R.S. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [PubMed]
- Köhr, G. NMDA receptor function: Subunit composition versus spatial distribution. Cell Tissue Res. 2006, 326, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.P.; Ventre, S.C.; Geddes-Klein, D.; Singh, P.K.; Meaney, D.F. Single-Neuron NMDA Receptor Phenotype Influences Neuronal Rewiring and Reintegration following Traumatic Injury. J. Neurosci. 2014, 34, 4200–4213. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, J.A.; Schepmann, D.; Frehland, B.; Thum, S.; Datunashvili, M.; Budde, T.; Hollmann, M.; Strutz-Seebohm, N.; Wünsch, B.; Seebohm, G. A common mechanism allows selective targeting of GluN2B subunit-containing N-methyl-D-aspartate receptors. Commun. Biol. 2019, 2, 420. [Google Scholar] [CrossRef]
- Lei, Y.; Tejani-Butt, S.M. N-methyl-D-aspartic acid receptors are altered by stress and alcohol in Wistar–Kyoto rat brain. Neuroscience 2010, 169, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. The use of total protein stains as loading controls: An alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 2008, 172, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Eaton, S.L.; Roche, S.L.; Llavero Hurtado, M.; Oldknow, K.J.; Farquharson, C.; Gillingwater, T.H.; Wishart, T.M. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS ONE 2013, 8, e72457. [Google Scholar] [CrossRef]
- Cominski, T.P.; Jiao, X.; Catuzzi, J.E.; Stewart, A.L.; Pang, K.C.H. The Role of the Hippocampus in Avoidance Learning and Anxiety Vulnerability. Front. Behav. Neurosci. 2014, 8, 273. [Google Scholar] [CrossRef] [Green Version]
- Gormley, S.; Rouine, J.; McIntosh, A.; Kerskens, C.; Harkin, A. Glial fibrillary acidic protein (GFAP) immunoreactivity correlates with cortical perfusion parameters determined by bolus tracking arterial spin labelling (bt-ASL) magnetic resonance (MR) imaging in the Wistar Kyoto rat. Physiol. Behav. 2016, 160, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, G.; Lippoldt, A.; Kempermann, G. Two genetic rat models of arterial hypertension show different mechanisms by which adult hippocampal neurogenesis is increased. Dev. Neurosci. 2007, 29, 124–133. [Google Scholar] [CrossRef]
- Kin, K.; Yasuhara, T.; Kameda, M.; Agari, T.; Sasaki, T.; Morimoto, J.; Okazaki, M.; Umakoshi, M.; Kuwahara, K.; Kin, I.; et al. Hippocampal neurogenesis of Wistar Kyoto rats is congenitally impaired and correlated with stress resistance. Behav. Brain Res. 2017, 329, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Chirumamilla, S.; Sun, D.; Bullock, M.R.; Colello, R.J. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma 2002, 19, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Mach, S.A.; Moore, A.N. Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J. Neurosci. Res. 2001, 63, 313–319. [Google Scholar] [CrossRef]
- Correll, E.A.; Ramser, B.J.; Knott, M.V.; McCullumsmith, R.E.; McGuire, J.L.; Ngwenya, L.B. Deficits in pattern separation and dentate gyrus proliferation after rodent lateral fluid percussion injury. IBRO Neurosci. Rep. 2021, 10, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Colello, R.J.; Daugherty, W.P.; Kwon, T.H.; McGinn, M.J.; Harvey, H.B.; Bullock, M.R. Cell proliferation and neuronal differentiation in the dentate gyrus in juvenile and adult rats following traumatic brain injury. J. Neurotrauma 2005, 22, 95–105. [Google Scholar] [CrossRef]
- Gaulke, L.J.; Horner, P.J.; Fink, A.J.; McNamara, C.L.; Hicks, R.R. Environmental enrichment increases progenitor cell survival in the dentate gyrus following lateral fluid percussion injury. Brain Res. Mol. Brain Res. 2005, 141, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Encinas, J.M.; Sierra, A. Neural stem cell deforestation as the main force driving the age-related decline in adult hippocampal neurogenesis. Behav. Brain. Res. 2012, 227, 433–439. [Google Scholar] [CrossRef]
- Neuberger, E.J.; Swietek, B.; Corrubia, L.; Prasanna, A.; Santhakumar, V. Enhanced Dentate Neurogenesis after Brain Injury Undermines Long-Term Neurogenic Potential and Promotes Seizure Susceptibility. Stem Cell Rep. 2017, 9, 972–984. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Rakoczy, S.; Brown-Borg, H. Assessment of spatial memory in mice. Life Sci. 2010, 87, 521–536. [Google Scholar] [CrossRef]
- McGuire, J.L.; Correll, E.A.; Lowery, A.C.; Rhame, K.; Anwar, F.N.; McCullumsmith, R.E.; Ngwenya, L.B. Pioglitazone improves working memory performance when administered in chronic TBI. Neurobiol. Dis. 2019, 132, 104611. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.L.; DePasquale, E.A.K.; Watanabe, M.; Anwar, F.; Ngwenya, L.B.; Atluri, G.; Romick-Rosendale, L.E.; McCullumsmith, R.E.; Evanson, N.K. Chronic Dysregulation of Cortical and Subcortical Metabolism after Experimental Traumatic Brain Injury. Mol. Neurobiol. 2018, 56, 2908–2921. [Google Scholar] [CrossRef] [PubMed]
- Au-Hagihara, H.; Au-Toyama, K.; Au-Yamasaki, N.; Au-Miyakawa, T. Dissection of Hippocampal Dentate Gyrus from Adult Mouse. J. Vis. Exp. 2009, e1543. [Google Scholar] [CrossRef] [Green Version]
- Schmued, L.C. A rapid, sensitive histochemical stain for myelin in frozen brain sections. J. Histochem. Cytochem. 1990, 38, 717–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngwenya, L.B.; Peters, A.; Rosene, D.L. Light and electron microscopic immunohistochemical detection of bromodeoxyuridine-labeled cells in the brain: Different fixation and processing protocols. J. Histochem. Cytochem. 2005, 53, 821–832. [Google Scholar] [CrossRef]
- Kirshner, Z.Z.; Gibbs, R.B. Use of the REVERT((R)) total protein stain as a loading control demonstrates significant benefits over the use of housekeeping proteins when analyzing brain homogenates by Western blot: An analysis of samples representing different gonadal hormone states. Mol. Cell Endocrinol. 2018, 473, 156–165. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knott, M.V.; Ngwenya, L.B.; Correll, E.A.; Bohnert, J.; Ziemba, N.J.; Allgire, E.; Hopkins, T.; McGuire, J.L. Lack of Glutamate Receptor Subunit Expression Changes in Hippocampal Dentate Gyrus after Experimental Traumatic Brain Injury in a Rodent Model of Depression. Int. J. Mol. Sci. 2021, 22, 8086. https://doi.org/10.3390/ijms22158086
Knott MV, Ngwenya LB, Correll EA, Bohnert J, Ziemba NJ, Allgire E, Hopkins T, McGuire JL. Lack of Glutamate Receptor Subunit Expression Changes in Hippocampal Dentate Gyrus after Experimental Traumatic Brain Injury in a Rodent Model of Depression. International Journal of Molecular Sciences. 2021; 22(15):8086. https://doi.org/10.3390/ijms22158086
Chicago/Turabian StyleKnott, Maxon V., Laura B. Ngwenya, Erika A. Correll, Judy Bohnert, Noah J. Ziemba, Emily Allgire, Tracy Hopkins, and Jennifer L. McGuire. 2021. "Lack of Glutamate Receptor Subunit Expression Changes in Hippocampal Dentate Gyrus after Experimental Traumatic Brain Injury in a Rodent Model of Depression" International Journal of Molecular Sciences 22, no. 15: 8086. https://doi.org/10.3390/ijms22158086