
NMDARs Containing NR2B Subunit Do Not Contribute to the LTP Form of Hippocampal Plasticity: In Vivo Pharmacological Evidence in Rats


Abstract
1. Introduction
2. Results
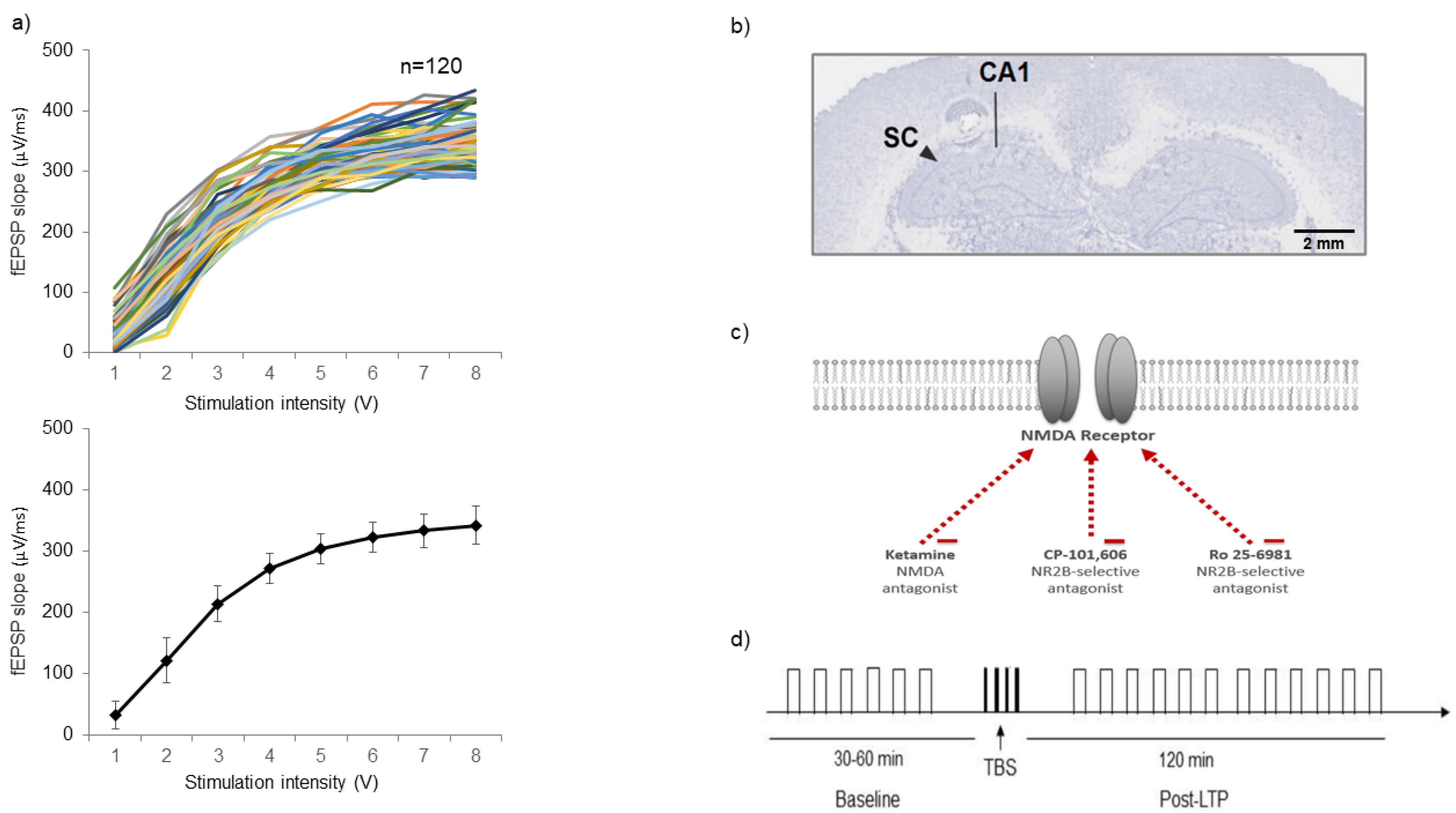
2.1. Input/Output (I/O) Criteria
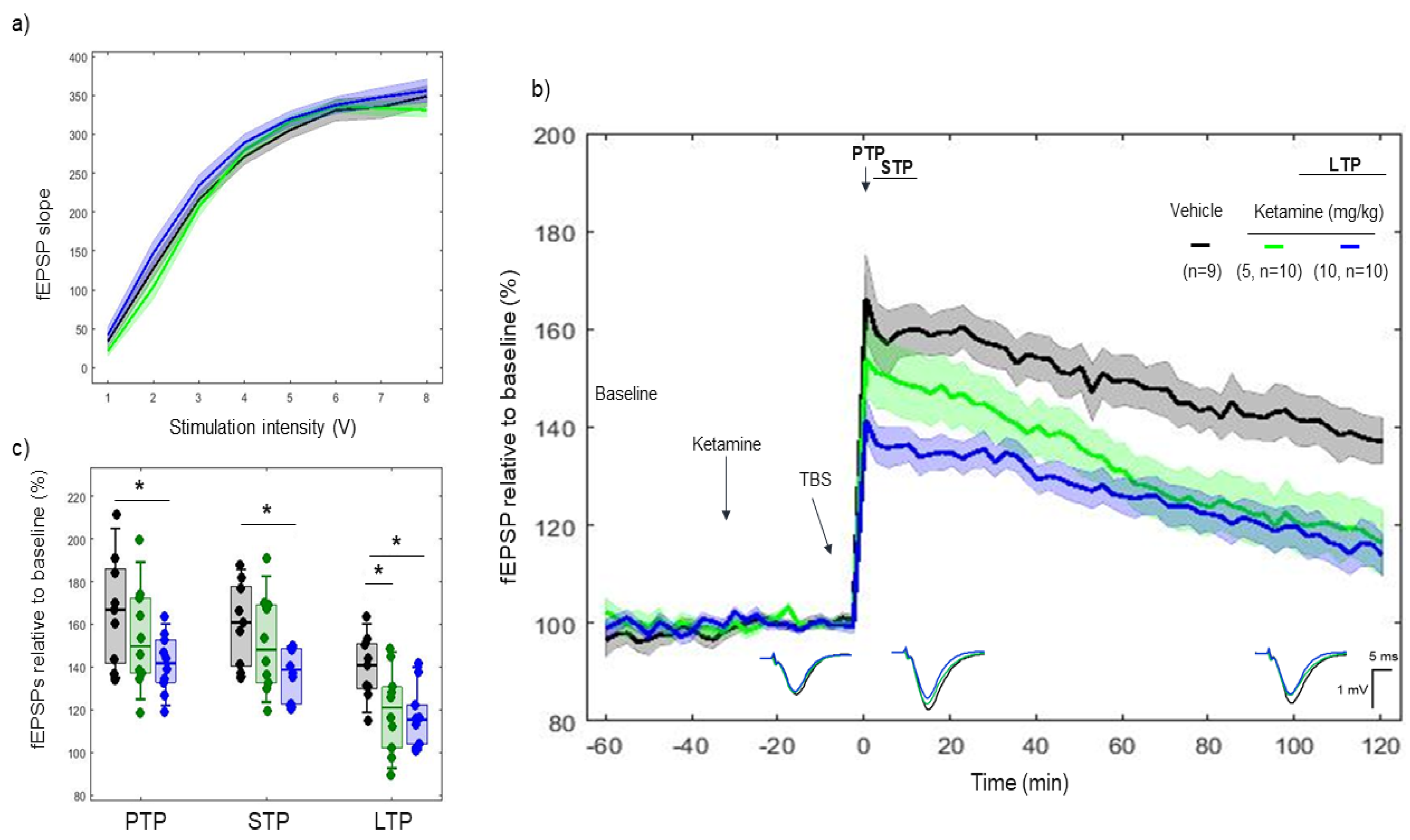
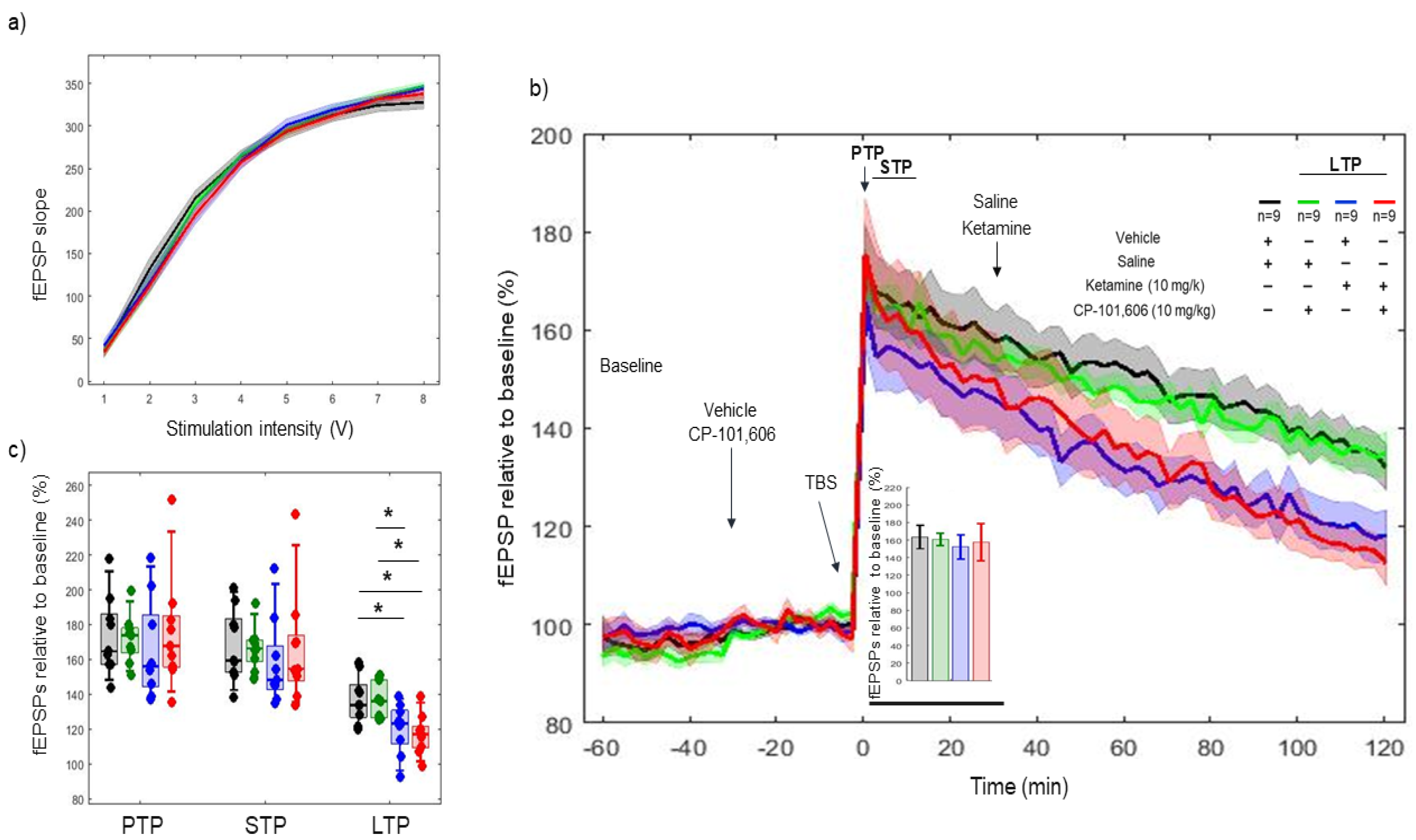
2.2. Effects of Ketamine on 5xTBS LTP Response
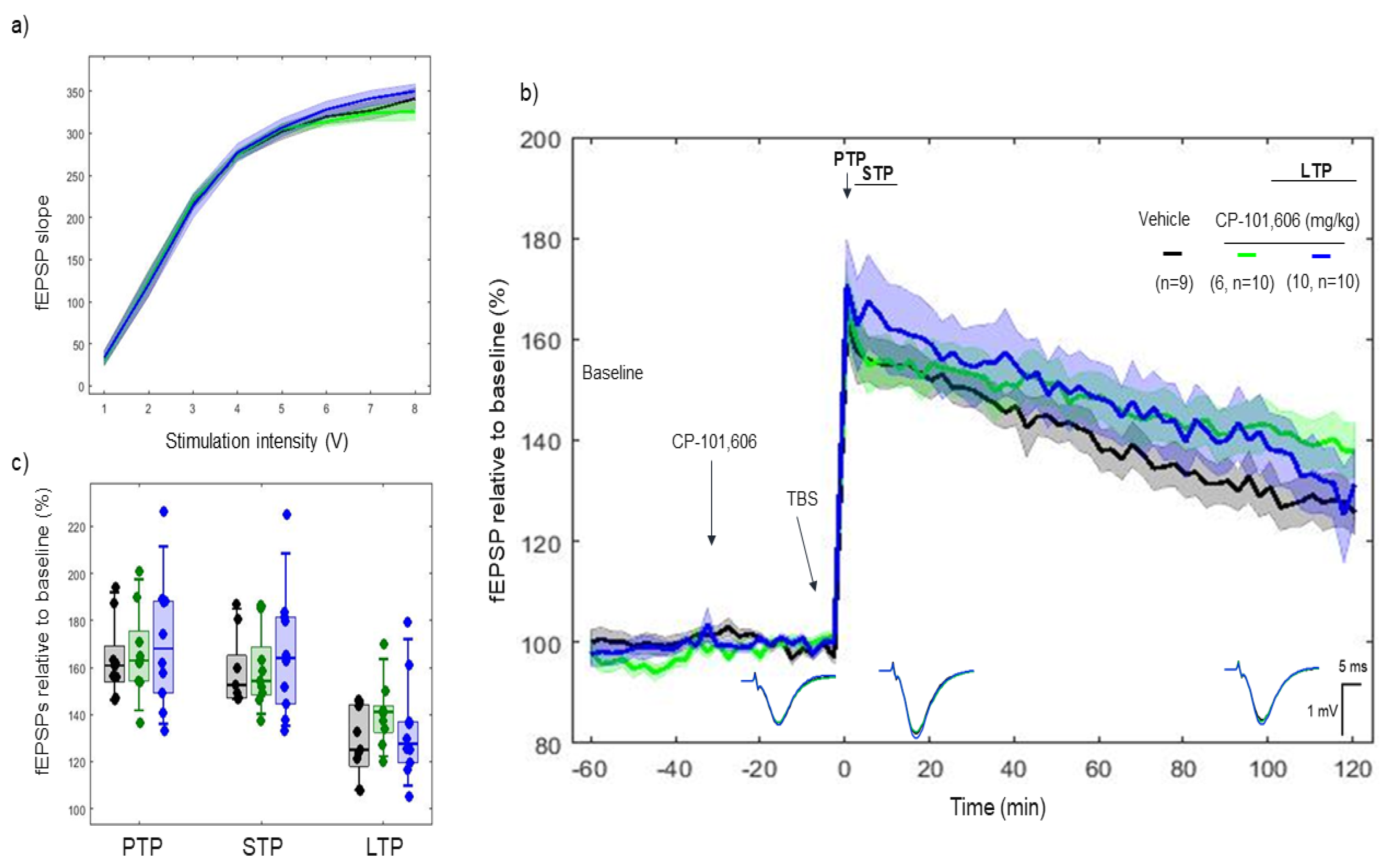
2.3. Effects of CP-101606 on 5xTBS LTP Response
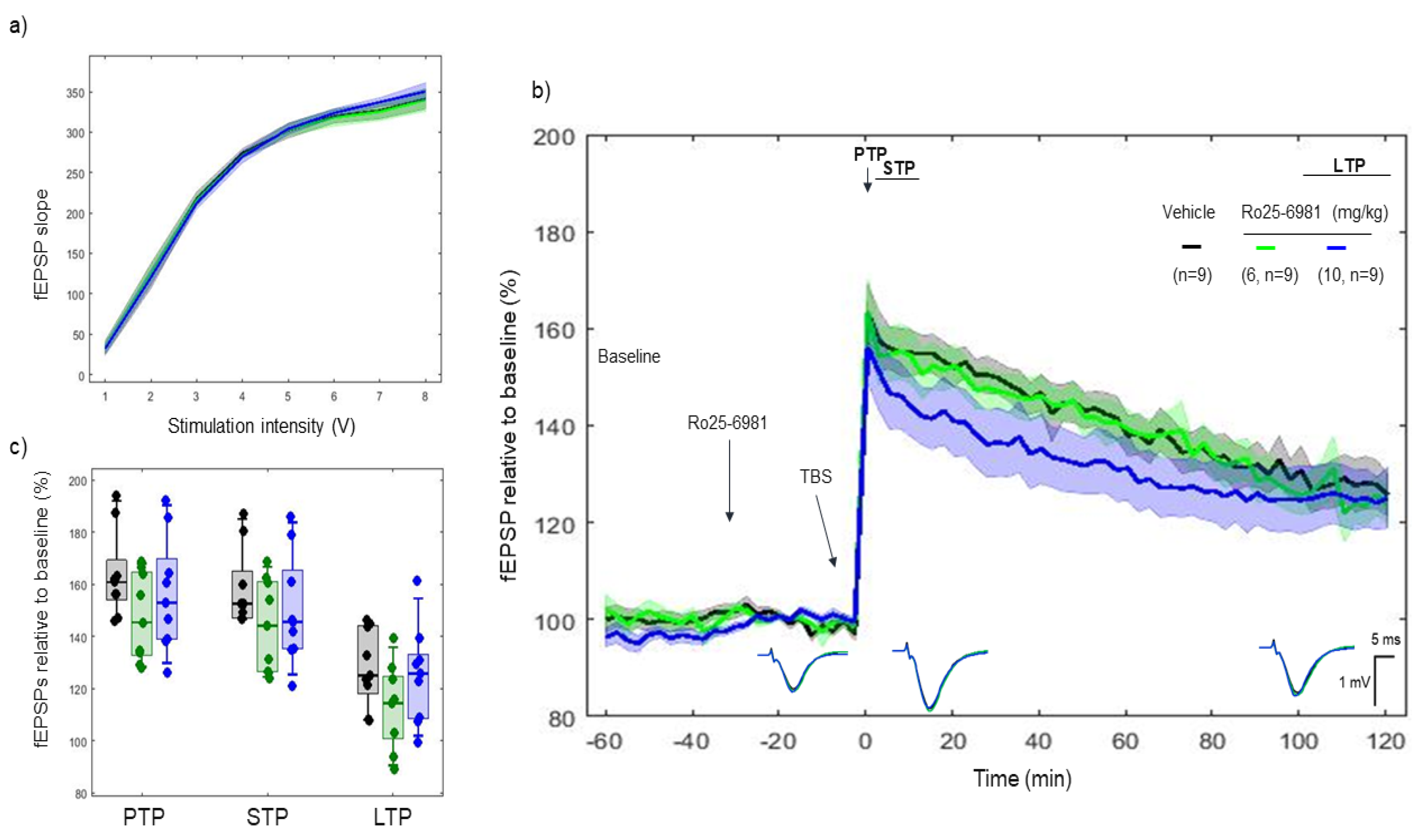
2.4. Effects of Ro 25-6981 on 5xTBS LTP Response
2.5. Effects of Combined Pharmacology of CP-101606 and Ketamine on 5xTBS LTP Response
3. Discussion
3.1. Ketamine, a Low-Affinity Antagonist of the NMDA Receptor, Decreased the LTP Response
3.2. CP-101606 and Ro 25-6981, High-Affinity and Selective NR2B Antagonists of the NR2B Subunit, Did Not Modify the LTP Response
3.3. In Combined Pharmacology, CP-101606 Did Not Influence the Effects of Ketamine
3.4. Perspective
3.5. Conclusion
4. Materials and Methods
4.1. Animals and Surgical Procedures
4.2. Input/Output Functions
4.3. In Vivo Electrophysiology LTP Protocol
4.4. Histology
4.5. Drugs
4.6. Statistical Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bliss, T.V.P.; Collingridge, G.L.; Morris, R.G.M. Synaptic plasticity in health and disease: Introduction and overview. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130129. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The dual role of glutamatergic neurotransmission in Alzheimer’s disease: From pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Cooke, S.F.; Bliss, T.V. Plasticity in the human central nervous system. Brain 2006, 129, 1659–1673. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Brickley, S.; Farrant, M. NMDA receptor subunits: Diversity, development and disease. Curr. Opin. Neurobiol. 2001, 11, 327–335. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Synaptic Plasticity, Dementia and Alzheimer Disease. CNS & Neurological Disorders-Drug Targets. CNS Neurol. Disord. Drug Targets 2017, 16, 220–233. [Google Scholar] [PubMed]
- Styr, B.; Slutsky, I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 2018, 21, 463–473. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Liu, L.; Wong, T.P.; Pozza, M.F.; Lingenhoehl, K.; Wang, Y.; Sheng, M.; Auberson, Y.P.; Wang, Y.T. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science 2004, 304, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Huang, F.S.; Abbas, A.K.; Wigström, H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007, 8, 55. [Google Scholar] [CrossRef]
- Ramirez, A.; Arbuckle, M.R. Synaptic Plasticity: The Role of Learning and Unlearning in Addiction and Beyond. Biol. Psychiatry 2016, 80, e73–e75. [Google Scholar] [CrossRef]
- Whitlock, J.R.; Heynen, A.J.; Shuler, M.G.; Bear, M.F. Learning induces long-term potentiation in the hippocampus. Science 2006, 313, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Neyton, J. NMDA receptor subunits: Function and pharmacology. Curr. Opin. Pharmacol. 2007, 7, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, T.E.; Bannister, N.J.; Collett, V.J.; Dargan, S.L.; Massey, P.V.; Bortolotto, Z.A.; Fitzjohn, S.M.; Bashir, Z.I.; Collingridge, G.L.; Lodge, D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 2007, 52, 60–70. [Google Scholar] [CrossRef]
- Chapman, D.E.; Keefe, K.A.; Wilcox, K.S. Evidence for functionally distinct synaptic NMDA receptors in ventromedial versus dorsolateral striatum. J. Neurophysiol. 2003, 89, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Landwehrmeyer, G.B.; Standaert, D.G.; Testa, C.M.; Penney, J.B., Jr.; Young, A.B. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J. Neurosci. 1995, 15, 5297–5307. [Google Scholar] [CrossRef]
- Morgan, S.L.; Teyler, T.J. Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J. Neurophysiol. 2001, 86, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Ohmura, T.; Komatsu, Y. Two forms of synaptic plasticity with distinct dependence on age, experience, and NMDA receptor subtype in rat visual cortex. J. Neurosci. 2003, 23, 6557–6566. [Google Scholar] [CrossRef]
- Hogsden, J.L.; Dringenberg, H.C. Decline of long-term potentiation (LTP) in the rat auditory cortex in vivo during postnatal life: Involvement of NR2B subunits. Brain Res. 2009, 1283, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Janssen, W.G.M.; Vissavajjhala, P.; Andrews, G.; Moran, T.; Hof, P.R.; Morrison, J.H. Cellular and synaptic distribution of NR2A and NR2B in macaque monkey and rat hippocampus as visualized with subunit-specific monoclonal antibodies. Exp. Neurol. 2005, 191 (Suppl. 1), S28–S44. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Law, A.J.; Weickert, C.S.; Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Harrison, P.J. Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. Eur. J. Neurosci. 2003, 18, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S.R.; Heresco-Levy, U.; Umbricht, D. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr. Bull. 2012, 38, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, B.; Brown, R.E.; McCarley, R.W.; Hajos, M. Impact of ketamine on neuronal network dynamics: Translational modeling of schizophrenia-relevant deficits. CNS Neurosci. Ther. 2013, 19, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Krystal, J.H. Capturing the angel in "angel dust": Twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr. Bull. 2012, 38, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Stoiljkovic, M.; Menniti, F.S.; Hajós, M. Differential effects of an NR2B NAM and ketamine on synaptic potentiation and gamma synchrony: Relevance to rapid-onset antidepressant efficacy. Neuropsychopharmacology 2016, 41, 1486–1494. [Google Scholar] [CrossRef][Green Version]
- Smalheiser, N.R. Ketamine: A neglected therapy for Alzheimer disease. Vol. 10, Frontiers in Aging Neuroscience. Front. Aging Neurosci. 2019, 11, 186. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Z.; Kumar, M.; Xu, G.; Zhang, M. Neuroprotective potential of ketamine prevents developing brain structure impairment and alteration of neurocognitive function induced via isoflurane through the PI3K/AKT/GSK-3β pathway. Drug Des. Dev. Ther. 2019, 13, 501–512. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Krystal, J.H.; Sanacora, G.; Duman, R.S. Rapid-acting glutamatergic antidepressants: The path to ketamine and beyond. Biol. Psychiatry 2013, 73113, 1133–1141. [Google Scholar] [CrossRef]
- McGirr, A.; Berlim, M.T.; Bond, D.J.; Fleck, M.P.; Yatham, L.N.; Lam, R.W. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol. Med. 2015, 45, 693–704. [Google Scholar] [CrossRef]
- Chazot, P.L. CP-101606 Pfizer Inc. Curr. Opin. Investig. Drugs 2000, 1, 370–374. [Google Scholar]
- Gardoni, F.; Mauceri, D.; Malinverno, M.; Polli, F.; Costa, C.; Tozzi, A.; Siliquini, S.; Picconi, B.; Cattabeni, F.; Calabresi, P.; et al. Decreased NR2B subunit synaptic levels cause impaired long-term potentiation but not long-term depression. J. Neurosci. 2009, 29, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Kopp, C.; Longordo, F.; Luthi, A. Experience-dependent changes in NMDA receptor composition at mature central synapses. Neuropharmacology 2007, 53, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Monfort, P.; Felipo, V. Hippocampal long-term potentiation is reduced in mature compared to young male rats but not in female rats. Neuroscience 2007, 146, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Laing, M.D.; Bashir, Z.I. Presynaptic NR2A-containing NMDARs are required for LTD between the amygdala and the perirhinal cortex: A potential mechanism for the emotional modulation of memory? eNeuro 2015, 2, ENEURO.0046–14.2015. [Google Scholar] [CrossRef]
- deBacker, J.; Hawken, E.R.; Normandeau, C.P.; Jones, A.A.; Di Prospero, C.; Mechefske, E.; Gardner Gregory, J.; Hayton, S.J.; Dumont, É.C. GluN2B-containing NMDA receptors blockade rescues bidirectional synaptic plasticity in the bed nucleus of the stria terminalis of cocaine self-administering rats. Neuropsychopharmacology 2015, 40, 394–405. [Google Scholar] [CrossRef]
- France, G.; Fernández-Fernández, D.; Burnell, E.S.; Irvine, M.W.; Monaghan, D.T.; Jane, D.E.; Bortolotto, Z.A.; Collingridge, G.L.; Volianskis, A. Multiple roles of GluN2B-containing NMDA receptors in synaptic plasticity in juvenile hippocampus. Neuropharmacology 2017, 112, 76–83. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, A.Q.; Luo, X.Q.; Guo, L.X.; Tang, Y.; Bao, C.J.; Lin, L.; Lin, C. Hippocampal NR2B-containing NMDA receptors enhance long-term potentiation in rats with chronic visceral pain. Brain Res. 2014, 1570, 43–53. [Google Scholar] [CrossRef]
- Fox, C.J.; Russell, K.I.; Wang, Y.T.; Christie, B.R. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus 2006, 16, 907–915. [Google Scholar] [CrossRef]
- Menniti, F.; Chenard, B.; Collins, M.; Ducat, M.; Shalaby, I.; White, F. CP-101,606, a potent neuroprotectant selective for forebrain neurons. Eur. J. Pharmacol. 1997, 331, 117–126. [Google Scholar] [CrossRef]
- Mott, D.D.; Doherty, J.J.; Zhang, S.; Washburn, M.S.; Fendley, M.J.; Lyuboslavsky, P.; Traynelis, S.F.; Dingledine, R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat. Neurosci. 1998, 1, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Simorowski, N.; Furukawa, H. Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 2011, 475, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Albensi, B.C.; Oliver, D.R.; Toupin, J.; Capocchi, G. Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability. Are they effective or relevant? Exp. Neurol. 2007, 204, 1–13. [Google Scholar] [CrossRef]
- Grover, L.M.; Teyler, T.J. Two components of long-term potentiation induced by different patterns of afferent activation. Nature 1990, 347, 477–479. [Google Scholar] [CrossRef]
- Hölscher, C.; Anwyl, R.; Rowan, M.J. Stimulation on the positive phase of hippocampal theta rhythm induces long-term potentiation that can Be depotentiated by stimulation on the negative phase in area CA1 in vivo. J. Neurosci. 1997, 17, 6470–6477. [Google Scholar] [CrossRef]
- Zucker, R.S. Short-term synaptic plasticity. Annu. Rev. Neurosci. 1989, 12, 13–31. [Google Scholar] [CrossRef]
- Ahnaou, A.; White, E.; Biermans, R.; Manyakov, N.V.; Drinkenburg, W.H.I.M. In Vivo Plasticity at Hippocampal Schaffer Collateral-CA1 Synapses: Replicability of the LTP Response and Pharmacology in the Long-Evans Rat. Neural Plast. 2020, 2020, 6249375. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.E.; Nicoll, R.A. Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu. Rev. Physiol. 2016, 78, 351–365. [Google Scholar] [CrossRef]
- Kuhnt, U.; Voronin, L.L. Interaction between paired-pulse facilitation and long-term potentiation in area ca1 of guinea-pig hippocampal slices: Application of quantal analysis. Neuroscience 1994, 62, 391–397. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Malenka, R.C. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 1995, 377, 115–118. [Google Scholar] [CrossRef]
- Volianskis, A.; Collingridge, G.L.; Jensen, M.S. The roles of STP and LTP in synaptic encoding. PeerJ 2013, 1, e3. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Shaffer, C.L.; Osgood, S.M.; Smith, D.L.; Liu, J.; Trapa, P.E. Enhancing ketamine translational pharmacology via receptor occupancy normalization. Neuropharmacology 2014, 86, 174–180. [Google Scholar] [CrossRef]
- Sleigh, J.; Harvey, M.; Voss, L.; Denny, B. Ketamine-more mechanisms of action than just NMDA blockade. Trends Anaesth. Crit. Care 2014, 4, 76–81. [Google Scholar] [CrossRef]
- Zorumski, C.F.; Izumi, Y.; Mennerick, S. Ketamine: NMDA receptors and beyond. J. Neurosci. 2016, 36, 11158–11164. [Google Scholar] [CrossRef] [PubMed]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Iadarola, N.D.; Niciu, M.J.; Richards, E.M.; Vande Voort, J.L.; Ballard, E.D.; Lundin, N.B.; Nugent, A.C.; Machado-Vieira, R.; Zarate, C.A., Jr. Ketamine and other N-methyl-D-aspartate receptor antagonists in the treatment of depression: A perspective review. Ther. Adv. Chronic Dis. 2015, 6, 97–114. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate CAJr Gould, T.D. Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef]
- Chazot, P.L.; Lawrence, S.; Thompson, C.L. Studies on the subtype selectivity of CP-101,606: Evidence for two classes of NR2B-selective NMDA receptor antagonists. Neuropharmacology 2002, 42, 319–324. [Google Scholar] [CrossRef]
- Nash, J.E.; Ravenscroft, P.; McGuire, S.; Crossman, A.R.; Menniti, F.S.; Brotchie, J.M. The NR2B-selective NMDA receptor antagonist CP-101,606 exacerbates L-DOPA-induced dyskinesia and provides mild potentiation of anti-parkinsonian effects of L-DOPA in the MPTP-lesioned marmoset model of Parkinson’s disease. Exp. Neurol. 2004, 188, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Sivarao, D.V.; Chen, P.; Yang, Y.; Li, Y.W.; Pieschl, R.; Ahlijanian, M.K. NR2B Antagonist CP-101,606 Abolishes Pitch-Mediated Deviance Detection in Awake Rats. Front. Psychiatry 2014, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Keavy, D.; Bristow, L.J.; Sivarao, D.V.; Batchelder, M.; King, D.; Thangathirupathy, S.; Macor, J.E.; Weed, M.R. The qEEG Signature of Selective NMDA NR2B Negative Allosteric Modulators; A Potential Translational Biomarker for Drug Development. PLoS ONE 2016, 11, e0152729. [Google Scholar] [CrossRef]
- Al-Hallaq, R.A.; Conrads, T.P.; Veenstra, T.D.; Wenthold, R.J. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 2007, 27, 8334–8343. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, R.Q.; Gu, Q.H.; Yan, J.Z.; Wang, S.H.; Liu, S.Y.; Lu, W. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 2009, 29, 8764–8773. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Nabavi, S.; Malinow, R. Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Graef, J.D.; Newberry, K.; Newton, A.; Pieschl, R.; Shields, E.; Luan, F.N.; Simmermacher, J.; Luchetti, D.; Schaeffer, E.; Li, Y.W.; et al. Effect of acute NR2B antagonist treatment on long-term potentiation in the rat hippocampus. Brain Res. 2015, 1609, 31–39. [Google Scholar] [CrossRef]
- Rauner, C.; Köhr, G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J. Biol. Chem. 2011, 286, 7558–7566. [Google Scholar] [CrossRef] [PubMed]
- Berberich, S.; Punnakkal, P.; Jensen, V.; Pawlak, V.; Seeburg, P.H.; Hvalby, Ø.; Köhr, G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J. Neurosci. 2005, 25, 6907–6910. [Google Scholar] [CrossRef] [PubMed]
- Kellermayer, B.; Ferreira, J.S.; Dupuis, J.; Levet, F.; Grillo-Bosch, D.; Bard, L.; Linares-Loyez, J.; Bouchet, D.; Choquet, D.; Rusakov, D.A.; et al. Differential Nanoscale Topography and Functional Role of GluN2-NMDA Receptor Subtypes at Glutamatergic Synapses. Neuron 2018, 100, 106–119.e7. [Google Scholar] [CrossRef]
- Müller, L.; Tokay, T.; Porath, K.; Köhling, R.; Kirschstein, T. Enhanced NMDA receptor-dependent LTP in the epileptic CA1 area via upregulation of NR2B. Neurobiol. Dis. 2013, 54, 183–193. [Google Scholar] [CrossRef]
- Prakash, C.; Cui, D.; Potchoiba, M.J.; Butler, T. Metabolism, distribution and excretion of a selective N-methyl-D-aspartate receptor antagonist, traxoprodil, in rats and dogs. Drug Metab. Dispos. 2007, 35, 1350–1364. [Google Scholar] [CrossRef]
- Taylor, T.J.; Diringer, K.; Russell, T.; Venkatakrishnan, K.; Wilner, K.; Crownover, P.H.; Benincosa, L.J.; Gibbs, M.A. Absolute oral bioavailability of traxoprodil in cytochrome P450 2D6 extensive and poor metabolisers. J. Clin. Psychopharmacol. 2008, 28, 631–637. [Google Scholar] [CrossRef]
- Taylor, T.J.; Diringer, K.; Russell, T.; Venkatakrishnan, K.; Wilner, K.; Crownover, P.H.; Benincosa, L.J.; Gibbs, M.A. Absolute oral bioavailability of traxoprodil in cytochrome P450 2D6 extensive and poor metabolisers. Clin. Pharmacokinet. 2006, 45, 989–1001. [Google Scholar] [CrossRef]
- Wang, Y.H.; Bosy, T.Z.; Yasuda, R.P.; Grayson, D.R.; Vicini, S.; Pizzorusso, T.; Wolfe, B.B. Characterization of NMDA Receptor Subunit-Specific Antibodies: Distribution of NR2A and NR2B Receptor Subunits in Rat Brain and Ontogenic Profile in the Cerebellum. J. Neurochem. 1995, 65, 176–183. [Google Scholar] [CrossRef]
- Flint, A.; Maisch, U.S.; Weishaupt, J.H.; Kriegstein, A.R.; Monyer, H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J. Neurosci. 1997, 17, 2469–2476. [Google Scholar] [CrossRef]
- Pandis, C.; Sotiriou, E.; Kouvaras, E.; Asprodini, E.; Papatheodoropoulos, C.; Angelatou, F. Differential expression of NMDA and AMPA receptor subunits in rat dorsal and ventral hippocampus. Neuroscience 2006, 140, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Sze, S.C.W.; Wong, C.K.C.; Yung, K.K.L. Modulation of the gene expression of N-methyl-D-aspartate receptor NR2B subunit in the rat neostriatum by a single dose of specific antisense oligodeoxynucleotide. Neurochem. Int. 2001, 39, 319–327. [Google Scholar] [CrossRef]
- Preskorn, S.H.; Baker, B.; Kolluri, S.; Menniti, F.S.; Krams, M.; Landen, J.W. An Innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective n-methyl-d-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 2008, 28, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Hayne, H. Infant memory development: Implications for childhood amnesia. Dev. Rev. 2004, 24, 33–73. [Google Scholar] [CrossRef]
- Newcombe, N.S.; Lloyd, M.E.; Ratliff, K.R. Development of episodic and autobiographical memory: A cognitive neuroscience perspective. Adv. Child Dev. Behav. 2007, 35, 37–85. [Google Scholar]
- Rovee-Collier, C.; Cuevas, K. The development of infant memory. In The Development of Memory in Infancy and Childhood; Courage, M.L., Cowan, N., Eds.; Psychology Press: New York, NY, USA, 2009. [Google Scholar]
- Rubin, D.C. The distribution of early childhood memories. Memory 2000, 8, 265–269. [Google Scholar] [CrossRef]
- Alberini, C.M.; Travaglia, A. Infantile Amnesia: A Critical Period of Learning to Learn and Remember. J. Neurosci. 2017, 37, 5783–5795. [Google Scholar] [CrossRef]
- Tovar, K.R.; McGinley, M.J.; Westbrook, G.L. Triheteromeric NMDA receptors at hippocampal synapses. J. Neurosci. 2007, 52, 60–70. [Google Scholar] [CrossRef]
- Travaglia, A.; Bisaz, R.; Sweet, E.S.; Blitzer, R.D.; Alberini, C.M. Infantile amnesia reflects a developmental critical period for hippocampal learning. Nat. Neurosci. 2016, 19, 1225–1233. [Google Scholar] [CrossRef]
- Rowland, L.H.; Astur, R.S.; Jung, R.E.; Bustillo, J.R.; Lauriello, J.; Yeo, R.A. Selective cognitive impairments associated with NMDA receptor blockade in humans. Neuropsychopharmacology 2005, 30, 633–639. [Google Scholar] [CrossRef]
- Ewald, R.C.; Cline, H.T. NMDA Receptors and Brain Development. Front. Mol. Neurosci. 2009, 2, 4. [Google Scholar]
- Abdou, K.; Shehata, M.; Choko, K.; Nishizono, H.; Matsuo, M.; Muramatsu, S.I.; Inokuchi, K. Synapse-specific representation of the identity of overlapping memory engrams. Science 2018, 360, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Yagishita, S.; Nakamura, M.; Shirai, F.; Wu, Y.I.; Loshbaugh, A.L.; Kuhlman, B.; Hahn, K.M.; Kasai, H. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 2015, 525, 333–338. [Google Scholar] [CrossRef]
- Nabavi, S.; Fox, R.; Proulx, C.D.; Lin, J.Y.; Tsien, R.Y.; Malinow, R. Engineering a memory with LTD and LTP. Nature 2014, 511, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Song, H.; Li, H.; Li, R.; Tao, X.; Zhan, X.; Yu, N.; Sun, N.; Lu, Y.; Mu, Y. Memory Susceptibility to Retroactive Interference Is Developmentally Regulated by NMDA Receptors. Cell Rep. 2019, 26, 2052–2063.e4. [Google Scholar] [CrossRef]
- Nutt, J.G.; Gunzler, S.A.; Kirchhoff, T.; Hogarth, P.; Weaver, J.L.; Krams, M.; Jamerson, B.; Menniti, F.S.; Landen, J.W. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and parkinsonism. Mov. Disord. 2008, 23, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Ahnaou, A.; Broadbelt, T.; Biermans, R.; Huysmans, H.; Manyakov, N.V.; Drinkenburg, W.H.I.M. The phosphodiesterase-4 and glycine transporter-1 inhibitors enhance in vivo hippocampal theta network connectivity and synaptic plasticity, whereas D-serine does not. Transl. Psychiatry 2020, 10, 197. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Type | Stimulation Protocol | Synaptic Plasticity—NMDA Receptors Subtype Relation | Reference |
|---|---|---|---|
| In vitro slices Hippocampus, Sprague Dawley rats | Theta-burst stimulation for LTP | A correlation between the NR2A subunit and synaptic plasticity form LTP | [37] |
| Hippocampus, Sprague Dawley rats | Theta-burst stimulation for LTP | A correlation between the NR2A subunit and synaptic plasticity form LTP | [37] |
| Hippocampus, Lister Hooded rats | Low-frequency stimulation for LTD | The NR2A subunit is responsible for subsequent mechanisms of LTD. | [38] |
| Hippocampus, Long Evans rats | Low-frequency stimulation for LTD | The involvement of the NR2B subunit in the synaptic plasticity form LTD. | [39] |
| Hippocampus, Wistar rats | High-frequency stimulation for LTP and low-frequency stimulation for LTD | The regulation of LTP and LTD by both NR2A and NR2B subunits. | [15] |
| Hippocampus, Sprague Dawley rats | Theta burst stimulation and paired low-frequency stimulation for LTP, and theta burst stimulation for LTD | Distinct functions of NR2B for LTP and NR2A for LTD seperately. | [19] |
| Hippocampal, Wistar rats | High-frequency stimulation for LTP and low-frequency stimulation for LTD | Cross-relation of subunits NR2A and NR2B for synaptic plasticity forms LTP and LTD. | [40] |
| In vivo Hippocampus, Sprague Dawley rats | High-frequency stimulation for LTP | Proof for the involvement of the NR2B subunit in LTP. | [41] |
| Hippocampus, Sprague Dawley rats | High-frequency and theta-burst stimulation for LTP, and low-frequency and paired-burst stimulation for LTD | This study proved the importance of both subunits, NR2A and NR2B, in any form LTP or LTD. | [42] |
| Baseline Configuration (−60–0 min) | TBS (at 0 min) | Post-Tetanization Configuration (0–120 min) | |
|---|---|---|---|
| Pulse source | Voltage | Voltage | Voltage |
| # pulse trains | 24× | 4× | 49× |
| pulse train interval | 150 s | 20 s | 150 s |
| pulse length | 200 µs | 200 µs | 200 µs |
| pulse current/voltage | Variable µA/mV (based on 50% IO curve) | Variable µA/mV (based on 50% IO curve) | Variable µA/mV (based on 50% IO curve) |
| burst trains frequency | / | 5 Hz | / |
| # burst trains | / | 5× | / |
| Burst frequency | / | 100 Hz | / |
| # burst pulses | / | 5× | / |
| pulse frequency | 0.033 Hz | / | 0.033 Hz |
| # pulses | 5× | / | 5× |
| post delay | 150 s | 10 s | 150 s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahnaou, A.; Heleven, K.; Biermans, R.; Manyakov, N.V.; Drinkenburg, W.H. NMDARs Containing NR2B Subunit Do Not Contribute to the LTP Form of Hippocampal Plasticity: In Vivo Pharmacological Evidence in Rats. Int. J. Mol. Sci. 2021, 22, 8672. https://doi.org/10.3390/ijms22168672
Ahnaou A, Heleven K, Biermans R, Manyakov NV, Drinkenburg WH. NMDARs Containing NR2B Subunit Do Not Contribute to the LTP Form of Hippocampal Plasticity: In Vivo Pharmacological Evidence in Rats. International Journal of Molecular Sciences. 2021; 22(16):8672. https://doi.org/10.3390/ijms22168672
Chicago/Turabian StyleAhnaou, Abdallah, Kobe Heleven, Ria Biermans, Nikolay V. Manyakov, and Wilhelmus H. Drinkenburg. 2021. "NMDARs Containing NR2B Subunit Do Not Contribute to the LTP Form of Hippocampal Plasticity: In Vivo Pharmacological Evidence in Rats" International Journal of Molecular Sciences 22, no. 16: 8672. https://doi.org/10.3390/ijms22168672
APA StyleAhnaou, A., Heleven, K., Biermans, R., Manyakov, N. V., & Drinkenburg, W. H. (2021). NMDARs Containing NR2B Subunit Do Not Contribute to the LTP Form of Hippocampal Plasticity: In Vivo Pharmacological Evidence in Rats. International Journal of Molecular Sciences, 22(16), 8672. https://doi.org/10.3390/ijms22168672