
Comprehensive Survey of Recent Drug Discovery Using Deep Learning


Abstract
:1. Introduction
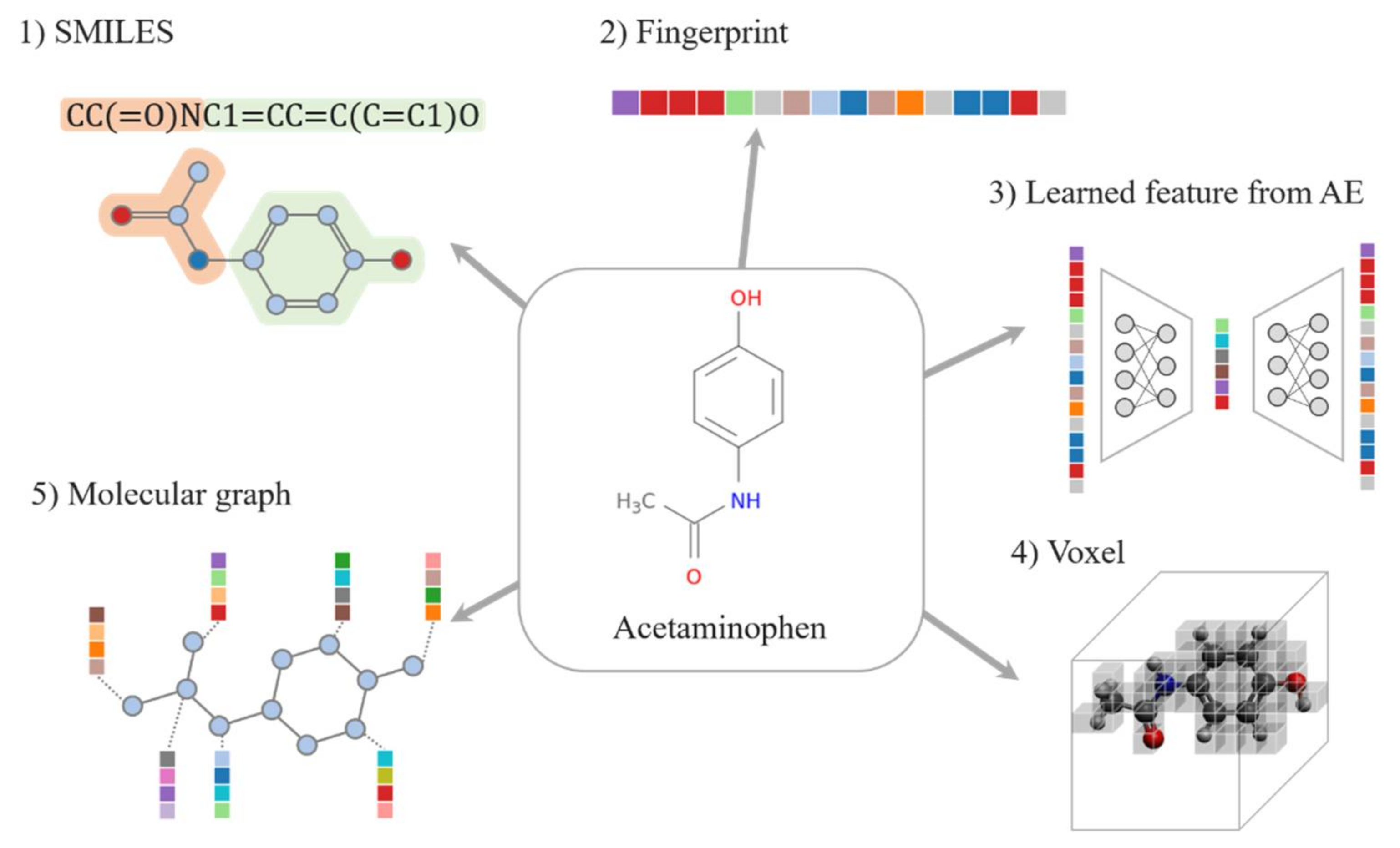
2. Data Representation
2.1. Drug Representations
2.1.1. SMILES
2.1.2. Fingerprint
2.1.3. Learned Representations
2.1.4. Voxel
2.1.5. Molecular Graph
2.2. Target Representations
2.2.1. Sequence-Based Feature
2.2.2. Structure-Based Feature
2.2.3. Relationship-Based Feature
3. Deep Learning Models
3.1. Multi-Layer Perceptron
3.2. Convolutional Neural Network
3.3. Graph Neural Network
3.4. Recurrent Neural Network
3.5. Attention-Based Model
3.6. Generative Adversarial Network
3.7. Autoencoder
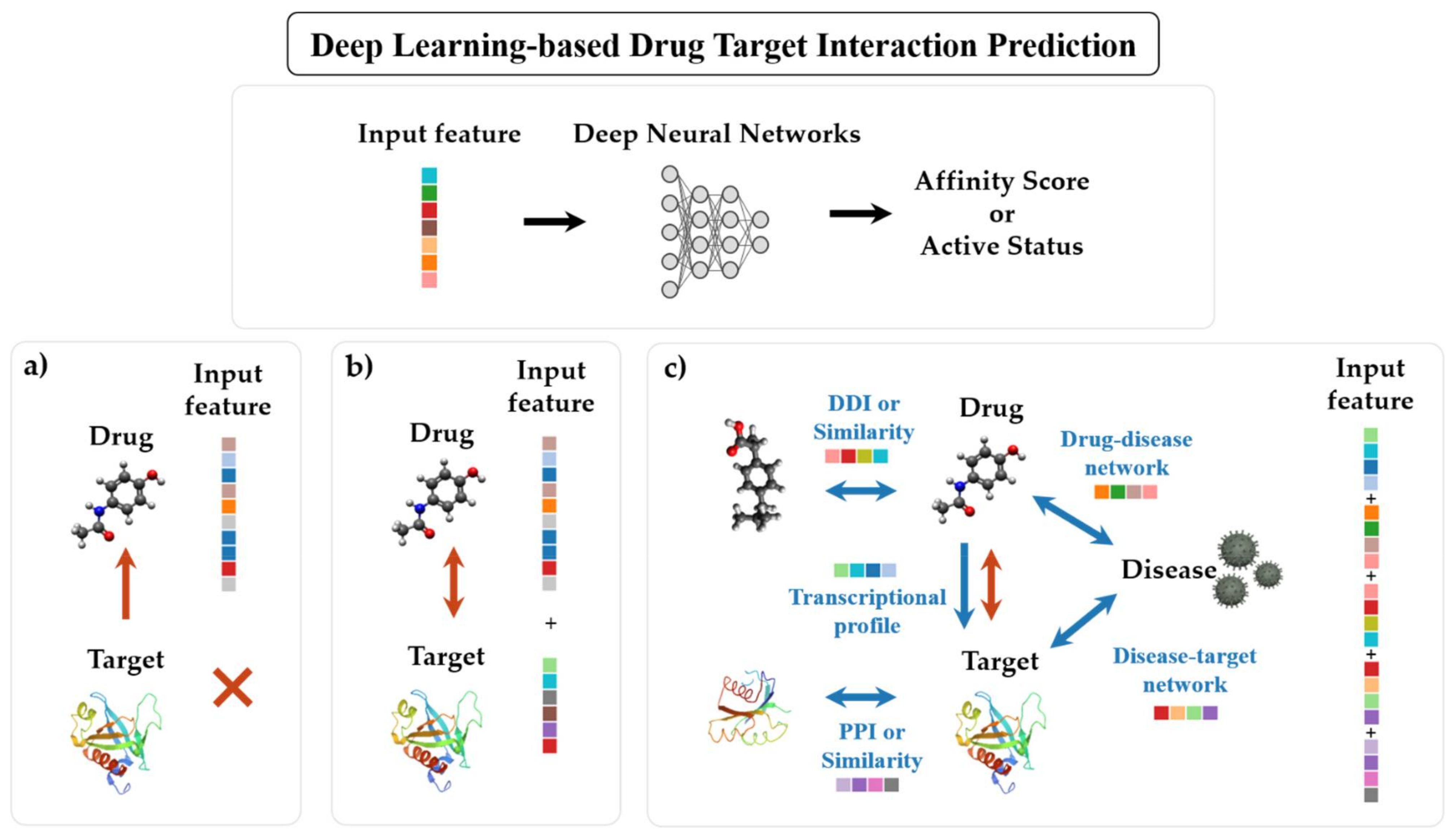
4. Deep Learning Methods for Drug–Target Interaction Prediction
4.1. Ligand-Based Approach
4.2. Structure-Based Approach
4.3. Relationship-Based Approach
5. Deep Learning Methods for De Novo Drug Design
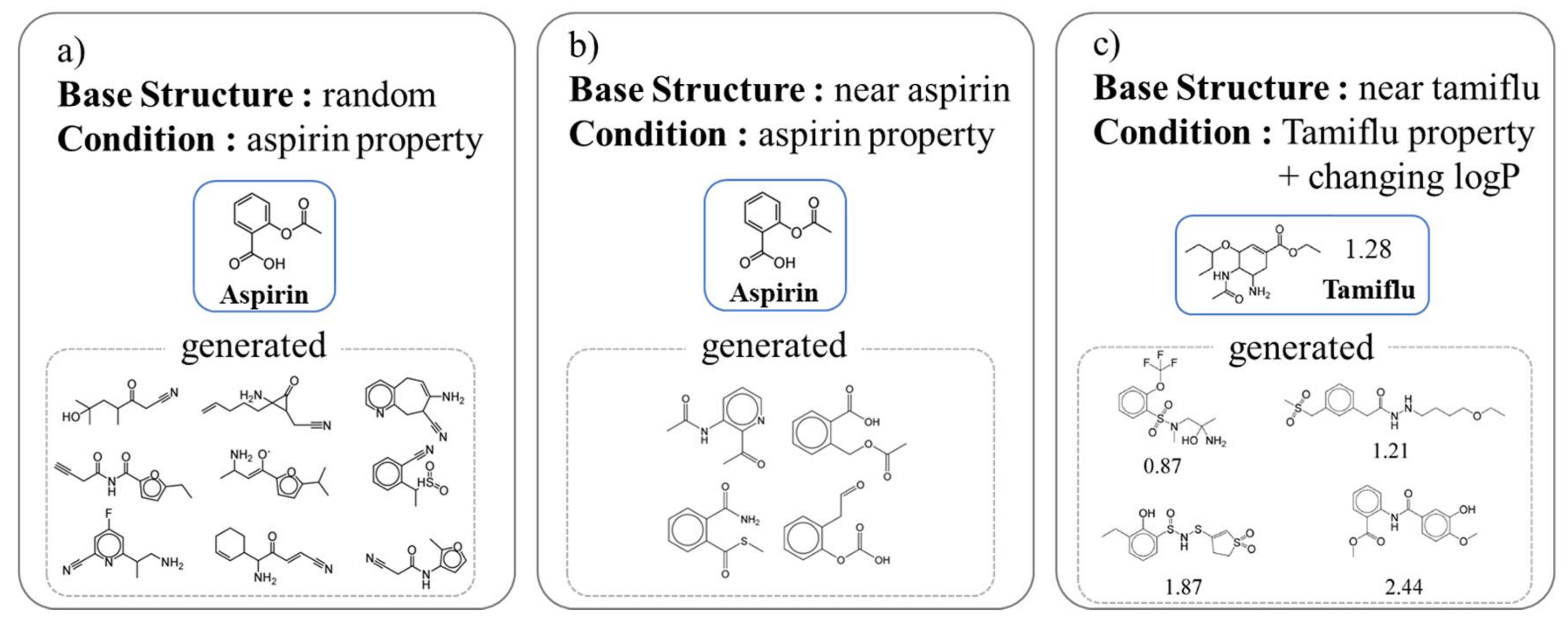
5.1. Chemical Latent Space
5.2. Condition Control of Compounds
5.3. Generation at Once or Sequentially
5.4. Fragment-Based Generation
5.5. Genetic Algorithm
6. Evaluation Method
6.1. Benchmarking Datasets and Tools
6.2. Evaluation Metrics for DTI Prediction
6.2.1. Classification Metrics
6.2.2. Regression Evaluation Metrics
6.3. Evaluation Metrics for De Novo Drug Design
6.3.1. Generation Metrics
6.3.2. Pharmacological Indicators
7. Limitation and Future Work
7.1. Current Challenges
7.1.1. Data Scarcity and Imbalance
7.1.2. Absence of Standard Benchmark
7.2. Promising Method
7.2.1. Transfer Learning
7.2.2. Data Augmentation
7.2.3. Uncertainty and Interpretation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AI | Artificial Intelligence |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| AUC | Area Under the Curve |
| AUPR | Area Under the Precision–Recall Curve |
| AE | Autoencoder |
| CNN | Convolutional Neural Networks |
| CI | Concordance Index |
| DDI | Drug–Drug Interaction |
| MAE | Mean Absolute Error |
| MCC | Matthews Correlation Coefficient |
| ML | Machine Learning |
| MLP | Multi-Layer Perceptron |
| DL | Deep learning |
| DTA | Drug–Target Affinity |
| DTI | Drug–Target Interaction |
| DTP | Drug–Target Pair |
| FP | Fingerprint |
| FPR | False Positive Rate |
| HBA | Hydrogen Bond Acceptor |
| HBD | Hydrogen Bond Donor |
| HTS | High-Throughput Screening |
| GAN | Generative Adversarial Networks |
| GCN | Graph Convolutional Networks |
| GO | Gene Ontology |
| LINCS | Library of Integrated Network-based Cellular Signatures |
| LSTM | Long Short-Term Memory |
| PPI | Protein–Protein Interaction |
| QSAR | Quantitative Structure-Activity Relationship |
| RMSE | Root Mean Square Error |
| RNN | Recurrent Neural Networks |
| SMILES | Simplified Molecular-Input Line-Entry System |
| TPR | True Positive Rate |
| TPSA | Topological Polar Surface Area |
| VAE | Variational AutoEncoder |
| VS | Virtual Screening |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Models | Input Drug Type | Datasets | Algorithm Type | Year | Evaluation Metrics |
|---|---|---|---|---|---|---|
| Gao et al. [33] | MLP; Multi-task | Fingerprint (ECFP; FP2; Estate1; Estate2; MACCS; ERG) | PDBbind | Regression | 2019 | Pearson correlation coefficient (R); RMSE |
| Wenzel et al. [115] | MLP; Multi-task | Atom pair; pharmacophoric donor–acceptor pairs | ChEMBL | Regression | 2019 | |
| Xie et al. [32] | MLP; LSTM | Fingerprint (MACCS+ECFP) | DrugBank; ChEMBL; PDBbind | Regression | 2020 | Pearson correlation coefficient (R); RMSE |
| Hirohara et al. [25] | CNN | SMILES convolution fingerprint | Tox21 | Classification | 2018 | AUC |
| Matsuzaka et al. [109] | CNN | 2D image | Tox21 | Classification | 2019 | AUC; Balanced accuracy; F-score; MCC |
| Rifaioglu et al. [19] | CNN | 2D image | ChEMBL; MUV; DUD-E | Classification | 2020 | AUC; Accuracy; Precision; Recall; F1-score; MCC |
| Liu et al. [81] | GCN; Multi-task | 3D molecular graph | Amgen’s internal dataset; ChEMBL | Regression | 2019 | ; Accuracy |
| Yang et al. [21] | GCN | SMILES | PDBbind; ChEMBL; PubChem Bioassay; MUV; Tox21; ToxCast; SIDER etc. | Classification; Regression | 2019 | MAE; RMSE; AUC; AUPR |
| Shang et al. [11] | GCN; Attention-based | Molecular graph | Tox21; HIV; Freesolv; Lipophilicity (MoleculeNet) | Regression | 2018 | AUC; RMSE |
| Reference | Models | Input Drug Type | Input Target Type | Datasets | Algorithm Type | Evaluation Metrics | Year |
|---|---|---|---|---|---|---|---|
| Wen et al. [37] | MLP | Fingerprint (ECFP) | PSC (protein sequence composition descriptor) | DrugBank | Classification | TPR; TNR; Accuracy; AUC | 2017 |
| Chen et al. [57] | MLP | Fingerprint (PubChemFP) | Various protein features * | DrugBank; Yamanishi | Classification | AUC; AUPR | 2020 |
| Öztürk et al. [24] | CNN | SMILES | Sequence | Davis; KIBA | Regression | CI; MSE | 2018 |
| Shin et al. [23] | CNN; attention | SMILES | Sequence | Davis; KIBA; | Regression | CI; RMSE; ; AUPR | 2019 |
| Zhao et al. [120] | CNN; attention | SMILES | Sequence | Davis; KIBA | Regression | CI; RMSE; ; AUPR | 2019 |
| Gonczarek et al. [202] | CNN | Atom pair | Atom pair | DUD-E; PDBBind | Regression | AUC | 2016 |
| Ragoza et al. [203] | CNN | Voxel | Voxel | DUD-E; CSAR | Regression; Classification | AUC | 2017 |
| Jiménez et al. [204] | CNN | Voxel | Voxel | PDBbind; CSAR2012 | Regression | RMSE; | 2018 |
| Kwon et al. [75] | CNN | Voxel | Voxel | CASF-2016 [205] | Regression | MAE; RMSE | 2020 |
| Pu et al. [51] | CNN; multi-classification | Voxel | Voxel | PDB; TOUGH-M1 [206] | Classification | MCC; AUC; Accuracy | 2019 |
| Lee et al. [31] | CNN | Fingerprint | Sequence | DrugBank; KEGG; IUPHAR; MATADOR; PubChem Bioassay; KinaseSARfari [189] | Classification | AUC; AUPR; Sensitivity; Specificity; Precision; Accuracy; F1-score | 2019 |
| Hasan Mahmud et al. [207] | CNN | SMILES; 193 features by Rcpi | Sequence; 1290 features by PROFEAT | DrugBank; Yamanishi | Regression | AUC; Accuracy; Sensitivity; Precision; F1 score; AUPR | 2020 |
| Wang et al. [34] | LSTM | Fingerprint (PubChemFP) | PSSM; Legendre Moment [208] | DrugBank; Yamanishi; KEGG; SuperTarget | Classification | AUC; Accuracy; TPR; Specificity; Precision; MCC | 2020 |
| Tsubaki et al. [209] | GNN; CNN; attention | Fingerprint (PubChemFP) | Sequence; Pfam domain | DUD-E; DrugBank; MATADOR | Classification | AUC; Precision; Recall | 2019 |
| Torng and Altman [118] | GCN | Molecular graph | Molecular graph | DUD-E; MUV | Classification | AUC | 2019 |
| Feng et al. [8] | GCN | Fingerprint (ECFP); 3D molecular graph | PSC (protein sequence composition descriptor) | Davis; Metz; KIBA; ToxCast | Regression | 2019 | |
| Jiang et al. [210] | GNN | 3D molecular graph | 3D molecular graph | KIBA; Davis | Regression | ; CI; MSE; Pearson correlation coefficient; Accuracy | 2020 |
| Reference | Models | Relationship Data Type | Datasets | Algorithm Type | Evaluation Metrics | Year |
|---|---|---|---|---|---|---|
| Xie et al. [71] | MLP | LINCS signature | DrugBank; CTD; DGIdb; STITCH | Regression | Accuracy; F-score; TPR | 2018 |
| Lee and Kim [63] | MLP; node2vec | LINCS signature; PPI (Protein-protein interaction); Pathway | LINCS; ChEMBL; TTD; MATADOR; KEGG; IUPHAR; PharmGKB; KiDB | Classification | AUC; Precision | 2019 |
| Gao et al. [119] | CNN; LSTM | LINCS signature; GO term | BindingDB | Regression | Accuracy; AUC; AUPR | 2018 |
| Shao and Zhang [147] | CNN; GCN | LINCS signature | LINCS; DrugBank | Classification | Accuracy; AUC | 2020 |
| Thafar et al. [67] | node2vec | Drug similarity (structure, side effects); Target similarity (sequence, GO); PPI | Yamanishi; KEGG; BRENDA; SuperTarget; DrugBank; BioGRID; SIDER | Classification | AUPR; AUC | 2020 |
| Zong et al. [13] | DeepWalk [130] | Drug-target association; Drug-disease association; Disease-target association | DrugBank; Human diseasome [211] | Classification | AUC | 2017 |
| Mongia and Majumdar [212] | Multi-graph deep matrix factorization | Drug similarity (structure); Target similarity (sequence) | Yamanishi; KEGG; BRENDA; SuperTarget; DrugBank | Classification | AUPR; AUC | 2020 |
| Wang et al. [59] | AE | Drug similarity (structure, side effects); Target similarity (sequence, GO); PPI | Yamanishi; KEGG; BRENDA; SuperTarget; DrugBank; SIDER | Classification | AUPR; AUC | 2020 |
| Zhao et al. [68] | CNN; AE | Drug similarity (structure); Target similarity (sequence); PPI | DrugBank; STRING | Classification | Accuracy; AUPR; AUC | 2020 |
| Peng et al. [97] | CNN; AE | Drug-target association; Drug-disease association; Disease-target association; Drug similarity (structure, side effects); Target similarity (sequence, GO); PPI | DrugBank; Human Protein Reference Database [2009]; CTD; SIDER; | Classification | AUPR; AUC | 2020 |
| Zhong et al. [213] | GCN | LINCS signature; PPI | ChEMBL; LINCS; STRING | Classification | Accuracy; F-score; AUPR; Precision; Recall; AUC | 2020 |
References
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, K.; Gupta, M.K. A comprehensive review of feature based methods for drug target interaction prediction. J. Biomed. Inform. 2019, 93, 103159. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Kimber, T.B.; Chen, Y.; Volkamer, A. Deep learning in virtual screening: Recent applications and developments. Int. J. Mol. Sci. 2021, 22, 4435. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.F.; Maltarollo, V.G.; Oliveira, P.R.; da Silva, A.B.F.; Honorio, K.M. Advances and Perspectives in Applying Deep Learning for Drug Design and Discovery. Front. Robot. AI 2019, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doǧan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Dueva, E.; Cherkasov, A.; Ester, M. PADME: A Deep Learning-Based Framework for Drug-Target Interaction Prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Skalic, M.; Varela-Rial, A.; Jiménez, J.; Martínez-Rosell, G.; De Fabritiis, G. LigVoxel: Inpainting binding pockets using 3D-convolutional neural networks. Bioinformatics 2019, 35, 243–250. [Google Scholar] [CrossRef]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, C.; Liu, Q.; Chen, K.-S.; Sun, J.; Lu, J.; Yi, J.; Bi, J. Edge Attention-based Multi-Relational Graph Convolutional Networks. arXiv 2018, arXiv:1802.04944. [Google Scholar]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Zong, N.; Kim, H.; Ngo, V.; Harismendy, O. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations. Bioinformatics 2017, 33, 2337–2344. [Google Scholar] [CrossRef]
- Husic, B.E.; Charron, N.E.; Lemm, D.; Wang, J.; Pérez, A.; Majewski, M.; Krämer, A.; Chen, Y.; Olsson, S.; De Fabritiis, G.; et al. Coarse graining molecular dynamics with graph neural networks. J. Chem. Phys. 2020, 153, 194101. [Google Scholar] [CrossRef] [PubMed]
- Hassan-Harrirou, H.; Zhang, C.; Lemmin, T. RosENet: Improving Binding Affinity Prediction by Leveraging Molecular Mechanics Energies with an Ensemble of 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2020, 60, 2791–2802. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Xue, L.; Bajorath, J. Molecular Descriptors in Chemoinformatics, Computational Combinatorial Chemistry, and Virtual Screening. Comb. Chem. High Throughput Screen. 2012, 3, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Redkar, S.; Mondal, S.; Joseph, A.; Hareesha, K.S. A Machine Learning Approach for Drug-target Interaction Prediction using Wrapper Feature Selection and Class Balancing. Mol. Inform. 2020, 39, 1900062. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atalay, V.; Martin, M.J.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations. bioRxiv 2018, 491365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Swanson, K.; Jin, W.; Coley, C.; Eiden, P.; Gao, H.; Guzman-Perez, A.; Hopper, T.; Kelley, B.; Mathea, M.; et al. Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 2019, 59, 3370–3388. [Google Scholar] [CrossRef] [Green Version]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackerman, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702.e13. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Park, S.; Kang, K.; Ho, J.C. Self-Attention Based Molecule Representation for Predicting Drug-Target Interaction. Proc. Mach. Learn. Res. 2019, 106, 1–18. [Google Scholar]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirohara, M.; Saito, Y.; Koda, Y.; Sato, K.; Sakakibara, Y. Convolutional neural network based on SMILES representation of compounds for detecting chemical motif. BMC Bioinformatics 2018, 19, 83–94. [Google Scholar] [CrossRef]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-art augmented NLP transformer models for direct and single-step retrosynthesis. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, R.; Zhang, C.; Wang, L.; Yao, C.; Ge, J.; Duan, H. Molecules Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level. Molecules 2020, 25, 2357. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Johansson, S.V.; Prykhodko, O.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.L.; Chen, H.; Engkvist, O. Randomized SMILES strings improve the quality of molecular generative models. J. Cheminform. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xu, L.; Kong, R.; Chang, S.; Xu, X. Improvement of Prediction Performance With Conjoint Molecular Fingerprint in Deep Learning. Front. Pharmacol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Duy Nguyen, D.; Sresht, V.; Mathiowetz, A.M.; Tu, M.; Wei, G.-W. Are 2D fingerprints still valuable for drug discovery? Phys. Chem. Chem. Phys. 2019, 22, 8373–8390. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.B.; You, Z.H.; Yang, S.; Yi, H.C.; Chen, Z.H.; Zheng, K. A deep learning-based method for drug-target interaction prediction based on long short-term memory neural network. BMC Med. Inform. Decis. Mak. 2020, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures—A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-Learning-Based Drug−Target Interaction Prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Moumbock, A.F.A.; Li, J.; Mishra, P.; Gao, M.; Günther, S. Current computational methods for predicting protein interactions of natural products. Comput. Struct. Biotechnol. J. 2019, 17, 1367–1376. [Google Scholar] [CrossRef]
- Wood, D.J.; De Vlieg, J.; Wagener, M.; Ritschel, T. Pharmacophore Fingerprint-Based Approach to Binding Site Subpocket Similarity and Its Application to Bioisostere Replacement. J. Chem. Inf. Model. 2012, 52, 2031–2043. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Wang, X.; Kaushik, A.C.; Chu, Y.; Shan, X.; Zhao, M.Z.; Xu, Q.; Wei, D.Q. SPVec: A Word2vec-Inspired Feature Representation Method for Drug-Target Interaction Prediction. Front. Chem. 2020, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.B.; Hodas, N.O.; Siegel, C.; Vishnu, A. SMILES2Vec: An Interpretable General-Purpose Deep Neural Network for Predicting Chemical Properties. arXiv 2017, arXiv:1712.02034. [Google Scholar] [CrossRef]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.; Dean, J. Distributed representations ofwords and phrases and their compositionality. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–10 December 2013; pp. 1–9. [Google Scholar]
- Asgari, E.; Mofrad, M.R.K. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef]
- Jaeger, S.; Fulle, S.; Turk, S. Mol2vec: Unsupervised Machine Learning Approach with Chemical Intuition. J. Chem. Inf. Model. 2018, 58, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminform. 2018, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.; Zhang, H.; Xiao, D.; Gong, Y.; Chuai, G.; Sun, Y.; Tian, H. X-MOL: Large-scale pre-training for molecular understanding and diverse molecular analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Li, P.; Wang, J.; Qiao, Y.; Chen, H.; Yu, Y. Learn molecular representations from large-scale unlabeled molecules for drug discovery. arXiv 2020, arXiv:2012.11175. [Google Scholar]
- Kuzminykh, D.; Polykovskiy, D.; Kadurin, A.; Zhebrak, A.; Baskov, I.; Nikolenko, S.; Shayakhmetov, R.; Zhavoronkov, A. 3D Molecular Representations Based on the Wave Transform for Convolutional Neural Networks. Mol. Pharm. 2018, 15, 4378–4385. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, S.; Song, G.; Cai, L. HamNet: Conformation-Guided Molecular Representation with Hamiltonian Neural Networks. arXiv 2021, arXiv:2105.03688. [Google Scholar]
- Amidi, A.; Amidi, S.; Vlachakis, D.; Megalooikonomou, V.; Paragios, N.; Zacharaki, E.I. EnzyNet: Enzyme classification using 3D convolutional neural networks on spatial representation. PeerJ 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Pu, L.; Govindaraj, R.G.; Lemoine, J.M.; Wu, H.-C.; Brylinski, M. DeepDrug3D: Classification of ligand-binding pockets in proteins with a convolutional neural network. PLoS Comput. Biol. 2019, 15, e1006718. [Google Scholar] [CrossRef] [Green Version]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodolà, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17, 184–192. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, S.; Duan, Y.; Huang, Y. A Point Cloud-Based Deep Learning Strategy for Protein-Ligand Binding Affinity Prediction. arXiv 2021, arXiv:2107.04340. [Google Scholar]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug-Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Green, W.H.; Jaakkola, T.S.; Jensen, K.F. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction. J. Chem. Inf. Model. 2017, 57, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Davari, M.D.; Li, W. Recent advances in the prediction of protein structural classes: Feature descriptors and machine learning algorithms. Crystals 2021, 11, 324. [Google Scholar] [CrossRef]
- Chen, C.; Shi, H.; Han, Y.; Jiang, Z.; Cui, X.; Yu, B. DNN-DTIs: Improved drug-target interactions prediction using XGBoost feature selection and deep neural network. bioRxiv 2020. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, J.; Dong, C.; Lian, Y.; Liu, D.; Yan, Z. A novel approach for drug-target interactions prediction based on multimodal deep autoencoder. Front. Pharmacol. 2020, 10, 1–19. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, W. Comparison of target features for predicting drug-target interactions by deep neural network based on large-scale drug-induced transcriptome data. Pharmaceutics 2019, 11, 377. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiong, H.; Li, X.; Wu, X.; Zhang, X.; Liu, J.; Bian, J.; Dou, D. Interpretable Deep Learning: Interpretation, Interpretability, Trustworthiness, and Beyond. arXiv 2021, arXiv:2103.10689. [Google Scholar]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Fan, K.; Song, D.; Lin, H. Graph-based prediction of Protein-protein interactions with attributed signed graph embedding. BMC Bioinformatics 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Thafar, M.A.; Thafar, M.A.; Olayan, R.S.; Olayan, R.S.; Ashoor, H.; Ashoor, H.; Albaradei, S.; Albaradei, S.; Bajic, V.B.; Gao, X.; et al. DTiGEMS+: Drug-target interaction prediction using graph embedding, graph mining, and similarity-based techniques. J. Cheminform. 2020, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, K.; Guan, B.; Guo, M.; Song, L.; Gao, J.; Qu, H.; Wang, Y.; Shi, D.; Zhang, Y. DLDTI: A learning-based framework for drug-target interaction identification using neural networks and network representation. J. Transl. Med. 2020, 18, 434. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Xie, L.; He, S.; Song, X.; Bo, X.; Zhang, Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genomics 2018, 19, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wang, J.; Wang, X.; Gao, M.; Guo, B.; Gao, M.; Liu, J.; Yu, Y.; Wang, L.; Kong, W.; et al. Prediction of drug efficacy from transcriptional profiles with deep learning. Nat. Biotechnol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, S. Deep learning-based imbalanced data classification for drug discovery. J. Chem. Inf. Model. 2020, 60, 4180–4190. [Google Scholar] [CrossRef]
- Townshend, R.J.L.; Powers, A.; Eismann, S.; Derry, A. ATOM3D: Tasks On Molecules in Three Dimensions. arXiv 2021, arXiv:2012.04035. [Google Scholar]
- Kwon, Y.; Shin, W.H.; Ko, J.; Lee, J. AK-score: Accurate protein-ligand binding affinity prediction using an ensemble of 3D-convolutional neural networks. Int. J. Mol. Sci. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformatics 2020, 36, 5545–5547. [Google Scholar] [CrossRef]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform. 2020, 12, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Networks 2009, 20, 61–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, X.; Wu, J. GCN-BMP: Investigating graph representation learning for DDI prediction task. Methods 2020, 179, 47–54. [Google Scholar] [CrossRef]
- Liu, K.; Sun, X.; Jia, L.; Ma, J.; Xing, H.; Wu, J.; Gao, H.; Sun, Y.; Boulnois, F.; Fan, J. Chemi-net: A molecular graph convolutional network for accurate drug property prediction. Int. J. Mol. Sci. 2019, 20, 3389. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.; Wu, M.; Liu, Y.; Kwoh, C.K.; Luo, J.; Li, X. Ensembling graph attention networks for human microbe-drug association prediction. Bioinformatics 2020, 36, I779–I786. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Z.; Hu, X.; Song, B. A novel graph attention adversarial network for predicting disease-related associations. Methods 2020, 179, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Hwang, S.Y.; Moon, S.; Kim, S.; Kim, W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2020, 11, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochreiter, S. The vanishing gradient problem during learning recurrent neural nets and problem solutions. Int. J. Uncertain. Fuzziness Knowlege-Based Syst. 1998, 6, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Si, X.; Hu, C.; Zhang, J. A review of recurrent neural networks: Lstm cells and network architectures. Neural Comput. 2019, 31, 1235–1270. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 2021, 22, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. In Proceedings of the EMNLP 2014 Conference on Empirical Methods in Natural Language Processing, Doha, Qatar, 25–29 October 2014; pp. 1724–1734. [Google Scholar] [CrossRef]
- Jastrzębski, S.; Leśniak, D.; Czarnecki, W.M. Learning to SMILE(S). arXiv 2016, arXiv:1602.06289. [Google Scholar]
- Guimaraes, G.L.; Sanchez-Lengeling, B.; Outeiral, C.; Farias, P.L.C.; Aspuru-Guzik, A. Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models. arXiv 2017, arXiv:1705.10843. [Google Scholar]
- De Cao, N.; Kipf, T. MolGAN: An implicit generative model for small molecular graphs. arXiv 2018, arXiv:1805.11973. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the NAACL HLT Annual Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Minneapolis, MN, USA, 2–7 June 2019; Volume 1, pp. 4171–4186. [Google Scholar]
- Lennox, M.; Robertson, N.M.; Devereux, B. Modelling Drug-Target Binding Affinity using a BERT based Graph Neural network. Annu. Rev. Biochem. 2021, 68, 559–581. [Google Scholar]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery. Molecules 2020, 25, 3250. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, W.; Wang, J.; Yu, Y. SeqGAN: Sequence generative adversarial nets with policy gradient. In Proceedings of the 31 AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; pp. 2852–2858. [Google Scholar]
- Peng, J.; Li, J.; Shang, X. A learning-based method for drug-target interaction prediction based on feature representation learning and deep neural network. BMC Bioinform. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Makhzani, A.; Shlens, J.; Jaitly, N.; Goodfellow, I.; Frey, B. Adversarial Autoencoders. arXiv 2015, arXiv:1511.05644. [Google Scholar]
- Vanhaelen, Q.; Lin, Y.C.; Zhavoronkov, A. The Advent of Generative Chemistry. ACS Med. Chem. Lett. 2020, 11, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883–10890. [Google Scholar] [CrossRef] [Green Version]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. DruGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Vetrov, D.; Ivanenkov, Y.; Aladinskiy, V.; Mamoshina, P.; Bozdaganyan, M.; Aliper, A.; Zhavoronkov, A.; Kadurin, A. Entangled Conditional Adversarial Autoencoder for de Novo Drug Discovery. Mol. Pharm. 2018, 15, 4398–4405. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, J.; López, M.; Gibert, E.; Herrero, E.; Luque, F.J. Merging Ligand-Based and Structure-Based Methods in Drug Discovery: An Overview of Combined Virtual Screening Approaches. Molecules 2020, 25, 4723. [Google Scholar] [CrossRef]
- Abbasi, K.; Razzaghi, P.; Poso, A.; Ghanbari-Ara, S.; Masoudi-Nejad, A. Deep Learning in Drug Target Interaction Prediction: Current and Future Perspectives. Curr. Med. Chem. 2020, 28, 2100–2113. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, S.; Prema, K.V.; Balaji, S. Machine learning models for drug–target interactions: Current knowledge and future directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug–target interaction: A survey paper. Brief. Bioinform. 2020, 22, 247–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thafar, M.; Bin Raies, A.; Albaradei, S.; Essack, M.; Bajic, V.B. Comparison Study of Computational Prediction Tools for Drug-Target Binding Affinities. Front. Chem. 2019, 7, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaka, Y.; Uesawa, Y. Prediction model with high-performance constitutive androstane receptor (CAR) using DeepSnap-deep learning approach from the tox21 10K compound library. Int. J. Mol. Sci. 2019, 20, 4855. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [Green Version]
- deepchem/deepchem: Democratizing Deep-Learning for Drug Discovery, Quantum Chemistry, Materials Science and Biology. Available online: https://github.com/deepchem/deepchem (accessed on 13 July 2021).
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Wenzel, J.; Matter, H.; Schmidt, F. Predictive Multitask Deep Neural Network Models for ADME-Tox Properties: Learning from Large Data Sets. J. Chem. Inf. Model. 2019, 59, 1253–1268. [Google Scholar] [CrossRef] [PubMed]
- Gilmer, J.; Schoenholz, S.S.; Riley, P.F.; Vinyals, O.; Dahl, G.E. Neural Message Passing for Quantum Chemistry. 34th Int. Conf. Mach. Learn. ICML 2017 2017, 3, 2053–2070. [Google Scholar]
- Withnall, M.; Lindelöf, E.; Engkvist, O.; Chen, H. Building attention and edge message passing neural networks for bioactivity and physical-chemical property prediction. J. Cheminform. 2020, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Torng, W.; Altman, R.B. Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model. 2019, 59, 4131–4149. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.Y.; Fokoue, A.; Luo, H.; Iyengar, A.; Dey, S.; Zhang, P. Interpretable Drug Target Prediction Using Deep Neural Representation. In Proceedings of the 2017 International Joint Conference on Artificial Intelligence, Melbourne, Australia, 19–25 August 2017. [Google Scholar]
- Zhao, Q.; Xiao, F.; Yang, M.; Li, Y.; Wang, J. AttentionDTA: Prediction of drug-target binding affinity using attention model. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine, San Diego, CA, USA, 18–21 November 2019; pp. 64–69. [Google Scholar]
- Cortés-Ciriano, I.; Ain, Q.U.; Subramanian, V.; Lenselink, E.B.; Méndez-Lucio, O.; IJzerman, A.P.; Wohlfahrt, G.; Prusis, P.; Malliavin, T.E.; van Westen, G.J.P.; et al. Polypharmacology modelling using proteochemometrics (PCM): Recent methodological developments, applications to target families, and future prospects. Medchemcomm 2015, 6, 24–50. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.C.A.; Prudêncio, R.B.C.; Costa, I.G. A multiple kernel learning algorithm for drug-target interaction prediction. BMC Bioinforma. 2016, 17, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug-target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Xia, Z.; Wu, L.Y.; Zhou, X.; Wong, S.T. Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. BMC Syst. Biol. 2010, 4, S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wu, M.; Miao, C.; Zhao, P.; Li, X.-L. Neighborhood Regularized Logistic Matrix Factorization for Drug-Target Interaction Prediction. PLoS Comput. Biol. 2016, 12, e1004760. [Google Scholar] [CrossRef] [PubMed]
- Metz, J.T.; Johnson, E.F.; Soni, N.B.; Merta, P.J.; Kifle, L.; Hajduk, P.J. Navigating the kinome. Nat. Chem. Biol. 2011, 7, 200–202. [Google Scholar] [CrossRef]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. DeepWalk: Online Learning of Social Representations. In Proceeding of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 24–27 August 2014; pp. 701–710. [Google Scholar] [CrossRef] [Green Version]
- Camacho, D.M.; Collins, K.M.; Powers, R.K.; Costello, J.C.; Collins, J.J. Next-Generation Machine Learning for Biological Networks. Cell 2018, 173, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [Green Version]
- David, L.; Arús-Pous, J.; Karlsson, J.; Engkvist, O.; Bjerrum, E.J.; Kogej, T.; Kriegl, J.M.; Beck, B.; Chen, H. Applications of Deep-Learning in Exploiting Large-Scale and Heterogeneous Compound Data in Industrial Pharmaceutical Research. Front. Pharmacol. 2019, 10, 1303. [Google Scholar] [CrossRef] [Green Version]
- Bleakley, K.; Yamanishi, Y. Supervised prediction of drug-target interactions using bipartite local models. Bioinformatics 2009, 25, 2397–2403. [Google Scholar] [CrossRef] [Green Version]
- Keum, J.; Nam, H. SELF-BLM: Prediction of drug-target interactions via self-training SVM. PLoS ONE 2017, 12, e0171839. [Google Scholar] [CrossRef] [Green Version]
- Hao, M.; Wang, Y.; Bryant, S.H. Improved prediction of drug-target interactions using regularized least squares integrating with kernel fusion technique. Anal. Chim. Acta 2016, 909, 41. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. Drug–target interaction prediction by random walk on the heterogeneous network. Mol. Biosyst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Bedi, P.; Sharma, C.; Vashisth, P.; Goel, D.; Dhanda, M. Handling cold start problem in Recommender Systems by using Interaction Based Social Proximity factor. In Proceeding of the 2015 International Conference on Advances in Computing, Communications and Informatics, Kerala, India, 10–13 August 2015; pp. 1987–1993. [Google Scholar] [CrossRef]
- Yu, H.; Choo, S.; Park, J.; Jung, J.; Kang, Y.; Lee, D. Prediction of drugs having opposite effects on disease genes in a directed network. BMC Syst. Biol. 2016, 10, S2. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef]
- Duda, M.; Zhang, H.; Li, H.-D.; Wall, D.P.; Burmeister, M.; Guan, Y. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl. Psychiatry 2018, 8, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.-P.; Hsieh, Y.-Y.; Chou, C.-J.; Yang, P.-M. Systematic polypharmacology and drug repurposing via an integrated L1000-based Connectivity Map database mining. R. Soc. Open Sci. 2018, 5, 181321. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Kim, S.; Lee, Y.-I.; Lee, J. Cellular Stress-Modulating Drugs Can Potentially Be Identified by in Silico Screening with Connectivity Map (CMap). Int. J. Mol. Sci. 2019, 20, 5601. [Google Scholar] [CrossRef] [Green Version]
- Hizukuri, Y.; Sawada, R.; Yamanishi, Y. Predicting target proteins for drug candidate compounds based on drug-induced gene expression data in a chemical structure-independent manner. BMC Med. Genomics 2015, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Sawada, R.; Iwata, M.; Tabei, Y.; Yamato, H.; Yamanishi, Y. Predicting inhibitory and activatory drug targets by chemically and genetically perturbed transcriptome signatures. Sci. Rep. 2018, 8, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arús-Pous, J.; Patronov, A.; Bjerrum, E.J.; Tyrchan, C.; Reymond, J.L.; Chen, H.; Engkvist, O. SMILES-based deep generative scaffold decorator for de-novo drug design. J. Cheminform. 2020, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Zhang, Z.; He, S.; Bo, X. DTIGCCN: Prediction of drug-target interactions based on GCN and CNN. In Proceedings of the 2020 IEEE 32nd International Conference on Tools with Artificial Intelligence (ICTAI), Baltimore, MD, USA, 9–11 November 2020; pp. 337–342. [Google Scholar] [CrossRef]
- Fefferman, C.; Mitter, S.; Narayanan, H. Testing the manifold hypothesis. J. Am. Math. Soc. 2016, 29, 983–1049. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning:Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xu, Y.; Yao, H.; Lin, K. Chemical space exploration based on recurrent neural networks: Applications in discovering kinase inhibitors. J. Cheminform. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- It, B.; Learning, M.; Autoen-, V.; Autoencoder, M.V.; Vae, E.; Vae, E.; Generative, N. Latent optimization variational autoencoder for conditional molecule generation. In Proceedings of the 8th International Conference on Learning Representations, Addis Ababa, Ethiopia, 26–30 April 2020; pp. 1–44. [Google Scholar]
- Aumentado-Armstrong, T. Latent molecular optimization for targeted therapeutic design. arXiv 2018, arXiv:1809.02032. [Google Scholar]
- Koge, D.; Ono, N.; Huang, M.; Altaf-Ul-Amin, M.; Kanaya, S. Embedding of Molecular Structure Using Molecular Hypergraph Variational Autoencoder with Metric Learning. Mol. Inform. 2021, 40, 1–7. [Google Scholar] [CrossRef]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of Generative Autoencoder in De Novo Molecular Design. Mol. Inform. 2018, 37, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Cho, K. Conditional Molecular Design with Deep Generative Models. J. Chem. Inf. Model. 2019, 59, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.H.; Ryu, S.; Lim, J.; Kim, W.Y. Molecular Generative Model Based on an Adversarially Regularized Autoencoder. J. Chem. Inf. Model. 2020, 60, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Bongini, P.; Bianchini, M.; Scarselli, F. Molecular graph generation with Graph Neural Networks. arXiv 2020, arXiv:2012.07397. [Google Scholar] [CrossRef]
- Bian, Y.; Xie, X.Q. (Sean) Computational Fragment-Based Drug Design: Current Trends, Strategies, and Applications. AAPS J. 2018, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mojica, M.F.; Bonomo, R.A.; Fast, W. B1-Metallo-β-Lactamases: Where Do We Stand? Curr. Drug Targets 2015, 17, 1029–1050. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Hierarchical Generation of Molecular Graphs using Structural Motifs. arXiv 2020, arXiv:2002.03230. [Google Scholar]
- Kawai, K.; Nagata, N.; Takahashi, Y. De novo design of drug-like molecules by a fragment-based molecular evolutionary approach. J. Chem. Inf. Model. 2014, 54, 49–56. [Google Scholar] [CrossRef]
- Spiegel, J.O.; Durrant, J.D. AutoGrow4: An open-source genetic algorithm for de novo drug design and lead optimization. J. Cheminform. 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Leguy, J.; Cauchy, T.; Glavatskikh, M.; Duval, B.; Da Mota, B. EvoMol: A flexible and interpretable evolutionary algorithm for unbiased de novo molecular generation. J. Cheminform. 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Li, Y.; Ooi, H.K.; Tchagang, A. Deep Evolutionary Learning for Molecular Design. arXiv 2020, arXiv:2102.01011. [Google Scholar]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J. Community benchmarks for virtual screening. J. Comput. Aided. Mol. Des. 2008, 22, 193–199. [Google Scholar] [CrossRef]
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef]
- Réau, M.; Langenfeld, F.; Zagury, J.F.; Lagarde, N.; Montes, M. Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 2018, 9, 11. [Google Scholar] [CrossRef]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0—A public library of challenging docking benchmark sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef]
- Xia, J.; Jin, H.; Liu, Z.; Zhang, L.; Wang, X.S. An Unbiased Method To Build Benchmarking Sets for Ligand-Based Virtual Screening and its Application To GPCRs. J. Chem. Inf. Model. 2014, 54, 1433–1450. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An unbiased data set for machine learning and virtual screening. J. Chem. Inf. Model. 2020, 60, 4263–4273. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2018, 11, 1931. [Google Scholar]
- Brown, N.; Fiscato, M.; Segler, M.H.S.; Vaucher, A.C. GuacaMol: Benchmarking Models for de Novo Molecular Design. J. Chem. Inf. Model. 2019, 59, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Grant, L.L.; Sit, C.S. De novo molecular drug design benchmarking. RSC Med. Chem. 2021, 12, 1273–1280. [Google Scholar] [CrossRef]
- García, V.; Mollineda, R.A.; Sánchez, J.S. Index of Balanced Accuracy: A Performance Measure for Skewed Class Distributions. In Proceedings of the Pattern Recognition and Image Analysis, 4th Iberian Conference, IbPRIA 2009, Póvoa de Varzim, Portugal, 10–12 June 2009; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2009; Volume 5524, pp. 441–448. [Google Scholar] [CrossRef] [Green Version]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genomics 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Carrington, A.M.; Fieguth, P.W.; Qazi, H.; Holzinger, A.; Chen, H.H.; Mayr, F.; Manuel, D.G. A new concordant partial AUC and partial c statistic for imbalanced data in the evaluation of machine learning algorithms. BMC Med. Inform. Decis. Mak. 2020, 20, 4. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.P.; Roy, K. On some aspects of variable selection for partial least squares regression models. QSAR Comb. Sci. 2008, 27, 302–313. [Google Scholar] [CrossRef]
- Playe, B.; Stoven, V. Evaluation of network architecture and data augmentation methods for deep learning in chemogenomics. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Kotsias, P.-C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- González-Medina, M.; Owen, J.R.; El-Elimat, T.; Pearce, C.J.; Oberlies, N.H.; Figueroa, M.; Medina-Franco, J.L. Scaffold diversity of fungal metabolites. Front. Pharmacol. 2017, 8, 180. [Google Scholar] [CrossRef] [Green Version]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. Explainable Deep Relational Networks for Predicting Compound-Protein Affinities and Contacts. J. Chem. Inf. Model. 2021, 61, 46–66. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Tang, K.; Ouyang, Q.; Lai, L.; Pei, J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Tran-Nguyen, V.K.; Rognan, D. Benchmarking data sets from pubchem bioassay data: Current scenario and room for improvement. Int. J. Mol. Sci. 2020, 21, 1–22. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017, 45, D955. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.-F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. ExCAPE-DB: An integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminform. 2017, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Sun, F.; Kong, T.; Zhang, W.; Yang, C.; Liu, C. A survey on deep transfer learning. In Proceedings of the Artificial Neural Networks and Machine Learning – ICANN 2018, 27th International Conference on Artificial Neural Networks, Rhodes, Greece, 4–7 October 2018; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2018; Volume 11141, pp. 270–279. [Google Scholar] [CrossRef] [Green Version]
- Shorten, C.; Khoshgoftaar, T.M. A survey on Image Data Augmentation for Deep Learning. J. Big Data 2019, 6, 60. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Bender, A. Improved Chemical Structure-Activity Modeling Through Data Augmentation. J. Chem. Inf. Model. 2015, 55, 2682–2692. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Awale, M.; Probst, D.; Reymond, J.L. Exploring chemical space with machine learning. Chimia 2019, 73, 1018–1023. [Google Scholar] [CrossRef]
- Cho, Y.R.; Kang, M. Interpretable machine learning in bioinformatics. Methods 2020, 179, 1–2. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Kim, B. Towards A Rigorous Science of Interpretable Machine Learning. arXiv 2017, arXiv:1702.08608. [Google Scholar]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Khosla, A.; Lapedriza, A.; Oliva, A.; Torralba, A. Learning Deep Features for Discriminative Localization. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 26 June–1 July 2016; pp. 2921–2929. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lee, C.-W.; Su, B.-H.; Tseng, Y.J. A new explainable graph convolution network based on discrete method: Using water solubility as an example Abstract. 2015, p. 2232. Available online: https://virtualman.ai/uploads/image/file/17/A_new_explainable_graph_convolution_network_based_on_discrete_method.pdf (accessed on 10 September 2021).
- Gonczarek, A.; Tomczak, J.M.; Zaręba, S.; Kaczmar, J.; Dąbrowski, P.; Walczak, M.J. Learning Deep Architectures for Interaction Prediction in Structure-based Virtual Screening. Comput. Biol. Med. 2016, 100, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2018, 59, 895–913. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Brylinski, M. Comparative assessment of strategies to identify similar ligand-binding pockets in proteins. BMC Bioinform. 2018, 19, 91. [Google Scholar] [CrossRef]
- Hasan Mahmud, S.M.; Chen, W.; Jahan, H.; Dai, B.; Din, S.U.; Dzisoo, A.M. DeepACTION: A deep learning-based method for predicting novel drug-target interactions. Anal. Biochem. 2020, 610, 113978. [Google Scholar] [CrossRef]
- Chong, C.W.; Raveendran, P.; Mukundan, R. Translation and scale invariants of Legendre moments. Pattern Recognit. 2004, 37, 119–129. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef]
- Jiang, M.; Li, Z.; Zhang, S.; Wang, S.; Wang, X.; Yuan, Q.; Wei, Z. Drug-target affinity prediction using graph neural network and contact maps. RSC Adv. 2020, 10, 20701–20712. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [Green Version]
- Mongia, A.; Majumdar, A. Drug-Target Interaction prediction using Multi-Graph Regularized Deep Matrix Factorization. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Wu, X.; Li, X.; Wang, D.; Fu, Z.; Liu, X.; Wan, X.; Yang, T.; Luo, X.; Chen, K.; et al. Computational target fishing by mining transcriptional data using a novel Siamese spectral-based graph convolutional network. bioRxiv 2020, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The Pharmacogenomics Knowledge Base. Methods Mol. Biol. 2013, 1015, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Backman, T.W.H.; Evans, D.S.; Girke, T. Large-scale bioactivity analysis of the small-molecule assayed proteome. PLoS ONE 2017, 12, e0171413. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Southan, C.; Sharman, J.L.; Campo, B.; Cavanagh, D.R.; Alexander, S.P.H.; Davenport, A.P.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: Extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 2020, 48, D1006–D1021. [Google Scholar] [CrossRef]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36, D919–D922. [Google Scholar] [CrossRef]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: Mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016, 44, D1036D1044. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic Target Database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [Green Version]
- KiBA—A Benchmark Dataset for Drug Target Prediction—Helsingin Yliopisto. Available online: https://researchportal.helsinki.fi/fi/datasets/kiba-a-benchmark-dataset-for-drug-target-prediction (accessed on 12 August 2021).
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Exploring ToxCast Data: Citing ToxCast Data | US EPA. Available online: https://www.epa.gov/chemical-research/exploring-toxcast-data-citing-toxcast-data (accessed on 12 August 2021).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Park, S.; Min, D.; Kim, W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. Int. J. Mol. Sci. 2021, 22, 9983. https://doi.org/10.3390/ijms22189983
Kim J, Park S, Min D, Kim W. Comprehensive Survey of Recent Drug Discovery Using Deep Learning. International Journal of Molecular Sciences. 2021; 22(18):9983. https://doi.org/10.3390/ijms22189983
Chicago/Turabian StyleKim, Jintae, Sera Park, Dongbo Min, and Wankyu Kim. 2021. "Comprehensive Survey of Recent Drug Discovery Using Deep Learning" International Journal of Molecular Sciences 22, no. 18: 9983. https://doi.org/10.3390/ijms22189983
APA StyleKim, J., Park, S., Min, D., & Kim, W. (2021). Comprehensive Survey of Recent Drug Discovery Using Deep Learning. International Journal of Molecular Sciences, 22(18), 9983. https://doi.org/10.3390/ijms22189983