
A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice

 , ,
, , 
Abstract
:1. Introduction
2. Results
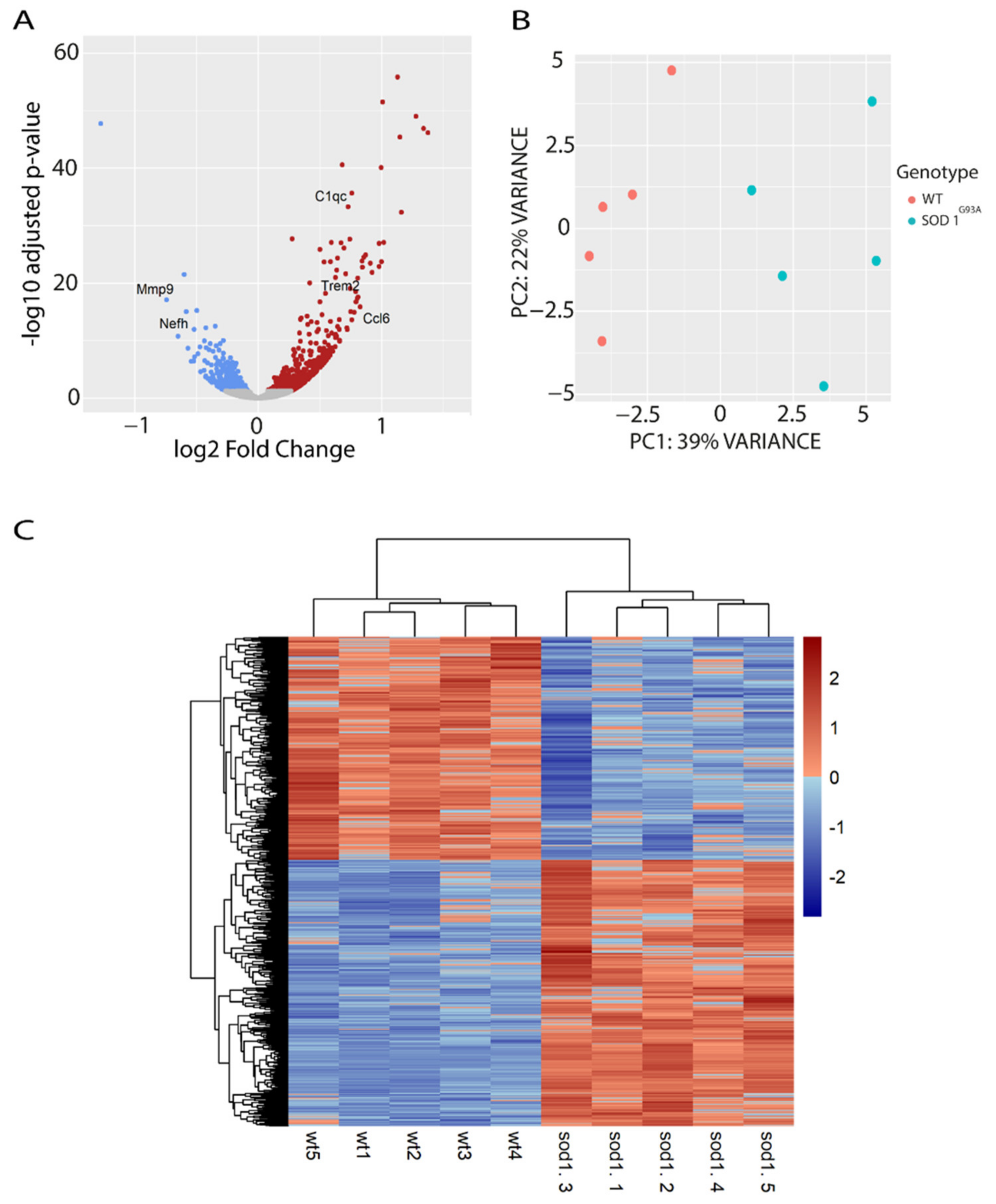
2.1. Analysis of Transcriptional Profile of the Spinal Cord from Early Symptomatic Disease Stage SOD1G93A Mice
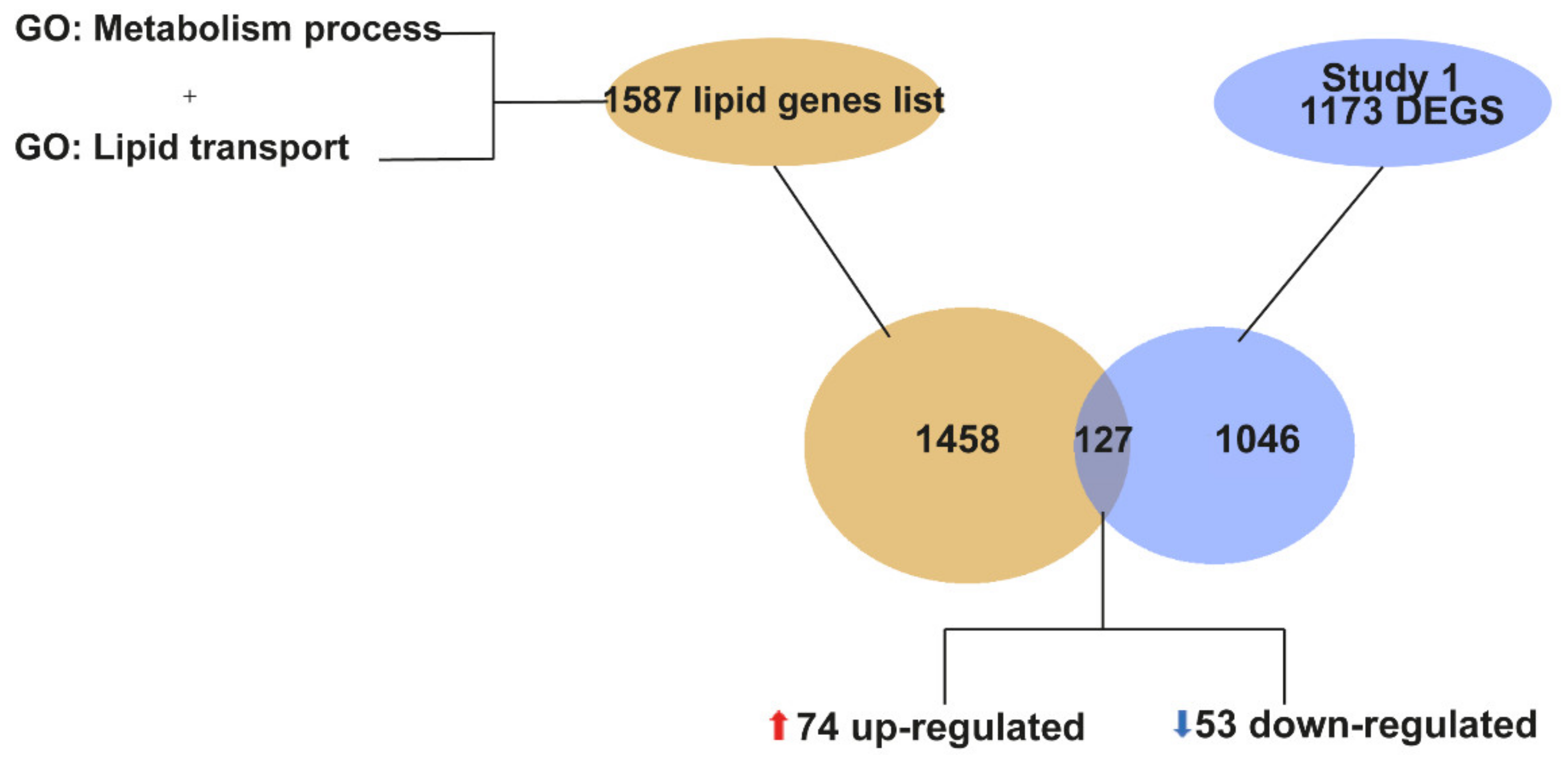
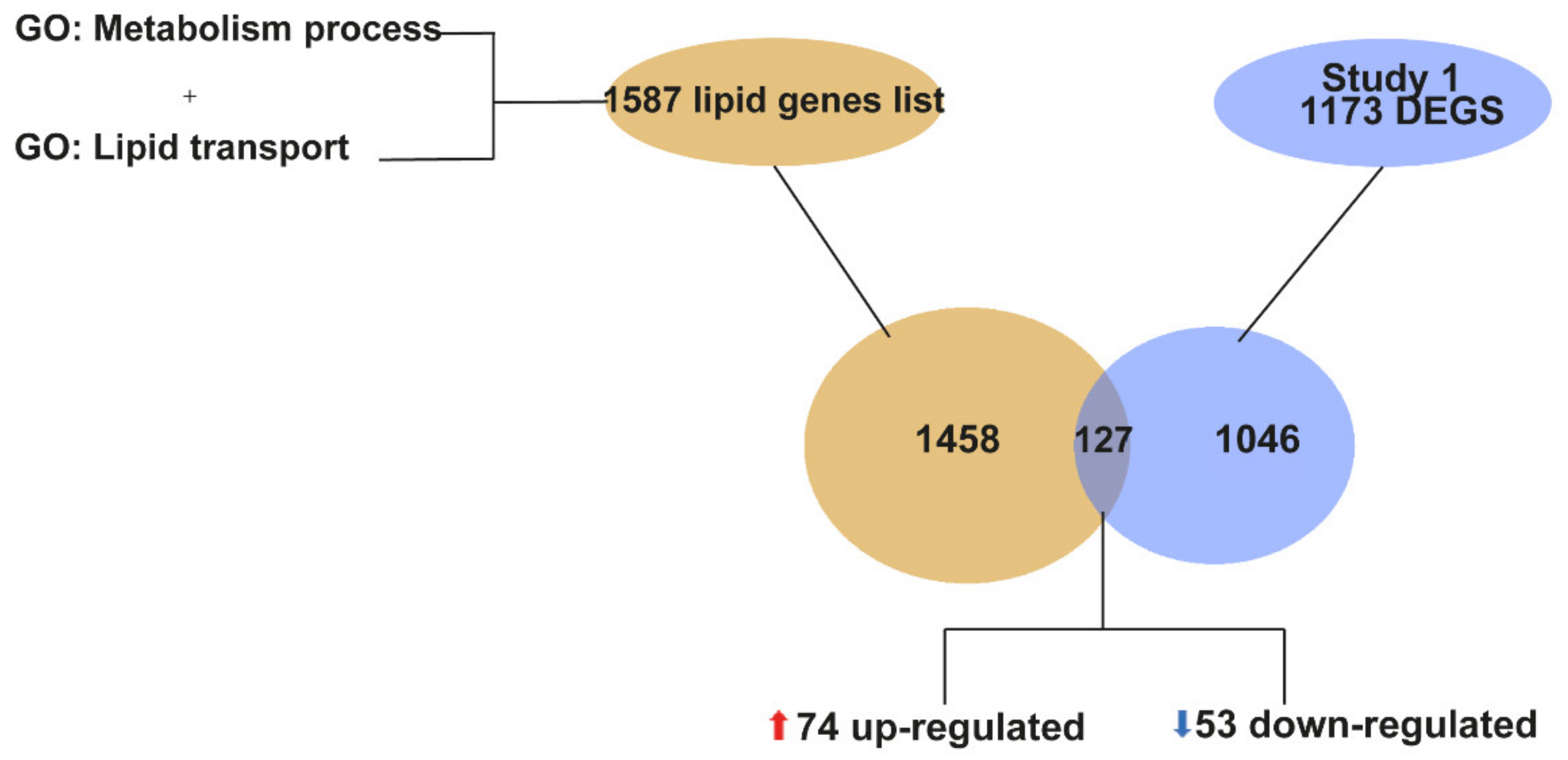
2.2. “Lipid Metabolism” Is a Biological Process That Is Transcriptionally Dysregulated in the Spinal Cord of SOD1G93A Mice at P90
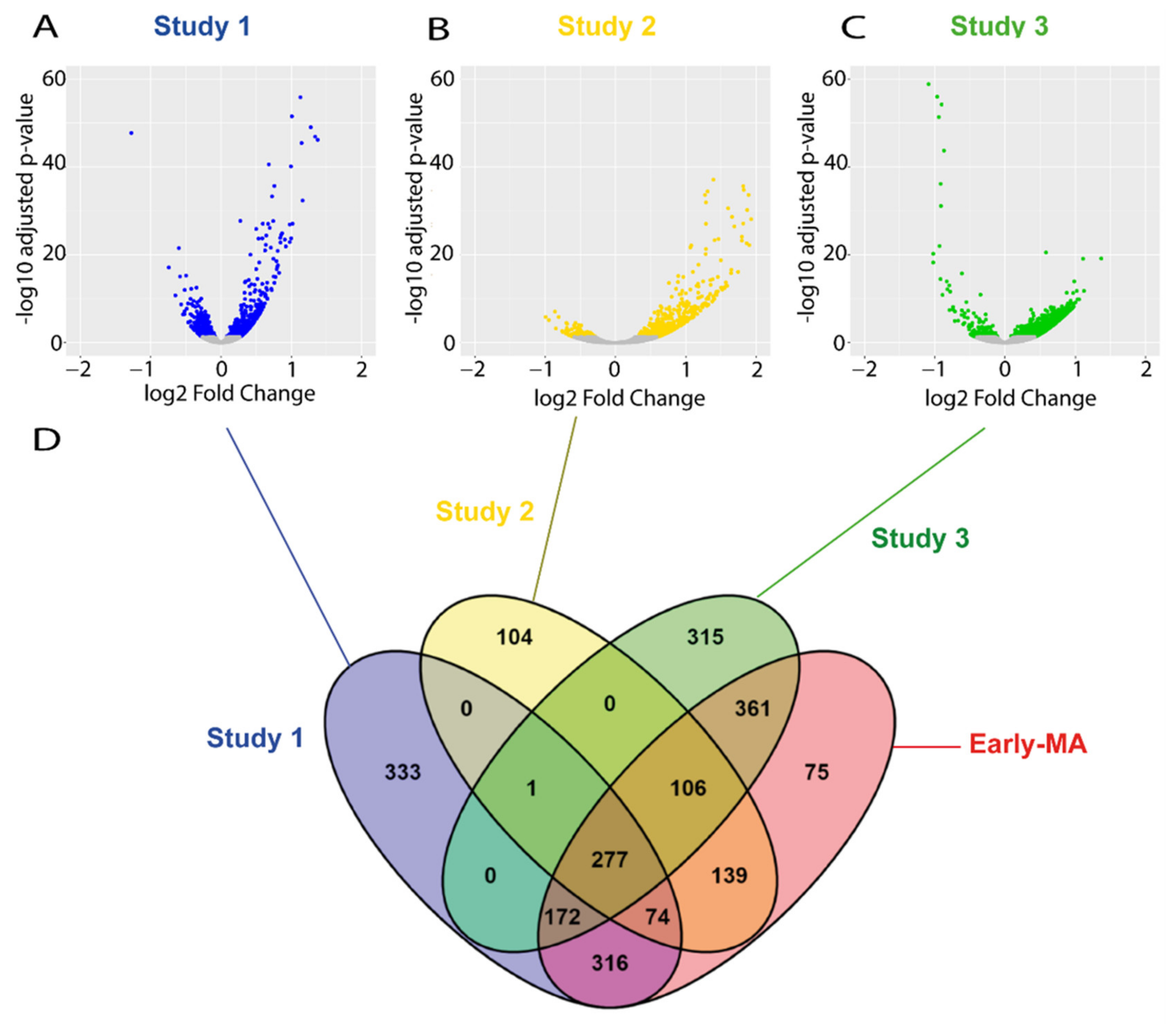
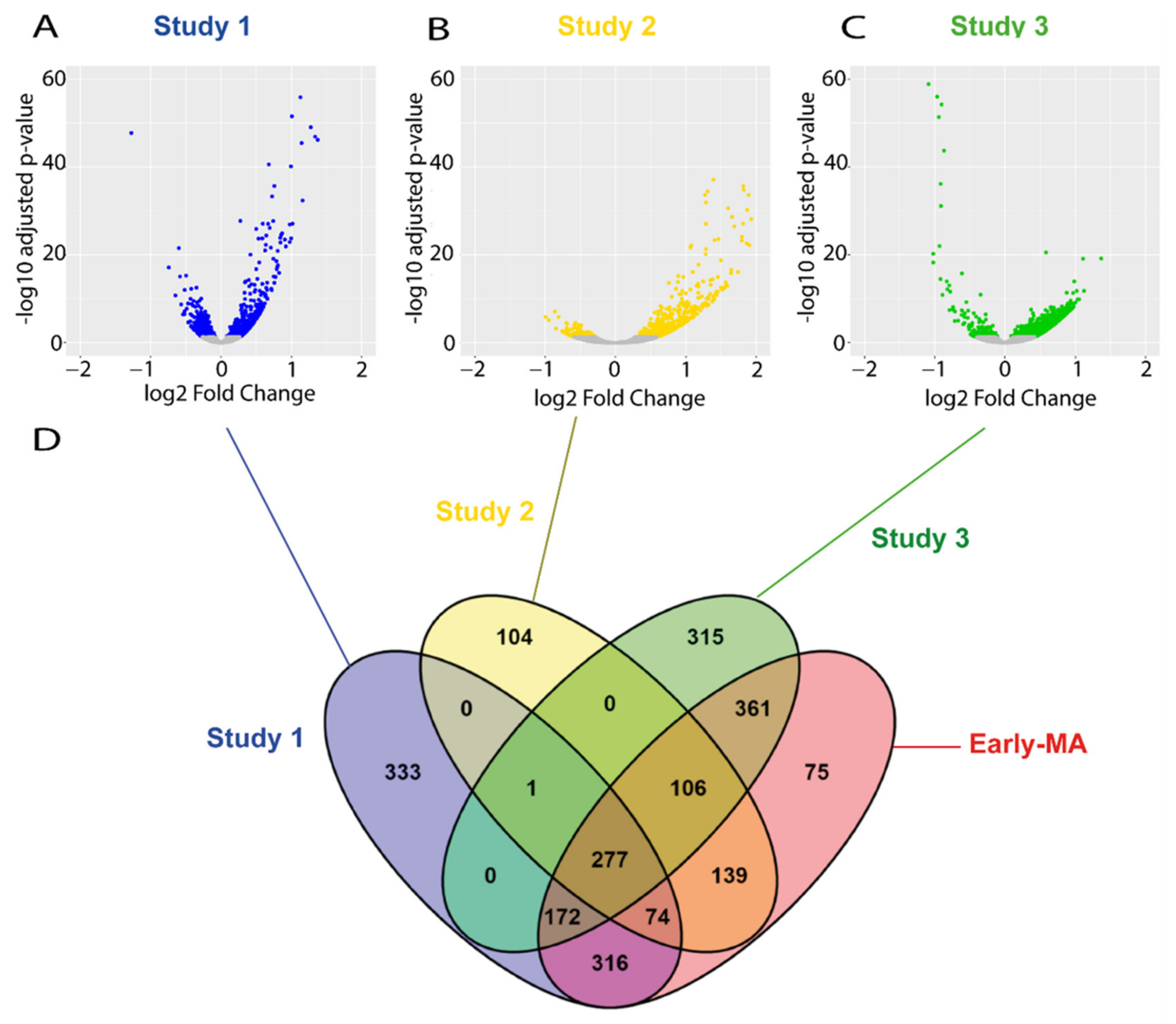
2.3. Meta-Analysis of RNA-seq Datasets from Spinal Cord of SOD1 Mice at Early and Late Symptomatic Disease Stages
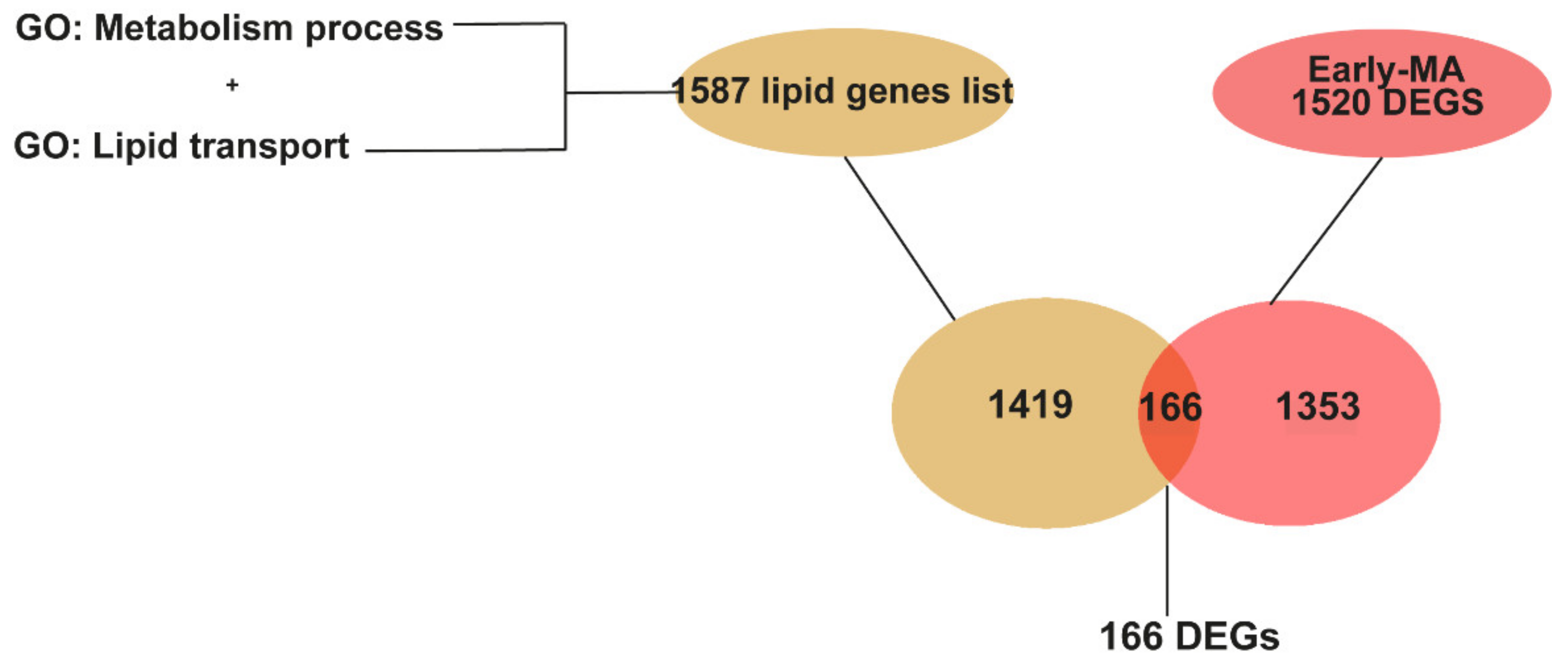
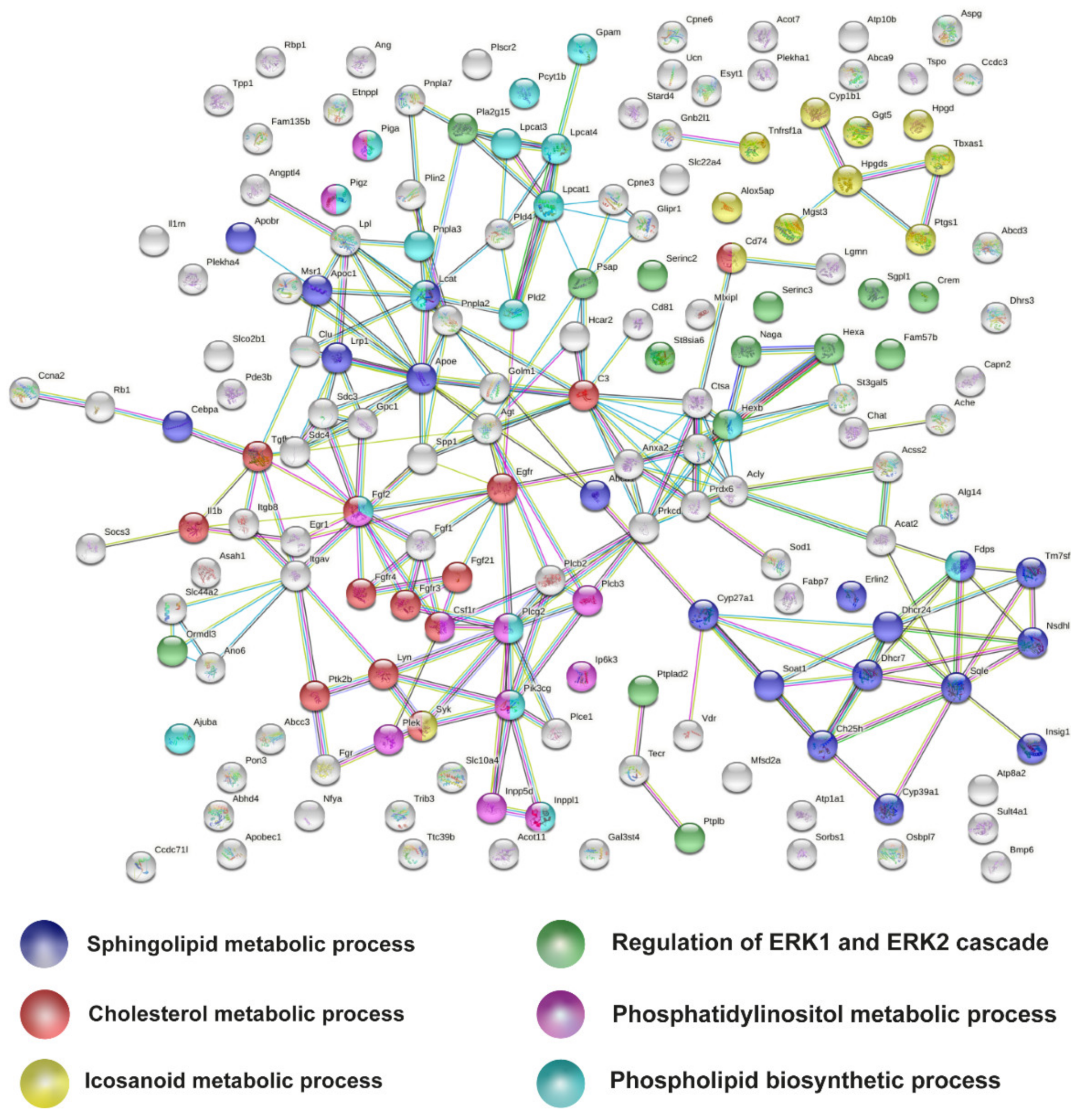
2.4. Meta-Analysis of RNA-seq Datasets Evidences Alterations in Lipid Metabolic Processes in the Spinal Cord of SOD1 Mice
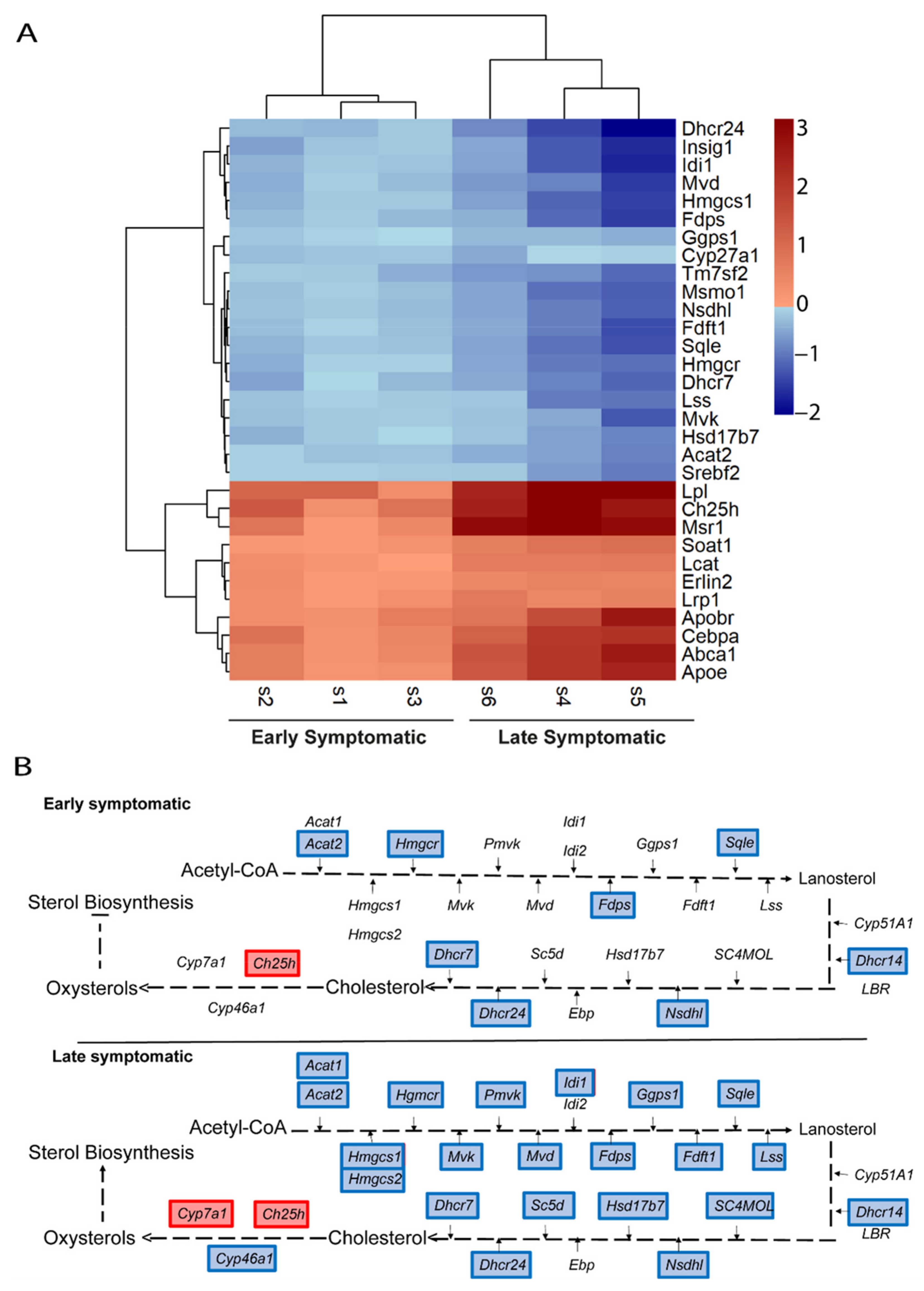
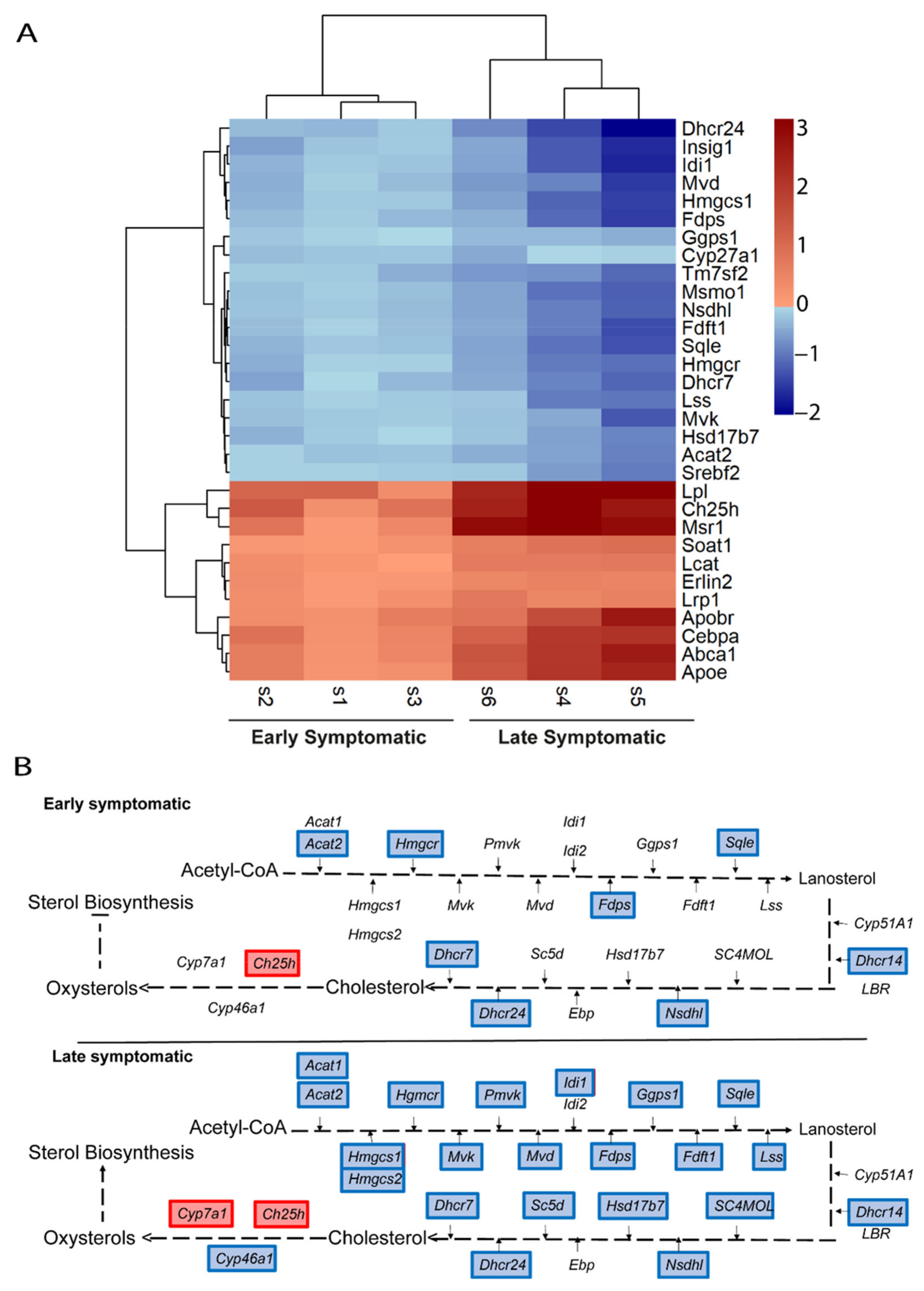
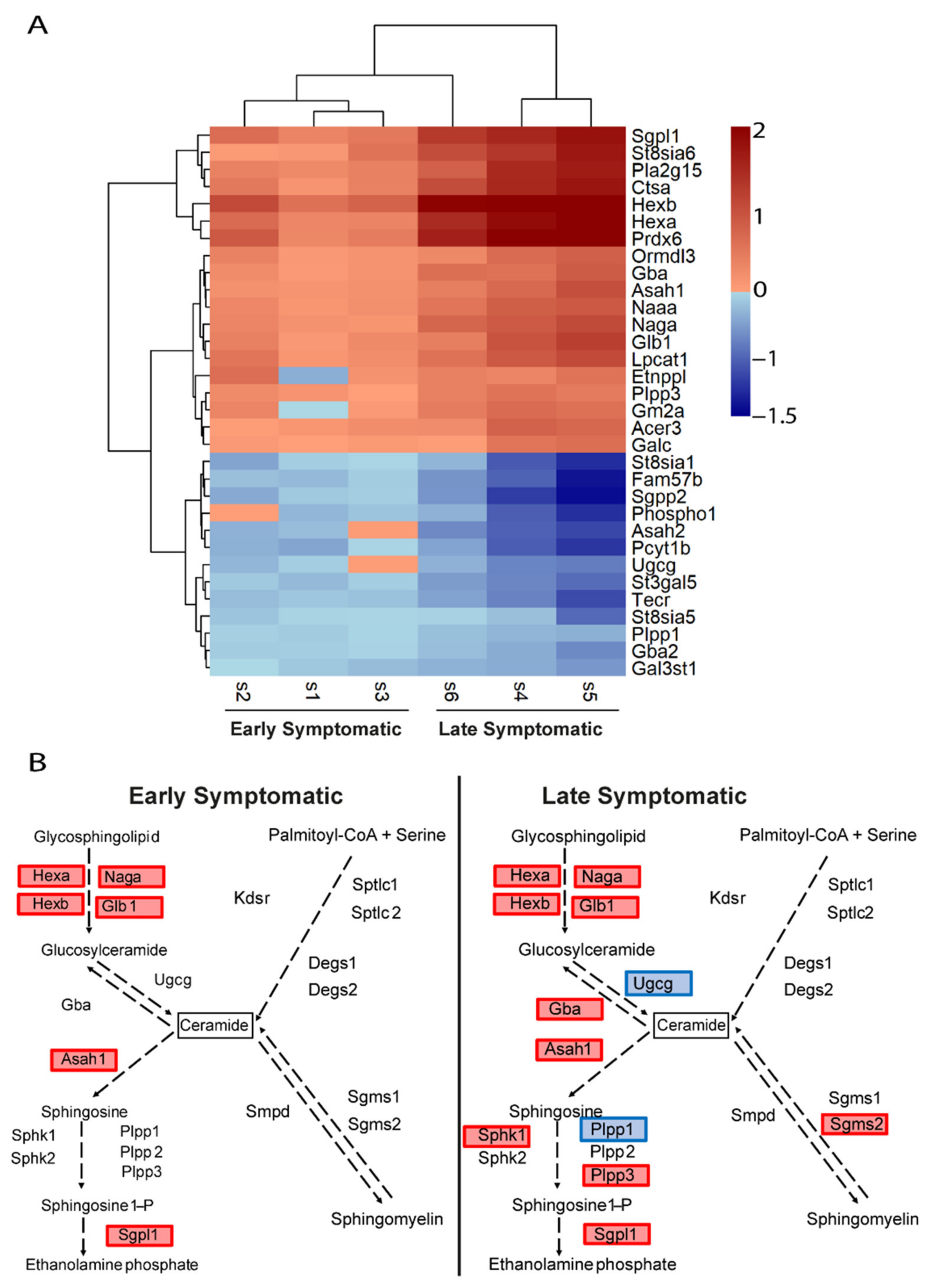
2.5. Metabolic Pathways of Cholesterol, Phospholipids, Ceramides, and Icosanoids Are Transcriptionally Altered at Early Symptomatic Disease Stage in the Spinal Cord of SOD1 Mice
2.6. The Transcriptional Dysregulation of Lipid Pathways in the Spinal Cord of SOD1 Mice Is Exacerbated at Late Disease Stage
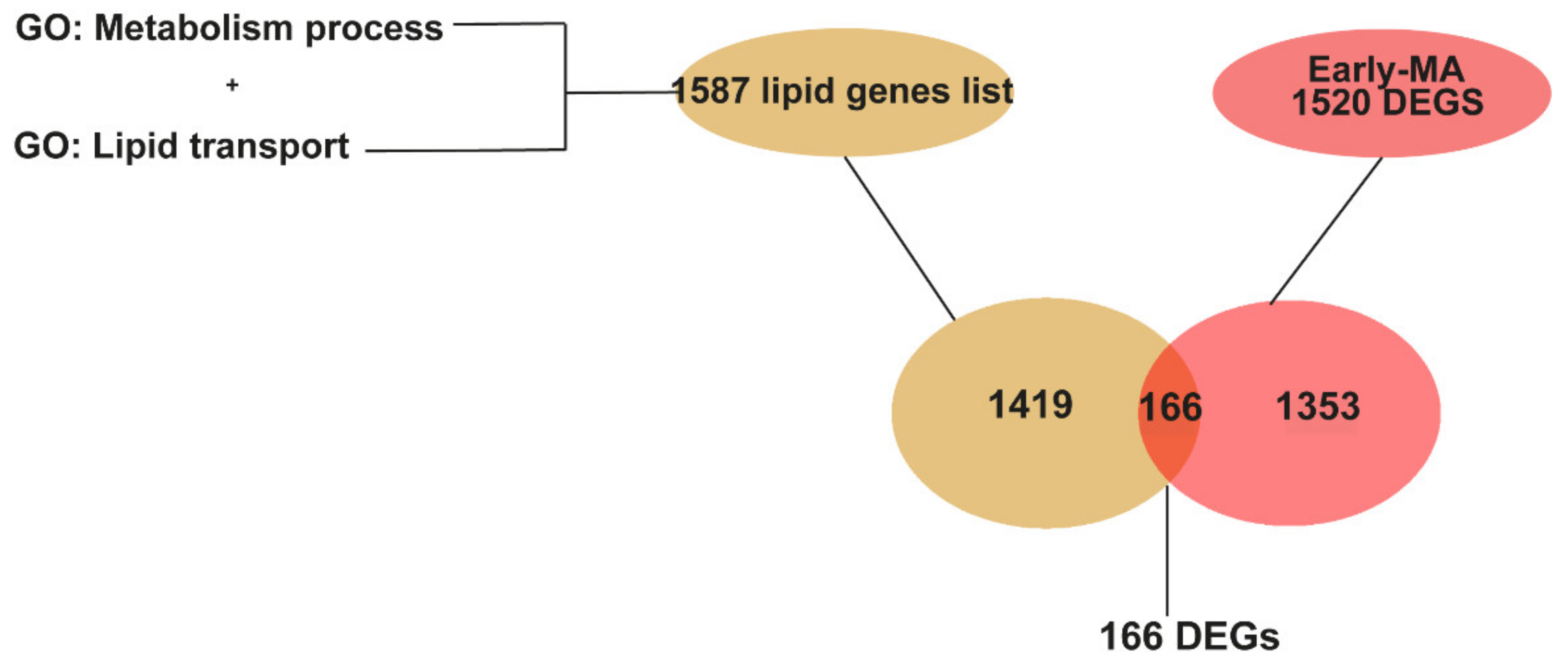
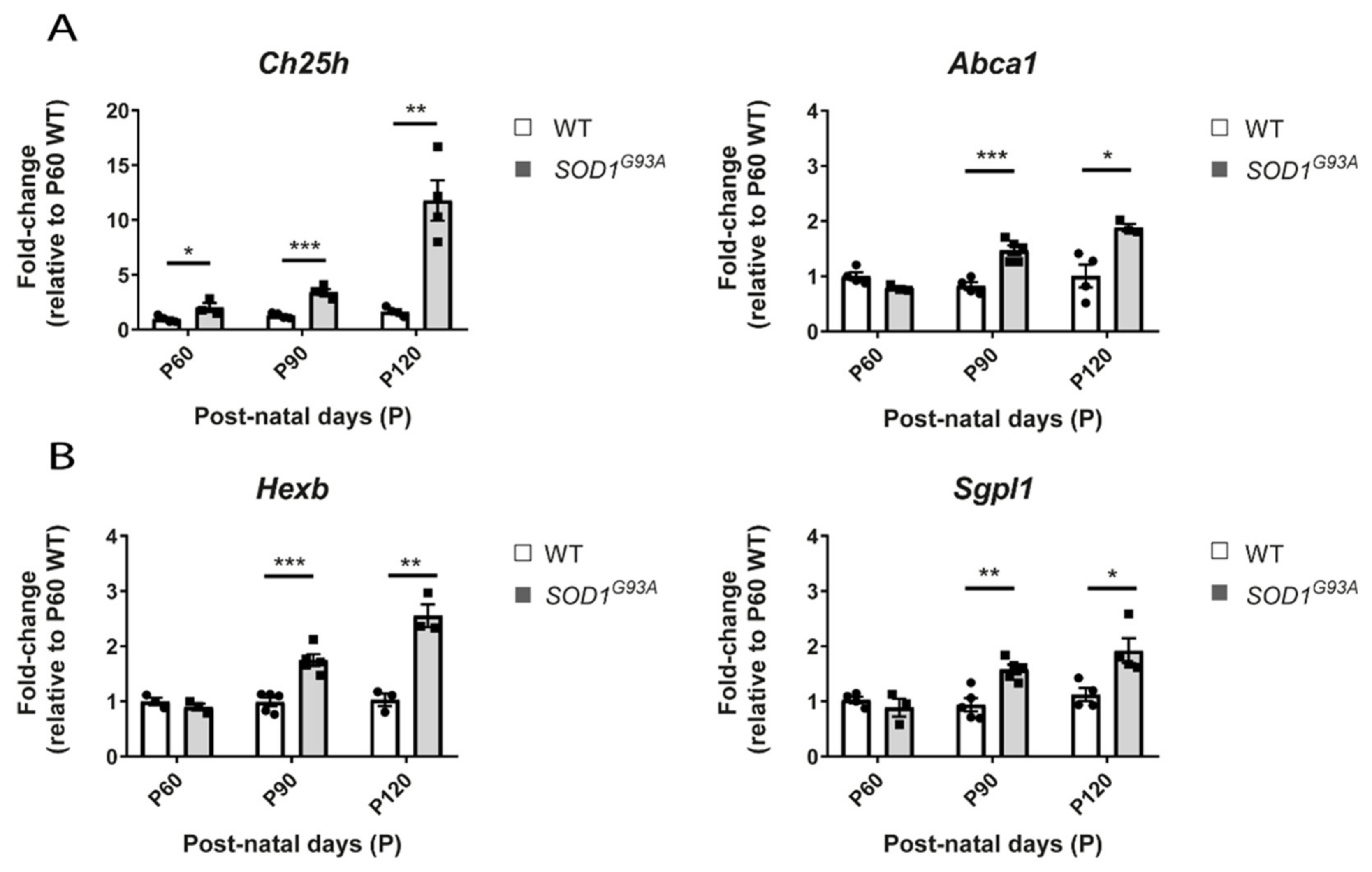
2.7. The Ch25h Gene Is an Early Marker of Lipid Alteratios in the Spinal Cord of SOD1 Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. RNA Extraction and Sequencing
4.3. RNA-seq Data Processing
4.4. Functional Enrichment Analysis
4.5. RNA-seq Databases Selected for the Meta-Analysis
4.6. Meta-Analysis of Combined RNA-seq Studies
4.7. Protein–Protein Interaction Network
4.8. Pathway Analysis
4.9. Quantitative PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Gregory, J.M.; Fagegaltier, D.; Phatnani, H.; Harms, M.B. Genetics of amyotrophic lateral sclerosis. Curr. Genet. Med. Rep. 2020, 8, 121–131. [Google Scholar] [CrossRef]
- Hamilton, J.; Hillard, C.J.; Spector, A.A.; Watkins, P.A. Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J. Mol. Neurosci. 2007, 33, 2–11. [Google Scholar] [CrossRef]
- Van Mantgem, M.R.J.; van Eijk, R.P.A.; van der Burgh, H.K.; Tan, H.H.G.; Westeneng, H.-J.; van Es, M.A.; Veldink, J.H.; van den Berg, L.H. Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2020, 91, 867–875. [Google Scholar] [CrossRef]
- Jawaid, A.; Murthy, S.B.; Wilson, A.M.; Qureshi, S.U.; Amro, M.J.; Wheaton, M.; Simpson, E.; Harati, Y.; Strutt, A.M.; York, M.K.; et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph. Lateral Scler. 2010, 11, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Moglia, C.; Calvo, A.; Grassano, M.; Canosa, A.; Manera, U.; D’Ovidio, F.; Bombaci, A.; Bersano, E.; Mazzini, L.; Mora, G.; et al. Early weight loss in amyotrophic lateral sclerosis: Outcome relevance and clinical correlates in a population-based cohort. J. Neurol. Neurosurg. Psychiatry 2019, 90, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; de Aguilar, J.-L.G.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Oudart, H.; Rene, F.; de Aguilar, J.-L.G.; Loeffler, J.-P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of high-caloric nutrition on survival in amyotrophic lateral sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef]
- Wills, A.-M.; Hubbard, J.; Macklin, E.; Glass, J.; Tandan, R.; Simpson, E.P.; Brooks, B.; Gelinas, D.; Mitsumoto, H.; Mozaffar, T.; et al. Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2014, 383, 2065–2072. [Google Scholar] [CrossRef] [Green Version]
- De Aguilar, J.-L.G. Lipid biomarkers for amyotrophic lateral sclerosis. Front. Neurol. 2019, 10, 284. [Google Scholar] [CrossRef]
- Dorst, J.; Kühnlein, P.; Hendrich, C.; Kassubek, J.; Sperfeld, A.D.; Ludolph, A.C. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J. Neurol. 2011, 258, 613–617. [Google Scholar] [CrossRef]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in lipid metabolism of spinal cord linked to amyotrophic lateral sclerosis. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cutler, R.G.; Pedersen, W.A.; Camandola, S.; Rothstein, J.D.; Mattson, M.P. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann. Neurol. 2002, 52, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Pharaoh, G.; Sataranatarajan, K.; Street, K.; Hill, S.; Gregston, J.; Ahn, B.; Kinter, C.; Kinter, M.; van Remmen, H. Metabolic and stress response changes precede disease onset in the spinal cord of mutant SOD1 ALS mice. Front. Neurosci. 2019, 13, 487. [Google Scholar] [CrossRef] [PubMed]
- Diekstra, F.P.; Saris, C.G.J.; van Rheenen, W.; Franke, L.; Jansen, R.C.; van Es, M.A.; van Vught, P.W.J.; Blauw, H.M.; Groen, E.J.N.; Horvath, S.; et al. Mapping of gene expression reveals CYP27A1 as a susceptibility gene for sporadic ALS. PLoS ONE 2012, 7, e35333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E. Erratum: Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation (Science (1772)). Science 1995, 269, 149. [Google Scholar] [CrossRef] [PubMed]
- Dodge, J.C.; Yu, J.; Sardi, S.P.; Shihabuddin, L.S. Sterol auto-oxidation adversely affects human motor neuron viability and is a neuropathological feature of amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 803. [Google Scholar] [CrossRef]
- Rau, A.; Marot, G.; Jaffrézic, F. Differential meta-analysis of RNA-seq data from multiple studies. BMC Bioinform. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutedja, N.A.; van der Schouw, Y.T.; Fischer, K.; Sizoo, E.M.; Huisman, M.H.B.; Veldink, J.H.; van den Berg, L.H. Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 638–642. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.W.; Kim, S.-M.; Kim, H.-J.; Kim, J.-E.; Park, K.S.; Kim, S.H.; Lee, K.-W.; Sung, J.-J. Hypolipidemia in patients with amyotrophic lateral sclerosis: A possible gender difference? J. Clin. Neurol. 2013, 9, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Kim, H.; Kim, J.-E.; Park, K.S.; Sung, J.-J.; Kim, S.H.; Lee, K.-W. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS ONE 2011, 6, e17985. [Google Scholar] [CrossRef]
- Zeng, P.; Zhou, X. Causal effects of blood lipids on amyotrophic lateral sclerosis: A Mendelian randomization study. Hum. Mol. Genet. 2019, 28, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuolikainen, A.; Ačimovič, J.; Lövgren-Sandblom, A.; Parini, P.; Andersen, P.M.; Björkhem, I. Cholesterol, oxysterol, triglyceride, and coenzyme Q homeostasis in ALS. Evidence against the hypothesis that elevated 27-hydroxycholesterol is a pathogenic factor. PLoS ONE 2014, 9, e113619. [Google Scholar] [CrossRef]
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.-A.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R.; et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res. 2017, 58, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodge, J.C.; Jensen, E.H.; Yu, J.; Sardi, S.P.; Bialas, A.R.; Taksir, T.V.; Bangari, D.S.; Shihabuddin, L.S. Neutral lipid cacostasis contributes to disease pathogenesis in amyotrophic lateral sclerosis. J. Neurosci. 2020, 40, 9137–9147. [Google Scholar] [CrossRef]
- Saeed, A.A.; Genové, G.; Li, T.; Lütjohann, D.; Olin, M.; Mast, N.; Pikuleva, I.; Crick, P.; Wang, Y.; Griffiths, W.; et al. Effects of a disrupted blood-brain barrier on cholesterol homeostasis in the brain. J. Biol. Chem. 2014, 289, 23712–23722. [Google Scholar] [CrossRef] [Green Version]
- Björkhem, I. Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Intern. Med. 2006, 260, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Björkhem, I.; Meaney, S. Brain Cholesterol: Long Secret Life Behind a Barrier. Arter. Thromb. Vasc. Biol. 2004, 24, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Nandar, W.; Neely, E.B.; Simmons, Z.; Connor, J.R. Statins accelerate disease progression and shorten survival in SOD1G93A mice. Muscle Nerve 2016, 54, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nefussy, B.; Hirsch, J.; Cudkowicz, M.E.; Drory, V.E. Gender-based effect of statins on functional decline in amyotrophic lateral sclerosis. J. Neurol. Sci. 2011, 300, 23–27. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.; McDonald, J.G.; Liang, G.; Russell, D.; Cyster, J.G. 25-hydroxycholesterol suppresses interleukin-1—Driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-M.; Noh, M.-Y.; Kim, H.; Cheon, S.-Y.; Lee, K.M.; Lee, J.; Cha, E.; Park, K.S.; Lee, K.-W.; Sung, J.-J.; et al. 25-hydroxycholesterol is involved in the pathogenesis of amyotrophic lateral sclerosis. Oncotarget 2017, 8, 11855–11867. [Google Scholar] [CrossRef] [Green Version]
- Marelli, C.; Lamari, F.; Rainteau, D.; Lafourcade, A.; Banneau, G.; Humbert, L.; Monin, M.-L.; Petit, E.; Debs, R.; Castelnovo, G.; et al. Plasma oxysterols: Biomarkers for diagnosis and treatment in spastic paraplegia type 5. Brain 2018, 141, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Canet-Pons, J.; Sen, N.-E.; Arsović, A.; Almaguer-Mederos, L.-E.; Halbach, M.V.; Key, J.; Döring, C.; Kerksiek, A.; Picchiarelli, G.; Cassel, R.; et al. Atxn2-CAG100-KnockIn mouse spinal cord shows progressive TDP43 pathology associated with cholesterol biosynthesis suppression. Neurobiol. Dis. 2021, 152, 105289. [Google Scholar] [CrossRef]
- Rickman, O.J.; Baple, E.L.; Crosby, A.H. Lipid metabolic pathways converge in motor neuron degenerative diseases. Brain 2020, 143, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- Dodge, J.C.; Treleaven, C.M.; Pacheco, J.; Cooper, S.; Bao, C.; Abraham, M.; Cromwell, M.; Sardi, S.P.; Chuang, W.-L.; Sidman, R.L.; et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2015, 112, 8100–8105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potenza, R.L.; de Simone, R.; Armida, M.; Mazziotti, V.; Pèzzola, A.; Popoli, P.; Minghetti, L. Fingolimod: A disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics 2016, 13, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Solomonov, Y.; Hadad, N.; Levy, R. Reduction of cytosolic phospholipase A2α upregulation delays the onset of symptoms in SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2016, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Shibata, N.; Kakita, A.; Takahashi, H.; Ihara, Y.; Nobukuni, K.; Fujimura, H.; Sakoda, S.; Kobayashi, M. Increased expression and activation of cytosolic phospholipase A2 in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2010, 119, 345–354. [Google Scholar] [CrossRef]
- Turner, M.R.; Goldacre, R.; Ramagopalan, S.; Talbot, K.; Goldacre, M.J. Autoimmune disease preceding amyotrophic lateral sclerosis: An epidemiologic study. Neurology 2013, 81, 1222–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Feature counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Rödel, E.; Fisher, R.A. Statistical methods for research workers, 14. aufl., Oliver & Boyd, Edinburgh, London 1970. XIII, 362 S., 12 abb., 74 tab., 40 s. Biom. Z. 1971, 13, 429–430. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated Genes | ||
| GO Term | Biological Process | FDR-Value 1 |
| GO:0002237 | response to molecule of bacterial origin | <1 × 10−13 |
| GO:0071216 | cellular response to biotic stimulus | <1 × 10−13 |
| GO:0002274 | myeloid leukocyte activation | <1 × 10−13 |
| GO:0002443 | leukocyte mediated immunity | 1.80 × 10−13 |
| GO:0001819 | positive regulation of cytokine production | 1.06 × 10−12 |
| GO:0006909 | phagocytosis | 1.37 × 10−12 |
| GO:0070661 | leukocyte proliferation | 5.78 × 10−12 |
| GO:0002250 | adaptive immune response | 2.01 × 10−11 |
| Downregulated Genes | ||
| GO Term | Biological Process | FDR-Value 1 |
| GO:0099177 | regulation of trans-synaptic signaling | 2.58 × 10−6 |
| GO:0042391 | regulation of membrane potential | 4.27 × 10−5 |
| GO:0051656 | establishment of organelle localization | 1.29 × 10−4 |
| GO:0061564 | axon development | 1.29 × 10−4 |
| GO:0051648 | vesicle localization | 1.57 × 10−4 |
| GO:0099504 | synaptic vesicle cycle | 1.64 × 10−4 |
| GO:0006836 | neurotransmitter transport | 3.4 × 10−4 |
| GO:0006887 | exocytosis | 3.99 × 10−4 |
| GO Term | Biological Process | FDR-Value 1 |
|---|---|---|
| GO:0071402 | cellular response to lipoprotein particle stimulus | 2.86 × 10−6 |
| GO:0055094 | response to lipoprotein particle | 1.11 × 10−5 |
| GO:0036314 | response to sterol | 5.13 × 10−4 |
| GO:0019216 | regulation of lipid metabolic process | 0.009 |
| GO:0006638 | neutral lipid metabolic process | 0.037 |
| GO:0046486 | glycerolipid metabolic process | 0.039 |
| Study | Dataset | Platform | Tissue | Age | Sample Size |
|---|---|---|---|---|---|
| Study 1 | Our study | Ilumina NovaSeq 6000 | Mouse spinal cord (lumbar) | 90 days | 5 WT 5 SOD1G93A (females) |
| Study 2 | GSE43879 | Ilumina Genome Analyzer II | Mouse spinal cord (unspecified region) | 95 days | 2 WT 2 SOD1G93A (both sexes) |
| Study 3 | GSE106364 | Ilumina HiSeq 4000 | Mouse spinal cord (lumbar) | 90 days | 5 WT 5 SOD1G86R (females) |
| Study 4 | GSE106803 | Ilumina HiSeq 2000 | Mouse spinal cord (unspecified region) | 5 months | 3 WT 3 SOD1G93A (both sexes) |
| Study 5 | GSE100888 | Ilumina HiSeq 2000 | Mouse spinal cord (lumbar) | 5 months | 4 WT 4 SOD1G93A (both sexes) |
| Study 6 | GSE433879 | Ilumina Genome Analyzer II | Mouse spinal cord (unspecified region) | 4 months | 2 WT 2 SOD1G93A (both sexes) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Beltrán, L.C.; Godoy-Corchuelo, J.M.; Losa-Fontangordo, M.; Williams, D.; Matias-Guiu, J.; Corrochano, S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. Int. J. Mol. Sci. 2021, 22, 9553. https://doi.org/10.3390/ijms22179553
Fernández-Beltrán LC, Godoy-Corchuelo JM, Losa-Fontangordo M, Williams D, Matias-Guiu J, Corrochano S. A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. International Journal of Molecular Sciences. 2021; 22(17):9553. https://doi.org/10.3390/ijms22179553
Chicago/Turabian StyleFernández-Beltrán, Luis C., Juan Miguel Godoy-Corchuelo, Maria Losa-Fontangordo, Debbie Williams, Jorge Matias-Guiu, and Silvia Corrochano. 2021. "A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice" International Journal of Molecular Sciences 22, no. 17: 9553. https://doi.org/10.3390/ijms22179553
APA StyleFernández-Beltrán, L. C., Godoy-Corchuelo, J. M., Losa-Fontangordo, M., Williams, D., Matias-Guiu, J., & Corrochano, S. (2021). A Transcriptomic Meta-Analysis Shows Lipid Metabolism Dysregulation as an Early Pathological Mechanism in the Spinal Cord of SOD1 Mice. International Journal of Molecular Sciences, 22(17), 9553. https://doi.org/10.3390/ijms22179553