
An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases




Abstract
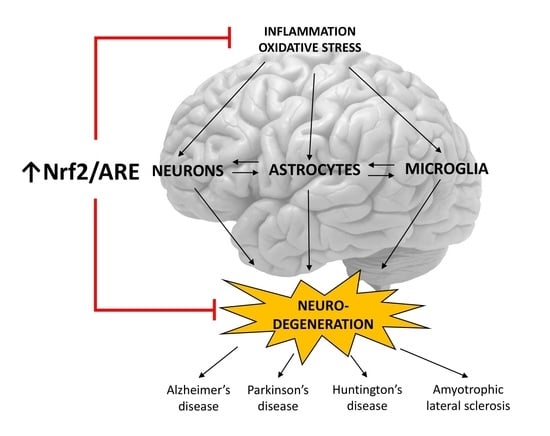
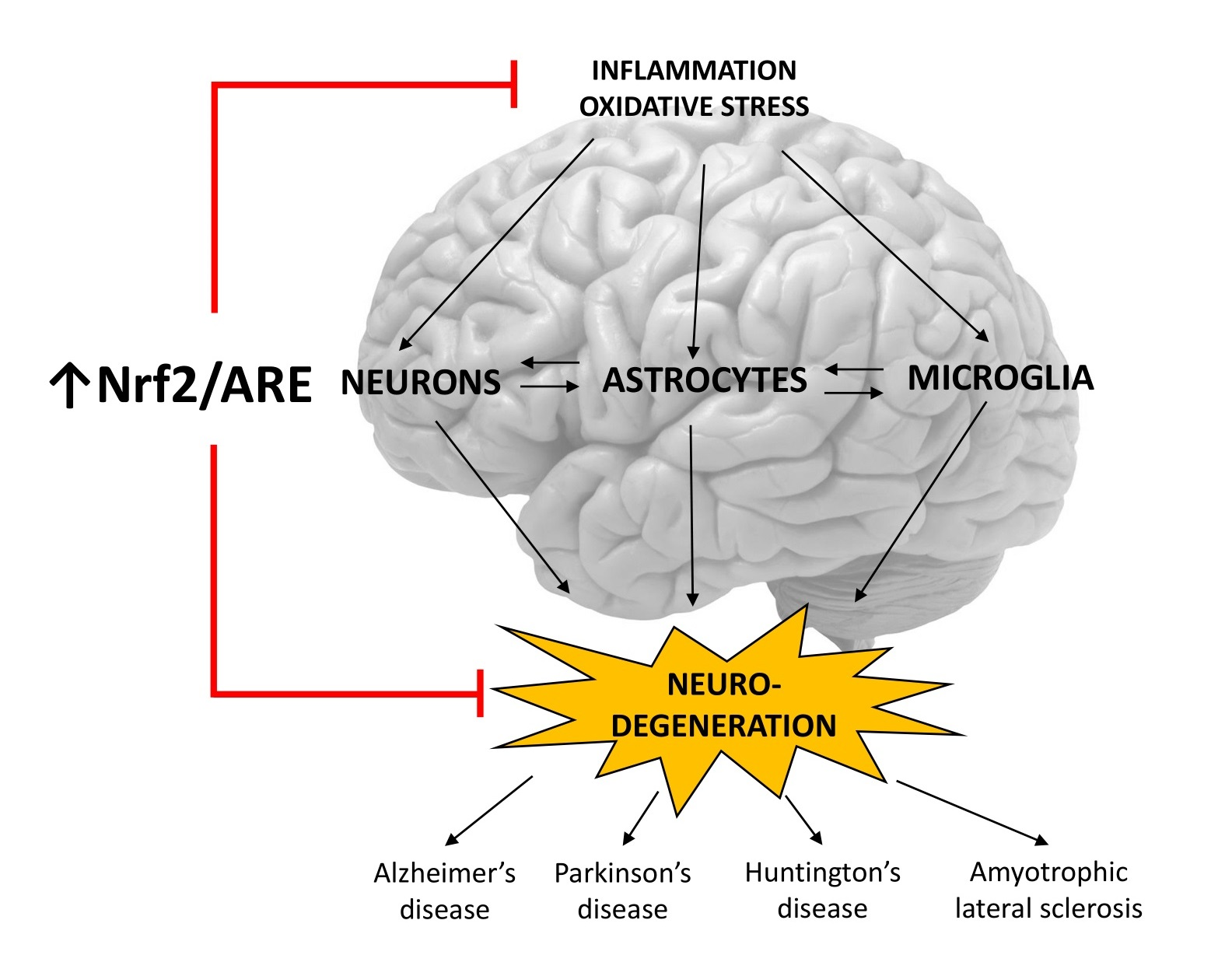
1. Introduction
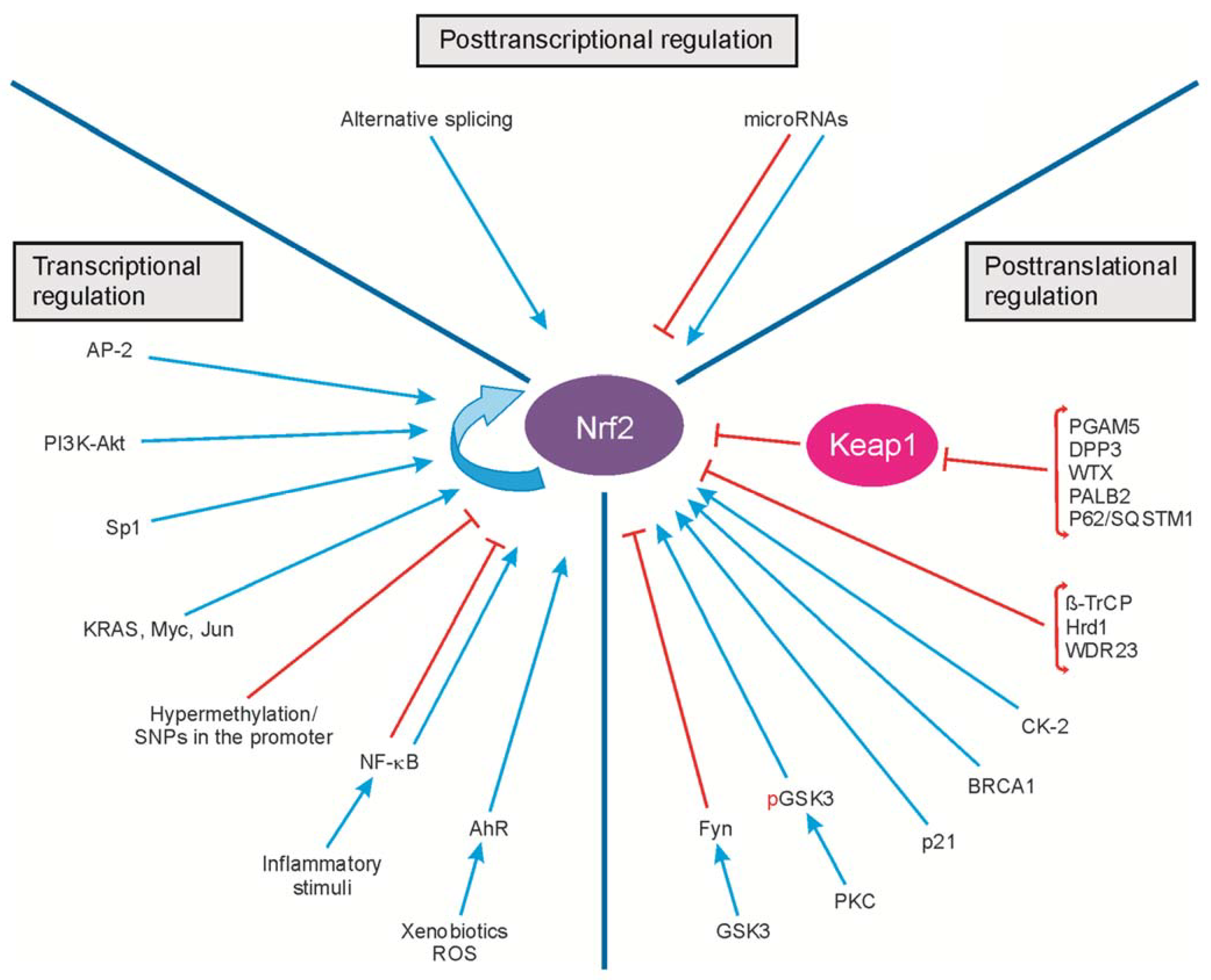
2. Regulation of Nrf2 Transcription
3. Regulation of Nrf2 Activity
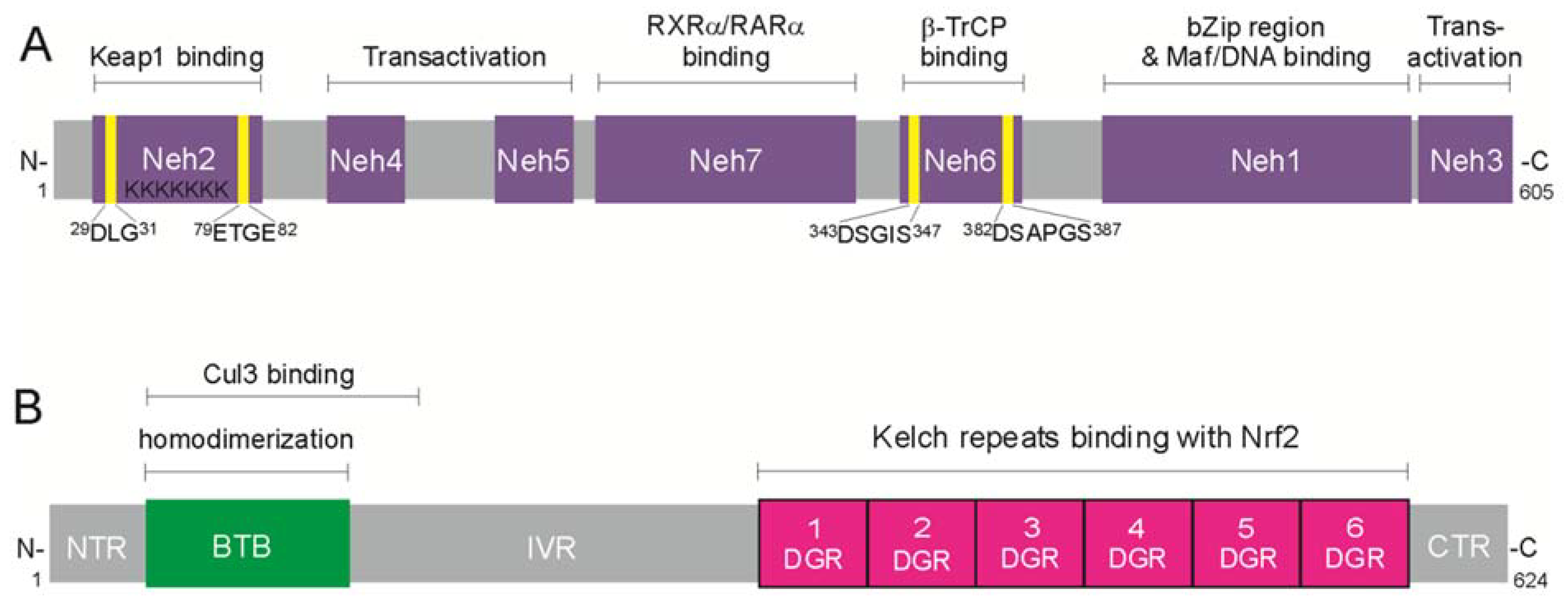
3.1. Structure of Nrf2 and Keap1 Inhibitor
3.2. Keap1-Dependent Nrf2 Activation
3.3. Keap1-Independent Nrf2 Activation
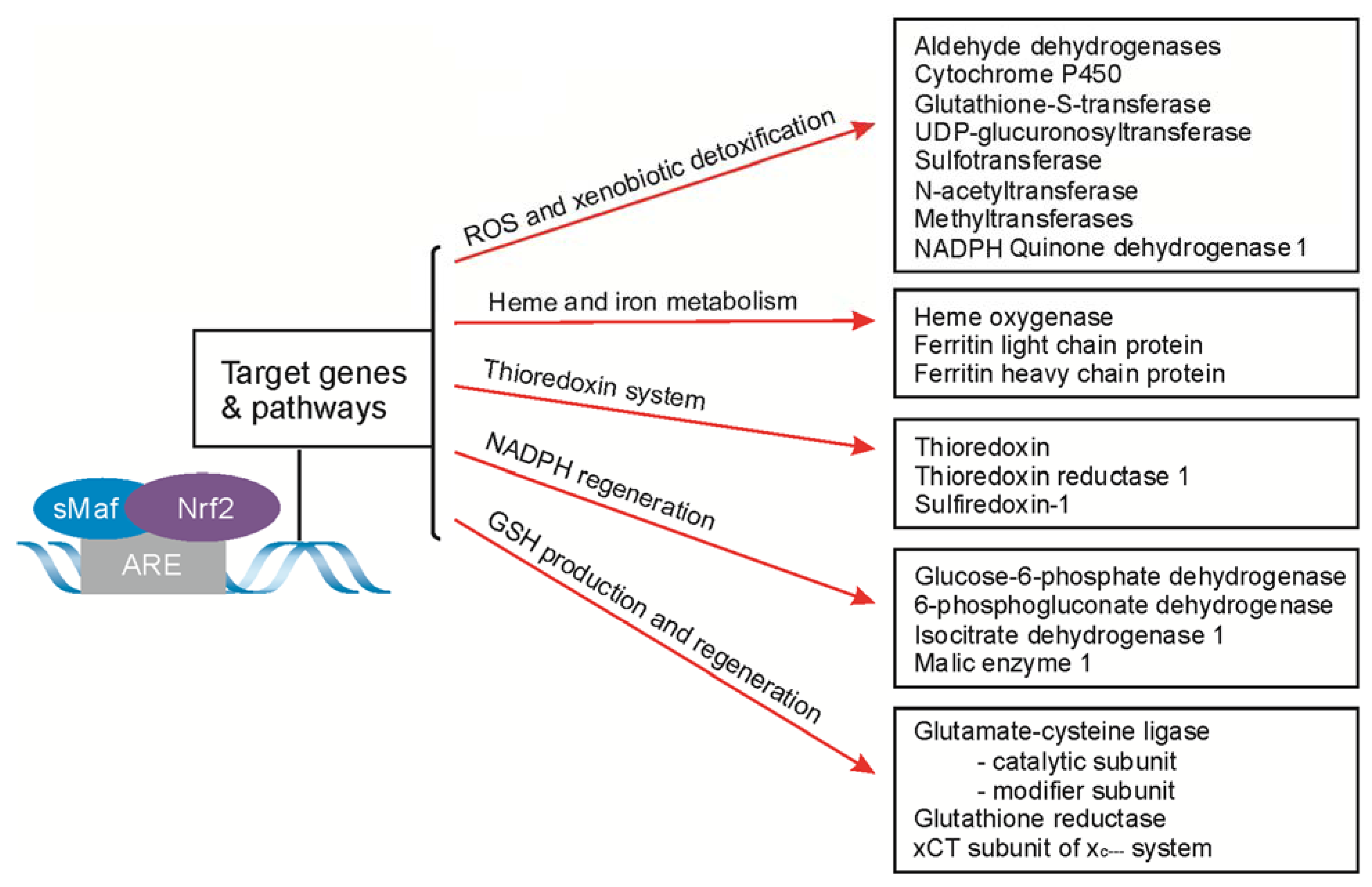
4. Nrf2-Signaling for ARE-Driven Genes
5. Role of the Nrf2/ARE Pathway in Neurodegenerative Diseases and Potential Therapeutic Targets
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Huntington’s Disease
5.4. Amyotrophic Lateral Sclerosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta 2015, 1849, 1260–1276. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharm. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 pathway in age-related neurological disorders: Insights into MicroRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Poganik, J.R.; Long, M.J.C.; Disare, M.T.; Liu, X.; Chang, S.H.; Hla, T.; Aye, Y. Post-transcriptional regulation of Nrf2-mRNA by the mRNA-binding proteins HuR and AUF1. FASEB J. 2019, 33, 14636–14652. [Google Scholar] [CrossRef]
- The National Center for Biotechnology Information, NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/4780/ (accessed on 16 August 2021).
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- UniProt Consortium. Available online: https://www.uniprot.org/uniprot/Q16236 (accessed on 16 August 2021).
- Kwak, M.-K.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a transcription factor for stress response and beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Yu, S.; Khor, T.O.; Cheung, K.L.; Li, W.; Wu, T.Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.N. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010, 5, e8579. [Google Scholar] [CrossRef]
- Cheng, D.; Wu, R.; Guo, Y.; Kong, A.-N.T. Regulation of Keap1-Nrf2 signaling: The role of epigenetics. Curr. Opin. Toxicol. 2016, 1, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Manolakou, S.D.; Ziros, P.G.; Sykiotis, G.P. NFE2L2 (nuclear factor, erythroid 2-like 2). Atlas Genet. Cytogenet. Oncol. Haematol. 2017, 2. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef]
- Eckert, D.; Buhl, S.; Weber, S.; Jäger, R.; Schorle, H. The AP-2 family of transcription factors. Genome Biol 2005, 6, 246. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Li, B.; Simon, M.C. Molecular Pathways: Targeting MYC-induced metabolic reprogramming and oncogenic stress in cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 5835–5841. [Google Scholar] [CrossRef]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol. 2006, 37, 668–674. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating NRF2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef]
- Kang, H.J.; Kim, H.J.; Kim, S.K.; Barouki, R.; Cho, C.-H.; Khanna, K.K.; Rosen, E.M.; Bae, I. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J. Biol. Chem. 2006, 281, 14654–14662. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, Q.; Xie, Y.; Shi, X.; Li, Y.; Peng, M.; Guo, H.; Sun, R.; Li, J.; Hong, Y.; et al. Breast cancer susceptibility protein 1 (BRCA1) rescues neurons from cerebral ischemia/reperfusion injury through NRF2-mediated antioxidant pathway. Redox Biol. 2018, 18, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Ryu, H.; Lee, J.; Zaman, K.; Kubilis, J.; Ferrante, R.J.; Ross, B.D.; Neve, R.; Ratan, R.R. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J. Neurosci. 2003, 23, 3597–3606. [Google Scholar] [CrossRef]
- Yan, L.J.; Fan, X.W.; Yang, H.T.; Wu, J.T.; Wang, S.L.; Qiu, C.G. MiR-93 inhibition ameliorates OGD/R induced cardiomyocyte apoptosis by targeting Nrf2. Eur. Rev. Med. Pharmacol. Sci 2017, 21, 5456–5461. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Y.; Hou, W.; Guo, L. MiR-153 regulates cardiomyocyte apoptosis by targeting Nrf2/HO-1 signaling. Chromosom. Res. 2019, 27, 167–178. [Google Scholar] [CrossRef]
- Zhao, Y.; Dong, D.; Reece, E.A.; Wang, A.R.; Yang, P. Oxidative stress-induced miR-27a targets the redox gene nuclear factor erythroid 2-related factor 2 in diabetic embryopathy. Am. J. Obs. Gynecol. 2018, 218, 136.e1–136.e10. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Jones, M.A.; Abdelrahman, A.A.; Powell, F.L.; Thounaojam, M.C.; Gutsaeva, D.; Bartoli, M.; Martin, P.M. Inhibiting microRNA-144 potentiates Nrf2-dependent antioxidant signaling in RPE and protects against oxidative stress-induced outer retinal degeneration. Redox Biol. 2020, 28, 101336. [Google Scholar] [CrossRef]
- Breving, K.; Esquela-Kerscher, A. The complexities of microRNA regulation: Mirandering around the rules. Int. J. Biochem. Cell Biol. 2010, 42, 1316–1329. [Google Scholar] [CrossRef]
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991. [Google Scholar] [CrossRef]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, M. miR-380-5p facilitates NRF2 and attenuates cerebral ischemia/reperfusion injury-induced neuronal cell death by directly targeting BACH1. Transl. Neurosci. 2021, 12, 210–217. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Qian, J. MicroRNA-152-3p protects neurons from oxygen-glucose-deprivation/reoxygenation-induced injury through upregulation of Nrf2/ARE antioxidant signaling by targeting PSD-93. Biochem. Biophys. Res. Commun. 2019, 517, 69–76. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef]
- Alam, J.; Killeen, E.; Gong, P.; Naquin, R.; Hu, B.; Stewart, D.; Ingelfinger, J.R.; Nath, K.A. Heme activates the heme oxygenase-1 gene in renal epithelial cells by stabilizing Nrf2. Am. J. Physiol. Ren. Physiol. 2003, 284, F743–F752. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef]
- Iso, T.; Suzuki, T.; Baird, L.; Yamamoto, M. Absolute Amounts and Status of the Nrf2-Keap1-Cul3 Complex within Cells. Mol. Cell. Biol. 2016, 36, 3100–3112. [Google Scholar] [CrossRef]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/NRF2 axis promotes human carcinogenesis. Antioxid. Redox Signal. 2014, 21, 2498–2514. [Google Scholar] [CrossRef]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J. Biol. Chem. 2006, 281, 12132–12142. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef]
- Lo, J.Y.; Spatola, B.N.; Curran, S.P. WDR23 regulates NRF2 independently of KEAP1. PLoS Genet. 2017, 13, e1006762. [Google Scholar] [CrossRef] [PubMed]
- Spatola, B.N.; Lo, J.Y.; Wang, B.; Curran, S.P. Nuclear and cytoplasmic WDR-23 isoforms mediate differential effects on GEN-1 and SKN-1 substrates. Sci. Rep. 2019, 9, 11783. [Google Scholar] [CrossRef]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response elemen. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [CrossRef]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell. Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Tamagawa, Y.; Kure, S.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Small Maf compound mutants display central nervous system neuronal degeneration, aberrant transcription, and Bach protein mislocalization coincident with myoclonus and abnormal startle response. Mol. Cell. Biol. 2003, 23, 1163–1174. [Google Scholar] [CrossRef][Green Version]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef]
- Muto, A.; Hoshino, H.; Madisen, L.; Yanai, N.; Obinata, M.; Karasuyama, H.; Hayashi, N.; Nakauchi, H.; Yamamoto, M.; Groudine, M.; et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3′ enhancer. EMBO J. 1998, 17, 5734–5743. [Google Scholar] [CrossRef]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef]
- Zhu, M.; Fahl, W.E. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem. Biophys. Res. Commun. 2001, 289, 212–219. [Google Scholar] [CrossRef]
- Liu, G.-H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, R.; Jaiswal, A.K. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 1998, 17, 3145–3156. [Google Scholar] [CrossRef] [PubMed]
- Sarcinelli, C.; Dragic, H.; Piecyk, M.; Barbet, V.; Duret, C.; Barthelaix, A.; Ferraro-Peyret, C.; Fauvre, J.; Renno, T.; Chaveroux, C.; et al. ATF4-Dependent. Cancers 2020, 12, 569. [Google Scholar] [CrossRef]
- Brown, S.L.; Sekhar, K.R.; Rachakonda, G.; Sasi, S.; Freeman, M.L. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008, 68, 364–368. [Google Scholar] [CrossRef]
- Hinoi, E.; Fujimori, S.; Wang, L.; Hojo, H.; Uno, K.; Yoneda, Y. Nrf2 negatively regulates osteoblast differentiation via interfering with Runx2-dependent transcriptional activation. J. Biol. Chem. 2006, 281, 18015–18024. [Google Scholar] [CrossRef]
- René, C.; Lopez, E.; Claustres, M.; Taulan, M.; Romey-Chatelain, M.C. NF-E2-related factor 2, a key inducer of antioxidant defenses, negatively regulates the CFTR transcription. Cell. Mol. Life Sci. 2010, 67, 2297–2309. [Google Scholar] [CrossRef]
- Liu, P.; de la Vega, M.; Sammani, S.; Mascarenhas, J.B.; Kerins, M.; Dodson, M.; Sun, X.; Wang, T.; Ooi, A.; Garcia, J.G.N.; et al. RPA1 binding to NRF2 switches ARE-dependent transcriptional activation to ARE-NRE-dependent repression. Proc. Natl. Acad. Sci. USA 2018, 115, E10352–E10361. [Google Scholar] [CrossRef]
- Chanas, S.A.; Jiang, Q.; McMahon, M.; McWalter, G.K.; McLellan, L.I.; Elcombe, C.R.; Henderson, C.J.; Wolf, C.R.; Moffat, G.J.; Itoh, K.; et al. Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 2002, 365, 405–416. [Google Scholar] [CrossRef]
- Vollrath, V.; Wielandt, A.M.; Iruretagoyena, M.; Chianale, J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 2006, 395, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef]
- Hawkes, H.-J.K.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef]
- Jayakumar, S.; Pal, D.; Sandur, S.K. Nrf2 facilitates repair of radiation induced DNA damage through homologous recombination repair pathway in a ROS independent manner in cancer cells. Mutat. Res. 2015, 779, 33–45. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Y.; Ji, K.; Liu, Y.; Kong, Y.; Nie, S.; Li, N.; Hao, J.; Xie, Y.; Xu, C.; et al. NRF2 preserves genomic integrity by facilitating ATR activation and G2 cell cycle arrest. Nucleic Acids Res. 2020, 48, 9109–9123. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef]
- Anedda, A.; López-Bernardo, E.; Acosta-Iborra, B.; Saadeh Suleiman, M.; Landázuri, M.O.; Cadenas, S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013, 61, 395–407. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Luna-Viramontes, N.I.; Campa-Córdoba, B.B.; Ontiveros-Torres, M.Á.; Harrington, C.R.; Villanueva-Fierro, I.; Guadarrama-Ortíz, P.; Garcés-Ramírez, L.; de la Cruz, F.; Hernandes-Alejandro, M.; Martínez-Robles, S.; et al. PHF-Core Tau as the Potential Initiating Event for Tau Pathology in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 247. [Google Scholar] [CrossRef]
- Delacourte, A.; David, J.P.; Sergeant, N.; Buée, L.; Wattez, A.; Vermersch, P.; Ghozali, F.; Fallet-Bianco, C.; Pasquier, F.; Lebert, F.; et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology 1999, 52, 1158–1165. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Ittner, L.M.; Götz, J. Amyloid-β and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef]
- Bahn, G.; Park, J.-S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef]
- Noble, W.; Hanger, D.P.; Miller, C.C.J.; Lovestone, S. The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 2013, 4, 83. [Google Scholar] [CrossRef]
- Williams, T.I.; Lynn, B.C.; Markesbery, W.R.; Lovell, M.A. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiol. Aging 2006, 27, 1094–1099. [Google Scholar] [CrossRef]
- Marcus, D.L.; Thomas, C.; Rodriguez, C.; Simberkoff, K.; Tsai, J.S.; Strafaci, J.A.; Freedman, M.L. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp. Neurol. 1998, 150, 40–44. [Google Scholar] [CrossRef]
- Zengi, O.; Karakas, A.; Ergun, U.; Senes, M.; Inan, L.; Yucel, D. Urinary 8-hydroxy-2′-deoxyguanosine level and plasma paraoxonase 1 activity with Alzheimer’s disease. Clin. Chem. Lab. Med. 2011, 50, 529–534. [Google Scholar] [CrossRef]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S.; et al. Nrf2 Ablation Promotes Alzheimer’s Disease-Like Pathology in APP/PS1 Transgenic Mice: The Role of Neuroinflammation and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Wang, Y.; Santa-Cruz, K.; DeCarli, C.; Johnson, J.A. NAD(P)H:quinone oxidoreductase activity is increased in hippocampal pyramidal neurons of patients with Aalzheimer’s disease. Neurobiol. Aging 2000, 21, 525–531. [Google Scholar] [CrossRef]
- Schipper, H.M.; Cissé, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- von Otter, M.; Landgren, S.; Nilsson, S.; Zetterberg, M.; Celojevic, D.; Bergström, P.; Minthon, L.; Bogdanovic, N.; Andreasen, N.; Gustafson, D.R.; et al. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech. Ageing Dev. 2010, 131, 105–110. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Moreira, P.I.; Siedlak, S.L.; Wang, X.; Santos, M.S.; Oliveira, C.R.; Tabaton, M.; Nunomura, A.; Szweda, L.I.; Aliev, G.; Smith, M.A.; et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy 2007, 3, 614–615. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Flint Beal, M. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef]
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The multifaceted role of Nrf2 in mitochondrial function. Curr. Opin. Toxicol. 2016, 1, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid beta formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef]
- Akhter, H.; Katre, A.; Li, L.; Liu, X.; Liu, R.M. Therapeutic potential and anti-amyloidosis mechanisms of tert-butylhydroquinone for Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Hui, Y.; Chengyong, T.; Cheng, L.; Haixia, H.; Yuanda, Z.; Weihua, Y. Resveratrol Attenuates the Cytotoxicity Induced by Amyloid-β. Neurochem. Res. 2018, 43, 297–305. [Google Scholar] [CrossRef]
- Solberg, N.O.; Chamberlin, R.; Vigil, J.R.; Deck, L.M.; Heidrich, J.E.; Brown, D.C.; Brady, C.I.; Vander Jagt, T.A.; Garwood, M.; Bisoffi, M.; et al. Optical and SPION-enhanced MR imaging shows that trans-stilbene inhibitors of NF-κB concomitantly lower Alzheimer’s disease plaque formation and microglial activation in AβPP/PS-1 transgenic mouse brain. J. Alzheimers Dis. 2014, 40, 191–212. [Google Scholar] [CrossRef]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.H.; Pipingas, A.; Scholey, A.B. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J. Psychopharmacol. 2015, 29, 642–651. [Google Scholar] [CrossRef]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. 2012, 4, 43. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.W.; Grossman, H.; Neugroschl, J.; Parker, S.; Burden, A.; Luo, X.; Sano, M. A randomized, double-blind, placebo-controlled trial of resveratrol with glucose and malate (RGM) to slow the progression of Alzheimer’s disease: A pilot study. Alzheimers Dement. 2018, 4, 609–616. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Oczkowska, A.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Mutations in PRKN and SNCA Genes Important for the Progress of Parkinson’s Disease. Curr. Genom. 2013, 14, 502–517. [Google Scholar] [CrossRef]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12. [Google Scholar] [CrossRef]
- Wei, Z.; Li, X.; Liu, Q.; Cheng, Y. Oxidative Stress in Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Colamartino, M.; Duranti, G.; Ceci, R.; Sabatini, S.; Testa, A.; Cozzi, R. A multi-biomarker analysis of the antioxidant efficacy of Parkinson’s disease therapy. Toxicol. In Vitro 2018, 47, 1–7. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.-X.; Dai, S.-X.; Guo, Y.-C.; Han, F.-F.; Zheng, J.-J.; Li, G.-H.; Huang, J.-F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimers. Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef]
- Williamson, T.P.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway by siRNA knockdown of Keap1 reduces oxidative stress and provides partial protection from MPTP-mediated neurotoxicity. Neurotoxicology 2012, 33, 272–279. [Google Scholar] [CrossRef]
- He, Q.; Song, N.; Jia, F.; Xu, H.; Yu, X.; Xie, J.; Jiang, H. Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2013, 45, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Jing, X.; Wei, X.; Shi, H.; Ren, D.; Zhang, X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology 2014, 79, 380–388. [Google Scholar] [CrossRef]
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on Nrf2 pathway. Biomed. Pharm. 2015, 75, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.E.; Mukhopadhyay, P.; Horváth, B.; Rajesh, M.; Tapia, E.; García-Torres, I.; Pedraza-Chaverri, J.; Pacher, P. Sulforaphane, a natural constituent of broccoli, prevents cell death and inflammation in nephropathy. J. Nutr. Biochem. 2012, 23, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Freeman, M.L.; Liebler, D.C. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Indu Viswanath, A.N.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef]
- Di Martino, R.M.C.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Pérez, C.; Martinez-Gonzalez, L.; Paglione, M.; Di Schiavi, E.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative damage in Huntington’s disease pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340. [Google Scholar] [CrossRef]
- Klepac, N.; Relja, M.; Klepac, R.; Hećimović, S.; Babić, T.; Trkulja, V. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: A cross-sectional study. J. Neurol. 2007, 254, 1676–1683. [Google Scholar] [CrossRef]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef]
- Pérez-Severiano, F.; Ríos, C.; Segovia, J. Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Res. 2000, 862, 234–237. [Google Scholar] [CrossRef]
- Jin, Y.N.; Yu, Y.V.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V.W. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Ganner, A.; Pfeiffer, Z.-C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902. [Google Scholar] [CrossRef]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef]
- Tsvetkov, A.S.; Arrasate, M.; Barmada, S.; Ando, D.M.; Sharma, P.; Shaby, B.A.; Finkbeiner, S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 2013, 9, 586–592. [Google Scholar] [CrossRef]
- Saito, Y.; Yako, T.; Otsu, W.; Nakamura, S.; Inoue, Y.; Muramatsu, A.; Nakagami, Y.; Shimazawa, M.; Hara, H. A triterpenoid Nrf2 activator, RS9, promotes LC3-associated phagocytosis of photoreceptor outer segments in a p62-independent manner. Free Radic. Biol. Med. 2020, 152, 235–247. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, K.; Sudhandiran, G. Naringin modulates oxidative stress and inflammation in 3-nitropropionic acid-induced neurodegeneration through the activation of nuclear factor-erythroid 2-related factor-2 signalling pathway. Neuroscience 2012, 227, 134–143. [Google Scholar] [CrossRef]
- Gao, Y.; Chu, S.; Li, J.; Zhang, Z.; Yan, J.; Wen, Z.; Xia, C.; Mou, Z.; Wang, Z.; He, W.; et al. Protopanaxtriol protects against 3-nitropropionic acid-induced oxidative stress in a rat model of Huntington’s disease. Acta Pharmacol. Sin. 2015, 36, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Moretti, D.; Tambone, S.; Cerretani, M.; Fezzardi, P.; Missineo, A.; Sherman, L.T.; Munoz-Sajuan, I.; Harper, S.; Dominquez, C.; Pacifici, R.; et al. NRF2 activation by reversible KEAP1 binding induces the antioxidant response in primary neurons and astrocytes of a Huntington’s disease mouse model. Free Radic. Biol. Med. 2021, 162, 243–254. [Google Scholar] [CrossRef]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; Ferreira E Vieira, T.H.; Macedo E Cordeiro, T.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef] [PubMed]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Ho, F.-M.; Kang, H.-C.; Lee, S.-T.; Chao, Y.; Chen, Y.-C.; Huang, L.-J.; Lin, W.-W. The anti-inflammatory actions of LCY-2-CHO, a carbazole analogue, in vascular smooth muscle cells. Biochem. Pharmacol. 2007, 74, 298–308. [Google Scholar] [CrossRef]
- Luo, J.-F.; Shen, X.-Y.; Lio, C.K.; Dai, Y.; Cheng, C.-S.; Liu, J.-X.; Yao, Y.-D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 Pathway by Nardochinoid C Inhibits Inflammation and Oxidative Stress in Lipopolysaccharide-Stimulated Macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [CrossRef]
- Quinti, L.; Dayalan Naidu, S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef]
- Huang, R.-Z.; Liang, G.-B.; Li, M.-S.; Fang, Y.-L.; Zhao, S.-F.; Zhou, M.-M.; Liao, Z.-X.; Sun, J.; Wang, H.-S. Synthesis and discovery of asiatic acid based 1,2,3-triazole derivatives as antitumor agents blocking NF-κB activation and cell migration. Medchemcomm 2019, 10, 584–597. [Google Scholar] [CrossRef]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; von Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Moujalled, D.; James, J.L.; Yang, S.; Zhang, K.; Duncan, C.; Moujalled, D.M.; Parker, S.J.; Caragounis, A.; Lidgerwood, G.; Turner, B.J.; et al. Phosphorylation of hnRNP K by cyclin-dependent kinase 2 controls cytosolic accumulation of TDP-43. Hum. Mol. Genet. 2015, 24, 1655–1669. [Google Scholar] [CrossRef]
- Wang, F.; Lu, Y.; Qi, F.; Su, Q.; Wang, L.; You, C.; Che, F.; Yu, J. Effect of the human SOD1-G93A gene on the Nrf2/ARE signaling pathway in NSC-34 cells. Mol. Med. Rep. 2014, 9, 2453–2458. [Google Scholar] [CrossRef] [PubMed]
- Pehar, M.; Vargas, M.R.; Robinson, K.M.; Cassina, P.; Díaz-Amarilla, P.J.; Hagen, T.M.; Radi, R.; Barbeito, L.; Beckman, J.S. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J. Neurosci. 2007, 27, 7777–7785. [Google Scholar] [CrossRef]
- Kraft, A.D.; Resch, J.M.; Johnson, D.A.; Johnson, J.A. Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 2007, 207, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.-L.; Bourefis, A.-R.; Kabashi, E. Diagnostic Challenge and Neuromuscular Junction Contribution to ALS Pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef]
- Hassanein, E.H.M.; Abd El-Ghafar, O.A.M.; Ahmed, M.A.; Sayed, A.M.; Gad-Elrab, W.M.; Ajarem, J.S.; Allam, A.A.; Mahmoud, A.M. Edaravone and Acetovanillone Upregulate Nrf2 and PI3K/Akt/mTOR Signaling and Prevent Cyclophosphamide Cardiotoxicity in Rats. Drug Des. Dev. Ther. 2020, 14, 5275–5288. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.K. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 2019, 39, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Teng, C.-H.; Wu, F.-F.; Ge, L.-Y.; Xiao, J.; Zhang, H.-Y.; Chen, D.-Q. Edaravone attenuates traumatic brain injury through anti-inflammatory and anti-oxidative modulation. Exp. Ther. Med. 2019, 18, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87. [Google Scholar] [CrossRef]
- Zhou, W.; Bercury, K.; Cummiskey, J.; Luong, N.; Lebin, J.; Freed, C.R. Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J. Biol. Chem. 2011, 286, 14941–14951. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Neymotin, A.; Calingasan, N.Y.; Wille, E.; Naseri, N.; Petri, S.; Damiano, M.; Liby, K.T.; Risingsong, R.; Sporn, M.; Beal, M.F.; et al. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2011, 51, 88–96. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanno, T.; Yanagisawa, Y.; Yasutake, K.; Inoue, S.; Hirayama, N.; Ikeda, J.-E. A novel acylaminoimidazole derivative, WN1316, alleviates disease progression via suppression of glial inflammation in ALS mouse model. PLoS ONE 2014, 9, e87728. [Google Scholar] [CrossRef]
- Kanno, T.; Tanaka, K.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Hirayama, N.; Ikeda, J.-E.E. A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2-ARE pathway: Therapeutic implications for ALS. Free Radic. Biol. Med. 2012, 53, 2028–2042. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Burton, N.C.; Kutzke, J.; Gan, L.; Johnson, D.A.; Schäfer, M.; Werner, S.; Johnson, J.A. Absence of Nrf2 or its selective overexpression in neurons and muscle does not affect survival in ALS-linked mutant hSOD1 mouse models. PLoS ONE 2013, 8, e56625. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mRNA | Protein | ||||||
|---|---|---|---|---|---|---|---|
| Transcript Variant | NCBI Reference Sequence | No of Base Pairs | Information | Isoform | NCBI Reference Sequence | No of Amino Acids | Information |
| 1 | NM_006164.5 | 2446 bp | The longest transcript variant encoding the longest isoform. | 1 | NP_006155.2 | 605 aa | The isoform has the canonical sequence. |
| 2 | NM_001145412.3 | 2988 bp | Uses an alternate promoter, 5’ UTR and a downstream start codon vs. var. 1. It has a shorter N-terminus than isoform 1. | 2 | NP_001138884.1 | 589 aa | Protein lacks the Keap1 interaction domain, resulting in Nrf2 stabilization. Found in the lung and head-and-neck cancers. |
| 3 | NM_001145413.3 | 2967 bp | Uses an alternate promoter, 5’ UTR, downstream start codon, and an alternate in-frame splice site in the 3’ coding region vs. var. 1. It has a shorter N-terminus and is missing an internal segment than. isoform 1. | 3 | NP_001138885.1 | 582 aa | Protein lacks the Keap1 interaction domain, resulting in Nrf2 stabilization. |
| 4 | NM_001313900.1 | 2862 bp | Uses an alternate promoter, 5’ UTR and a downstream start codon vs. var. 1. It has a shorter N-terminus than isoform 1. | 2 | NP_001138884.1 | 589 aa | Protein lacks the Keap1 interaction domain, resulting in Nrf2 stabilization. Found in the lung and head-and-neck cancers. |
| 5 | NM_001313901.1 | 2954 bp | Uses an alternate promoter, 5’ UTR and a downstream start codon vs. var. 1. It has a shorter N-terminus than isoform 1. | 2 | NP_001138884.1 | 589 aa | Protein lacks the Keap1 interaction domain, resulting in Nrf2 stabilization. Found in the lung and head-and-neck cancers. |
| 6 | NM_001313902.1 | 2769 bp | Lacks an alternate in-frame exon in the 3’ coding region vs. var. 1. It is shorter than isoform 1. | 4 | NP_001300831.1 | 575 aa | Computationally mapped isoform. |
| 7 | NM_001313903.1 | 2640 bp | Uses an alternate in-frame splice site in the 3’ coding region vs. var. 1. It is shorter than isoform 1. | 5 | NP_001300832.1 | 532 aa | Computationally mapped isoform. |
| 8 | NM_001313904.1 | 2917 bp | Uses an alternate promoter, 5’ UTR and an alternate in-frame splice site in the 3’ coding region, vs. variant 1. It is shorter than isoform 1. | 6 | NP_001300833.1 | 505 aa | Computationally mapped isoform. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zgorzynska, E.; Dziedzic, B.; Walczewska, A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 9592. https://doi.org/10.3390/ijms22179592
Zgorzynska E, Dziedzic B, Walczewska A. An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2021; 22(17):9592. https://doi.org/10.3390/ijms22179592
Chicago/Turabian StyleZgorzynska, Emilia, Barbara Dziedzic, and Anna Walczewska. 2021. "An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases" International Journal of Molecular Sciences 22, no. 17: 9592. https://doi.org/10.3390/ijms22179592
APA StyleZgorzynska, E., Dziedzic, B., & Walczewska, A. (2021). An Overview of the Nrf2/ARE Pathway and Its Role in Neurodegenerative Diseases. International Journal of Molecular Sciences, 22(17), 9592. https://doi.org/10.3390/ijms22179592