
In Silico Methods for the Identification of Diagnostic and Favorable Prognostic Markers in Acute Myeloid Leukemia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
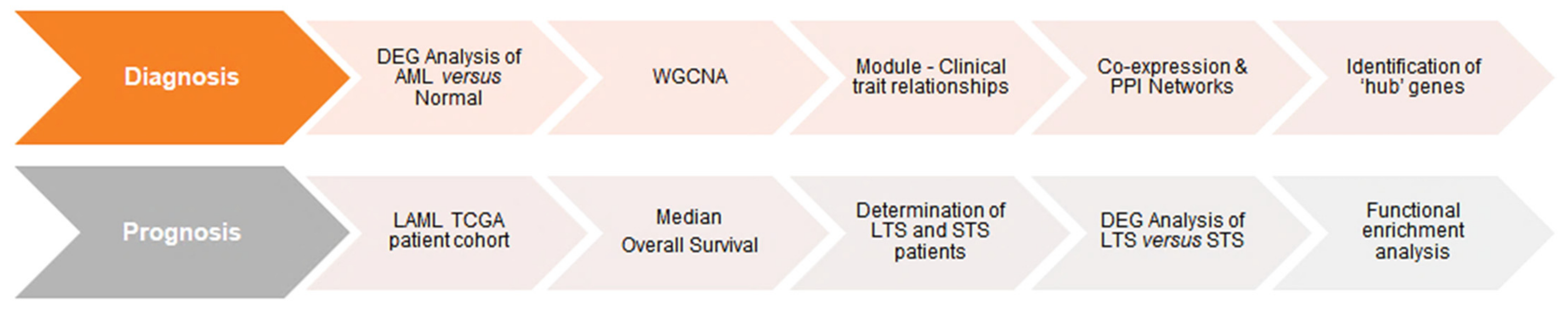
2.1. Identification of Diagnostic Markers in AML
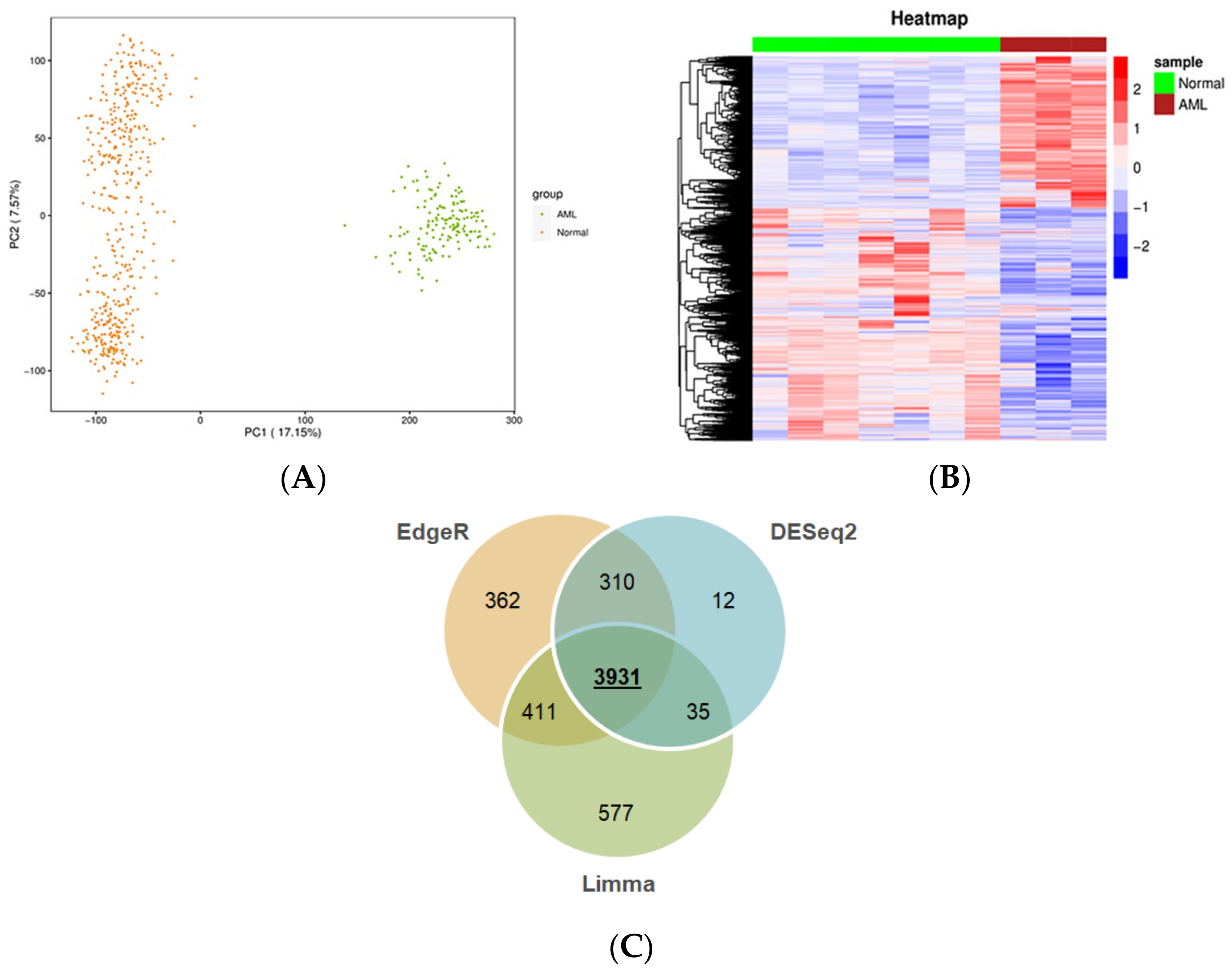
2.1.1. Comprehensive Characterization of Consensus AML Transcriptomes Compared to Normal Blood
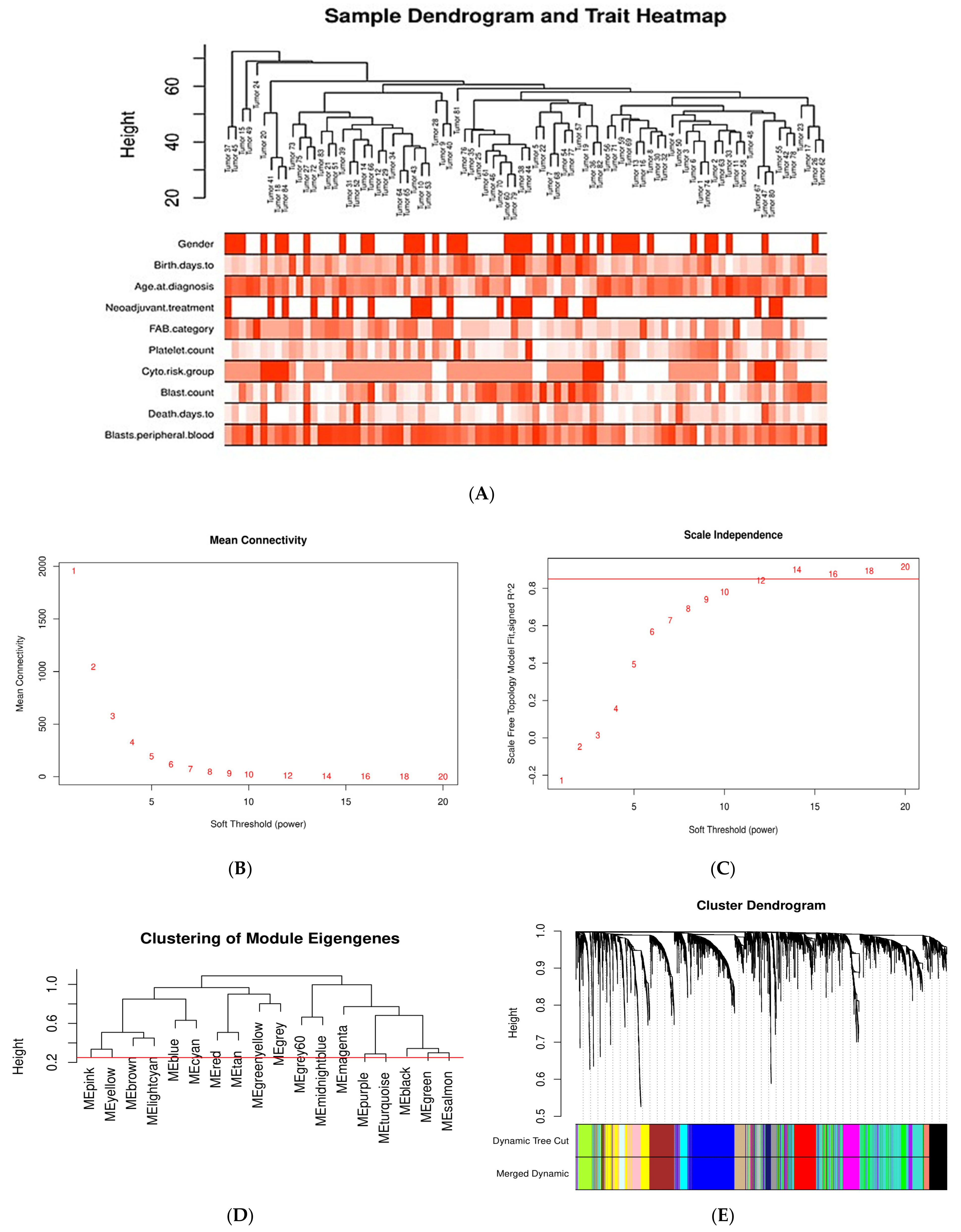
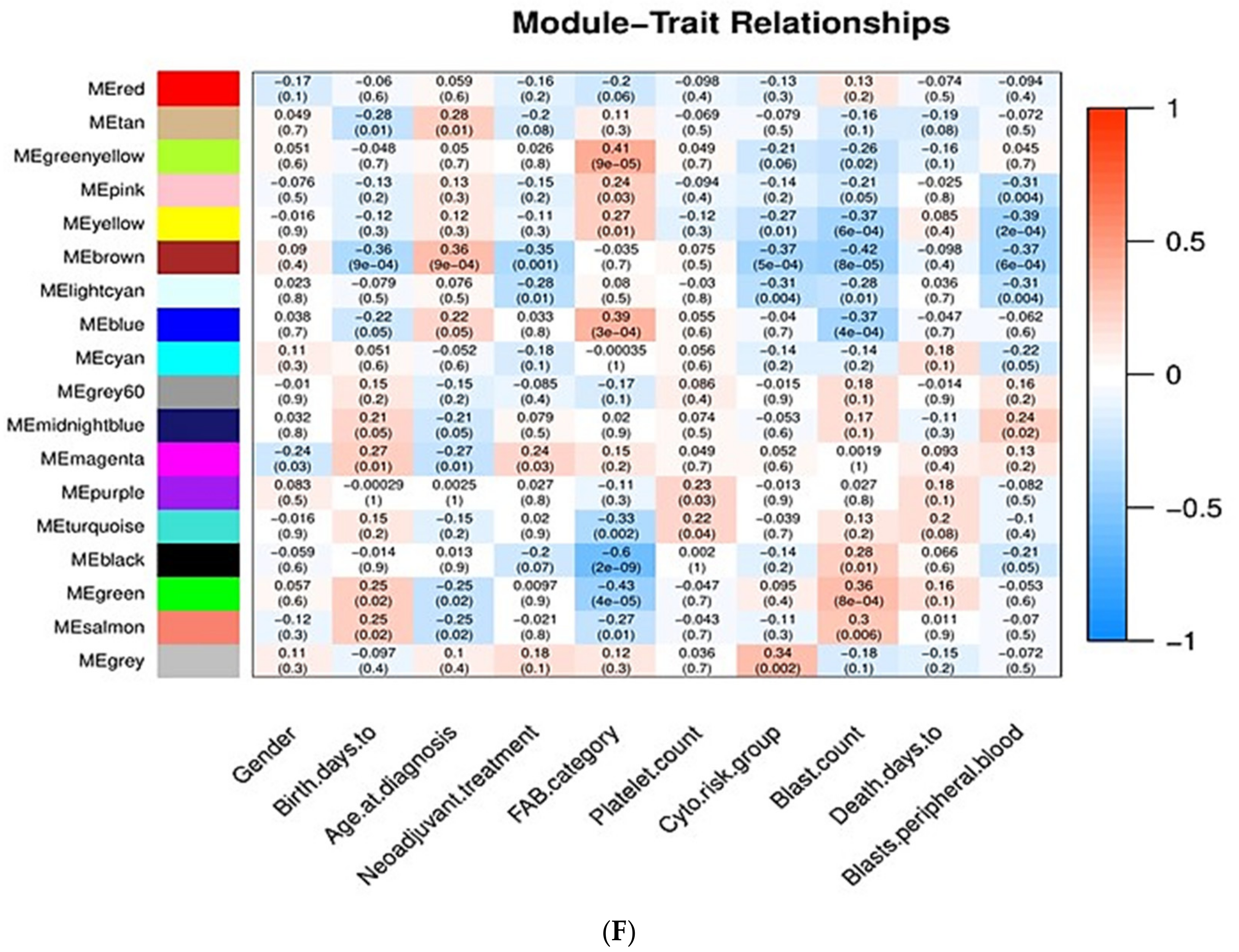
2.1.2. Weighted Gene Co-Expression Network Analysis of DEGs
Sample Clustering for Detecting Outliers
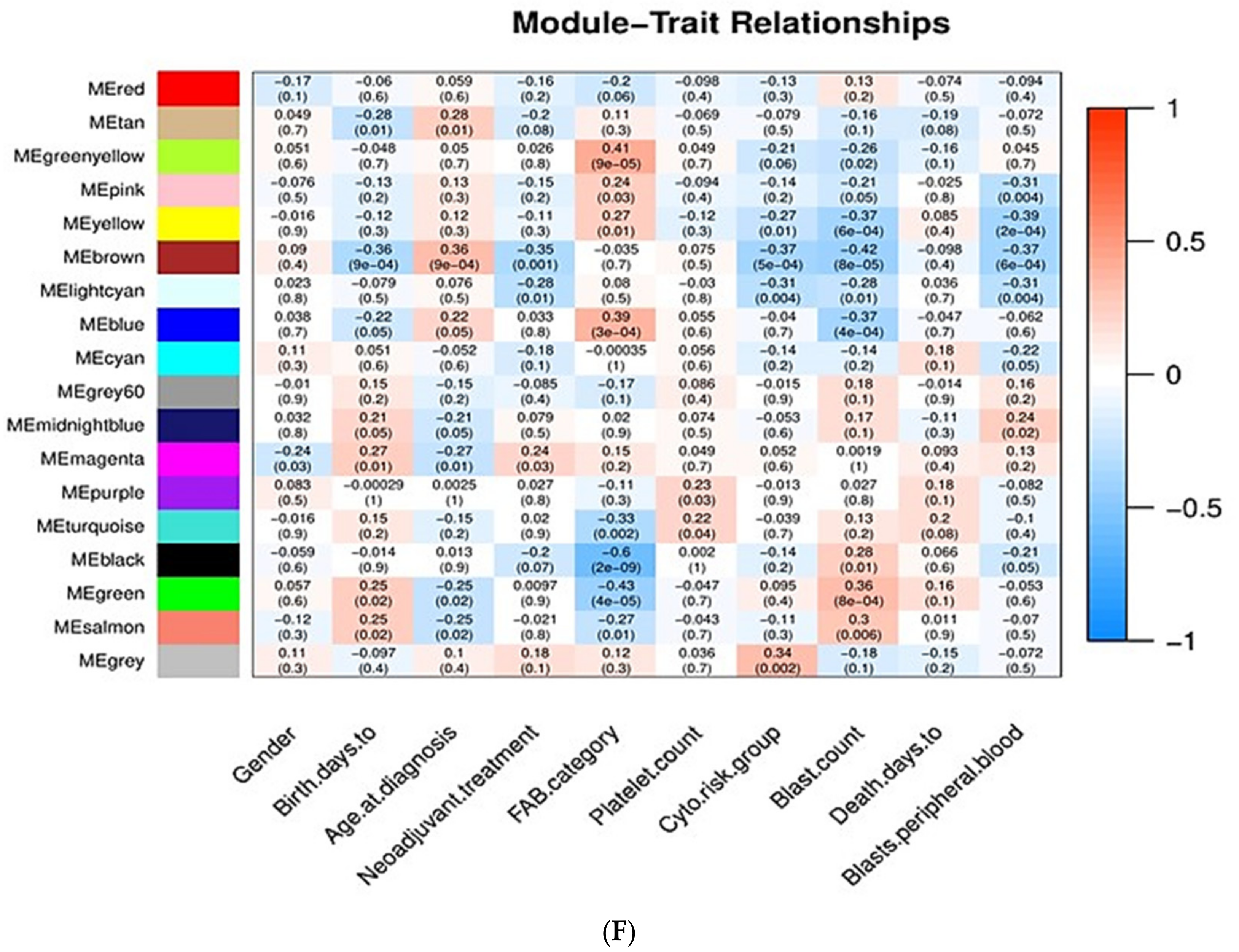
Associations of Gene Modules with Clinicopathological Traits of AML
- Gender: Men are more prone to leukemia than women [22].
- Chronological age: Elderly people are more prone to AML than young people [23].
- Neoadjuvant treatment: Whether patients have previously received chemotherapy or not; patients who have previously received chemotherapy are more prone to AML [26].
- FAB category: Based on the type of cell from which leukemia developed and the degree of maturity of these cells. FAB category corresponds to the French–American–British classification scheme which is useful for classifying AML into subtypes from M0 (undifferentiated AML or AML with minimal differentiation) to M7 (acute megakaryoblastic leukemia, AMegL) [27].
- Cytogenetic risk group: Cytogenetic analysis is recognized as being the most important prognostic indicator in acute myeloid leukemia. According to the cytogenetic risk group, AML is divided into three groups: favorable risk, intermediate risk, and unfavorable risk. Cytogenetic tests help predict the response of cancer to treatment and allow physicians to design a more effective therapy [30,31].
- Blast count and peripheral blood blast: For the diagnosis and classification of AML, the percentage of peripheral blood (PB) and bone marrow (BM) blasts is especially important. BM blasts normally represent 1% to 5% of marrow cells. Generally, a percentage of 20% blasts is required for AML diagnosis. Most patients with AML have a higher percentage of BM blasts compared to PB blasts [32,33,34].
- Days to death: Related to overall survival, which is the survival time after initial diagnosis. Measuring the overall survival is required to assess how well a new treatment works in a clinical trial. In AML, the 5-year overall survival is less than 50%; regarding elderly patients, only 20% survive 2 years after diagnosis [35,36].
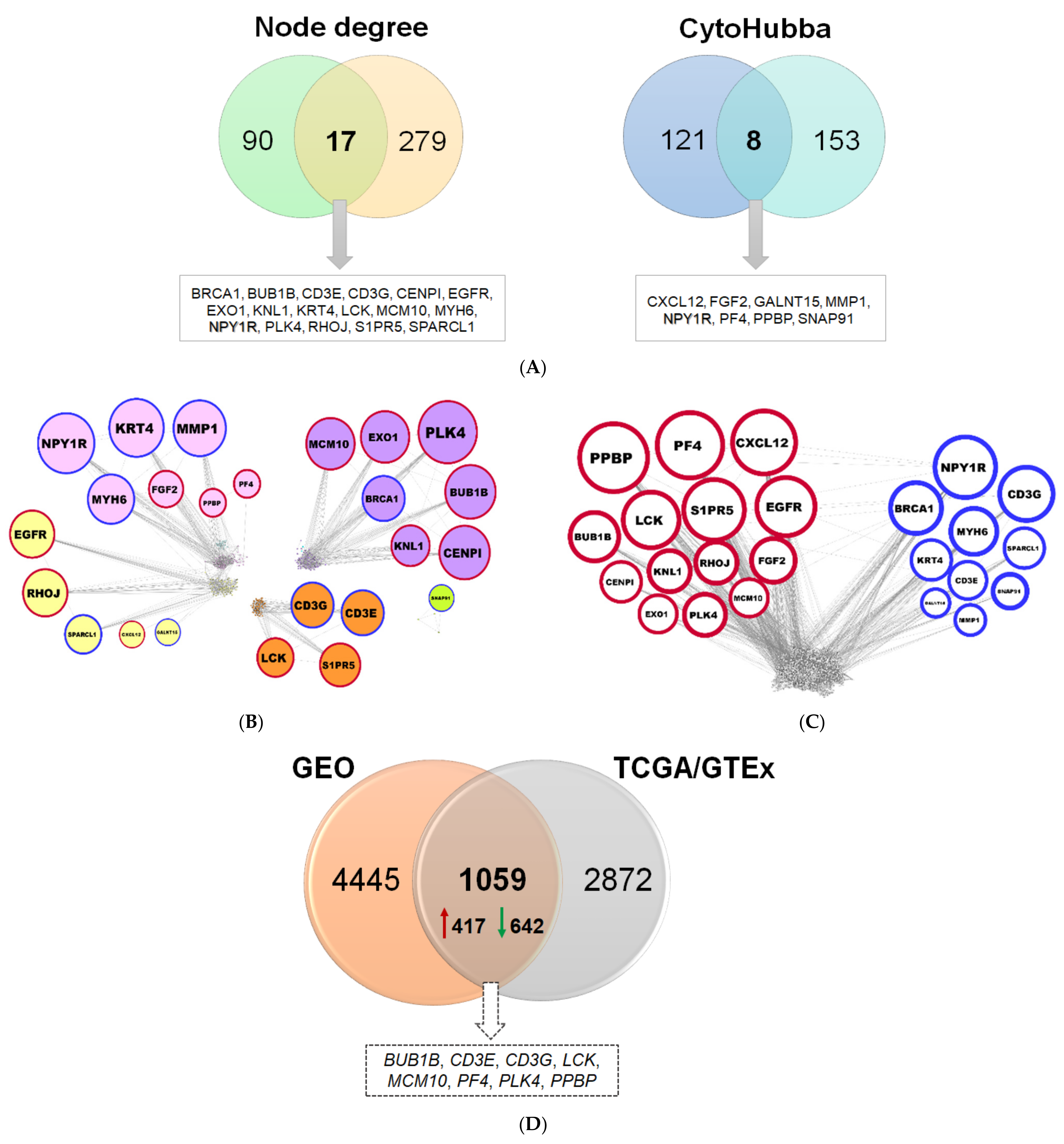
2.1.3. Reconstruction of AML Molecular Networks
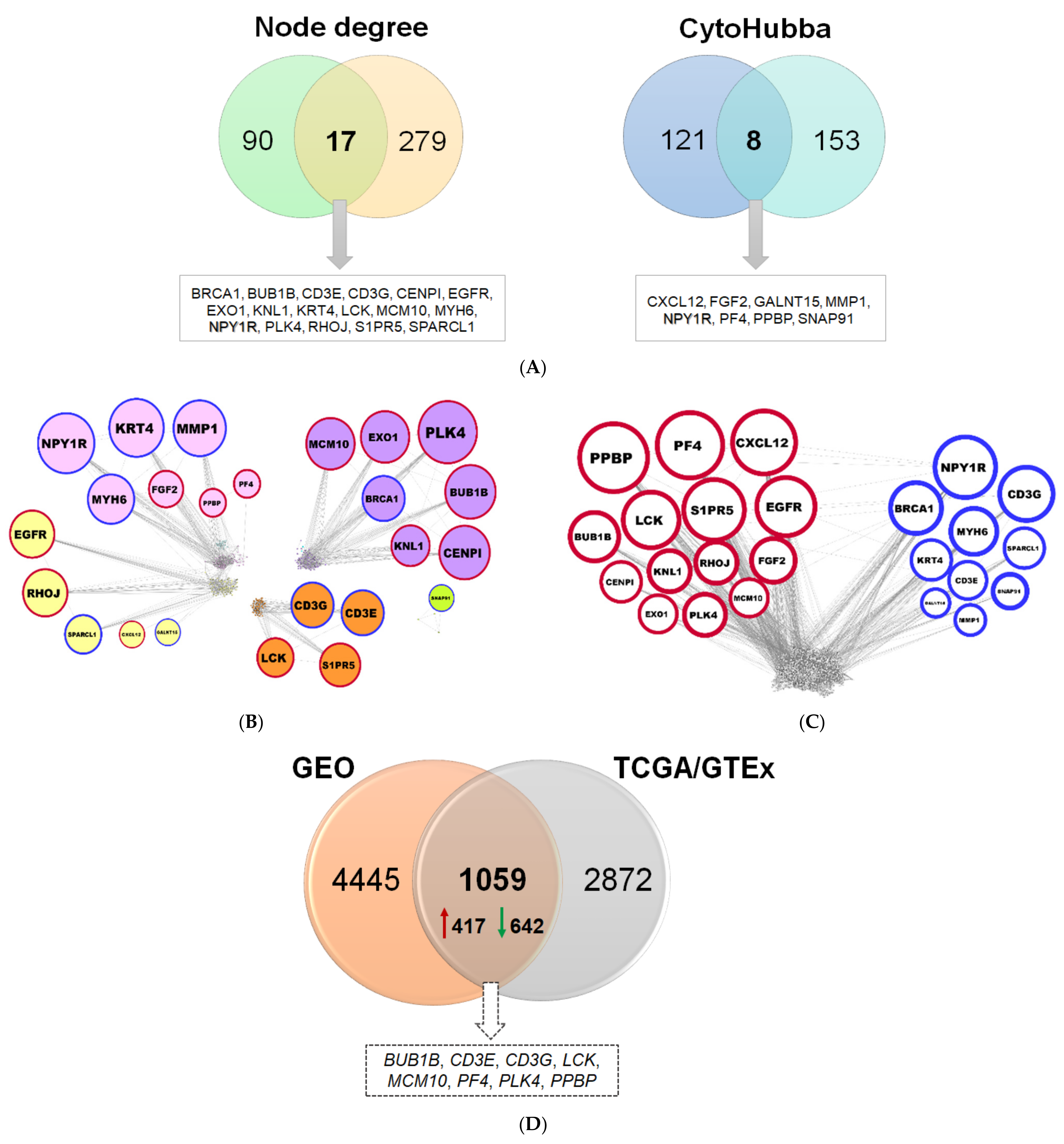
2.1.4. Comparison of Diagnostic Signatures with Independent AML Datasets
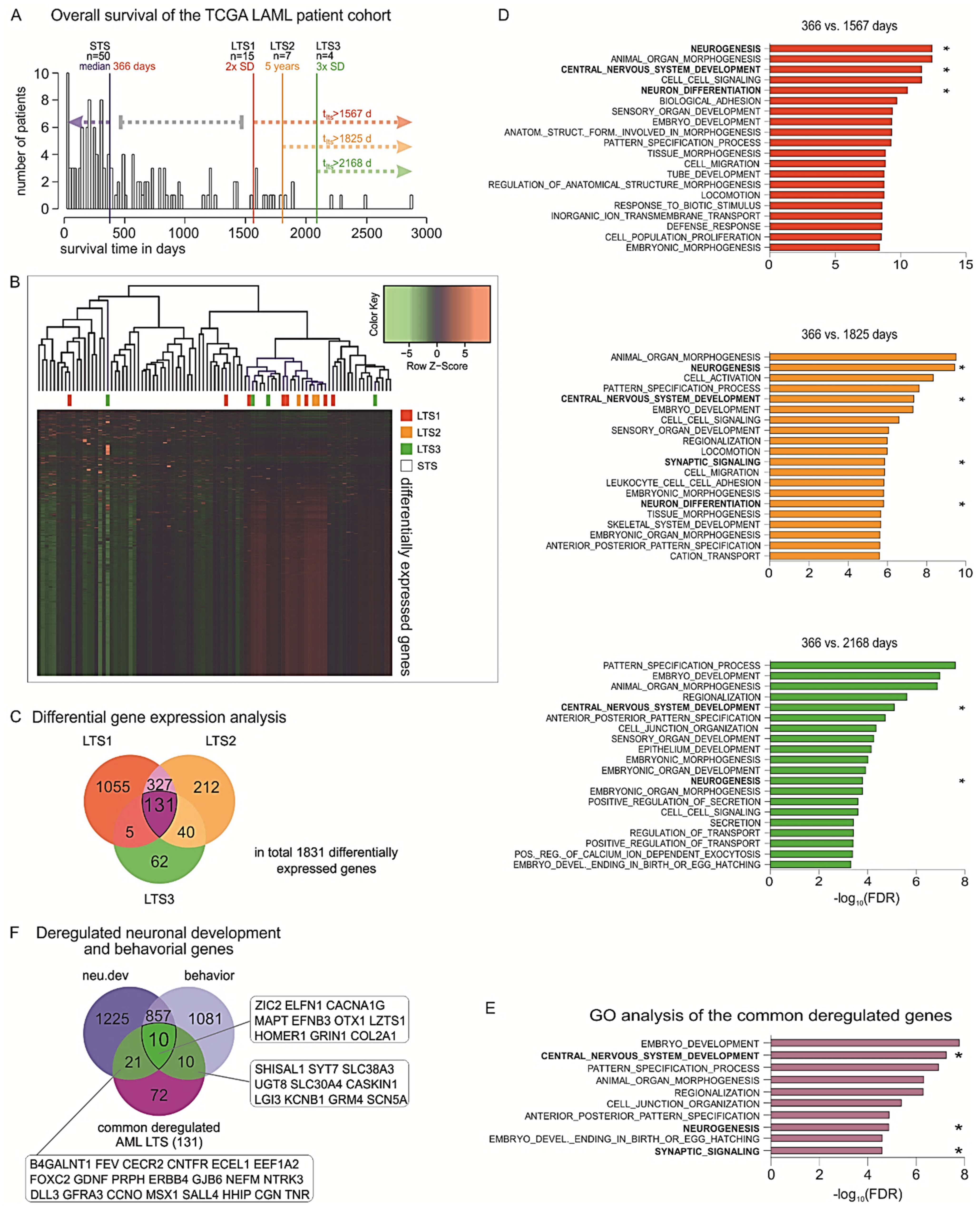
2.2. Inferring Favorable Prognostic Markers from the Transcriptomic Profiles of Long-Term Survivors of AML
3. Discussion
4. Materials and Methods
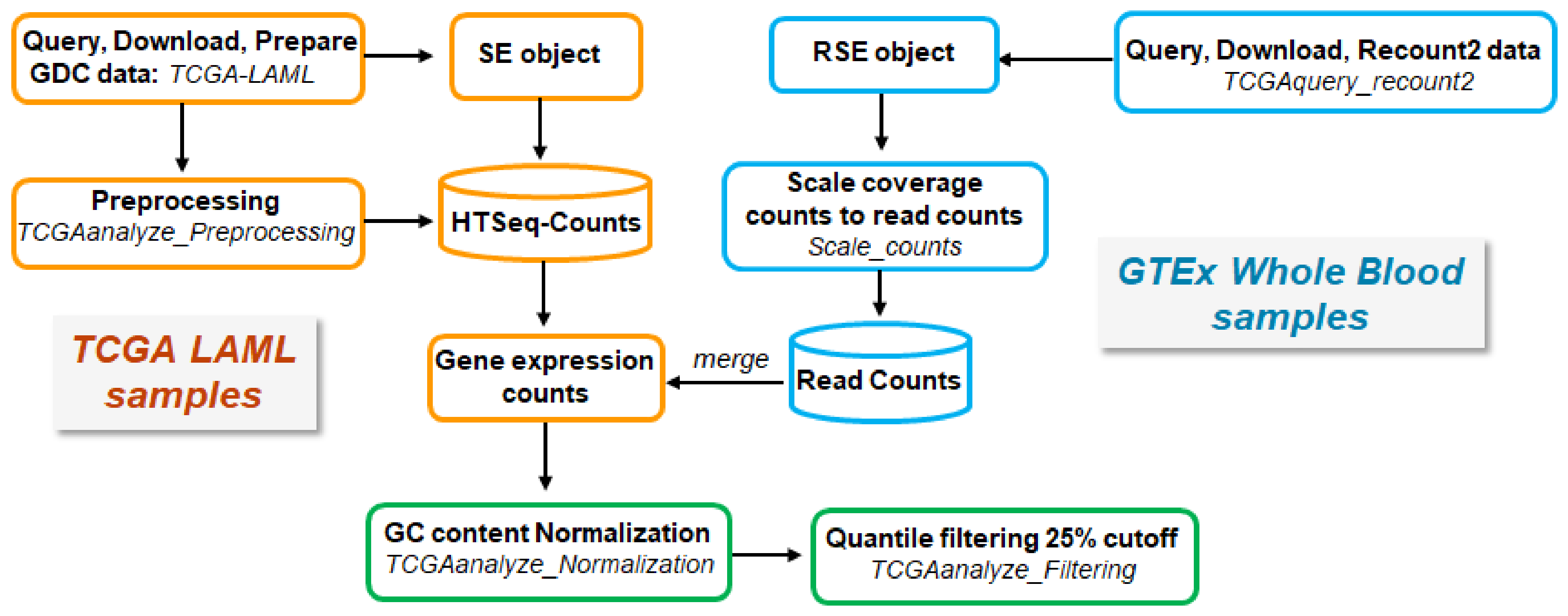
4.1. Data Retrieval, Processing, and Analysis
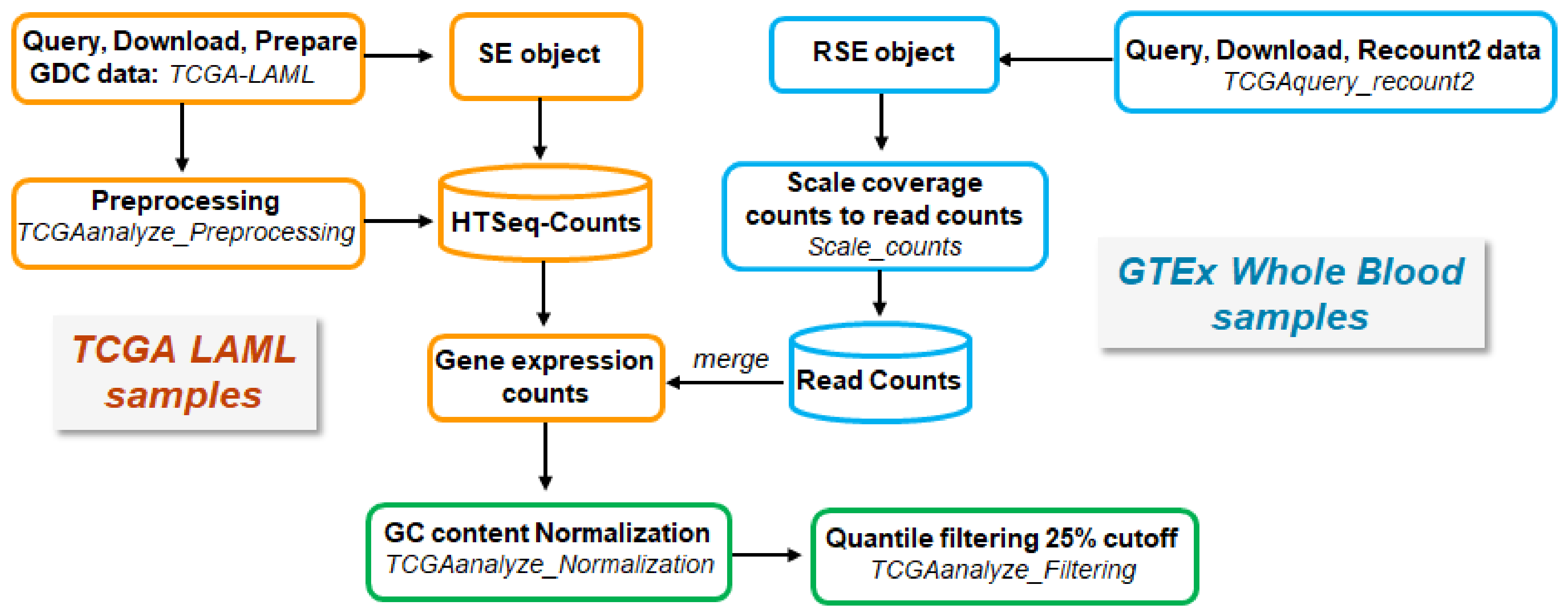
4.1.1. RNA-seq Data Acquisition and Processing
AML Samples from TCGA
Normal Tissue Samples from GTEx
Processing and Merging of TCGA- and GTEx-Derived Data
Principal Component Analysis
4.2. Identification of Differentially Expressed Genes
4.3. Construction of Weighted Gene Co-Expression Network
4.4. Identification of Clinically Important Modules
4.5. Co-Expression and Protein–Protein Interaction (PPI) Network Analysis
4.6. NCBI GEO Gene Expression Datasets
4.6.1. Microarray-Based Transcriptomic Data Analysis
4.6.2. RNA-seq-based Transcriptomic Data Analysis
4.7. Identification of DEGs Correlated with Long-Term Survival
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leukemia-acute Myeloid-aml: Statistics. Available online: https://www.cancer.net/cancer-types/leukemia-acute-myeloid-aml/statistics (accessed on 7 June 2021).
- Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia (aml). Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 7 June 2021).
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Shipley, J.L.; Butera, J.N. Acute myelogenous leukemia. Exp. Hematol. 2009, 37, 649–658. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the european leukemianet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.cancer.gov/news-events/press-releases/2020 (accessed on 7 June 2021).
- Pulte, D.; Gondos, A.; Brenner, H. Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st century. Blood 2009, 113, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Farag, S.S.; Ruppert, A.S.; Mrozek, K.; Mayer, R.J.; Stone, R.M.; Carroll, A.J.; Powell, B.L.; Moore, J.O.; Pettenati, M.J.; Koduru, P.R.; et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: A cancer and leukemia group b study. J. Clin. Oncol. 2005, 23, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Klepin, H.D.; Balducci, L. Acute myelogenous leukemia in older adults. Oncology 2009, 14, 222–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Paez, D.; Labonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.J. Cancer dormancy: A model of early dissemination and late cancer recurrence. Clin. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Benton, C.B.; Ravandi, F. A mind map for managing minimal residual disease in acute myeloid leukemia. Clin. Adv. Hematol. Oncol. HO 2017, 15, 859–867. [Google Scholar]
- Lauber, C.; Correia, N.; Trumpp, A.; Rieger, M.A.; Dolnik, A.; Bullinger, L.; Roeder, I.; Seifert, M. Survival differences and associated molecular signatures of dnmt3a-mutant acute myeloid leukemia patients. Sci. Rep. 2020, 10, 12761. [Google Scholar] [CrossRef]
- Guo, C.; Gao, Y.Y.; Ju, Q.Q.; Zhang, C.X.; Gong, M.; Li, Z.L. The landscape of gene co-expression modules correlating with prognostic genetic abnormalities in aml. J. Transl. Med. 2021, 19, 228. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, Q.; Deng, X.; An, N.; Du, X.; Liu, J. Gene coexpression network analysis revealed biomarkers correlated with blast cells and survival in acute myeloid leukemia. Mol. Clin. Oncol. 2020, 12, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifenberger, G.; Weber, R.G.; Riehmer, V.; Kaulich, K.; Willscher, E.; Wirth, H.; Gietzelt, J.; Hentschel, B.; Westphal, M.; Simon, M.; et al. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int. J. Cancer 2014, 135, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Hu, J.; Wu, J.; Luo, X.; Li, Y.; Li, J. Molecular characterization of long-term survivors of hepatocellular carcinoma. Aging 2021, 13, 7517–7537. [Google Scholar] [CrossRef] [PubMed]
- Saner, F.A.M.; Herschtal, A.; Nelson, B.H.; deFazio, A.; Goode, E.L.; Ramus, S.J.; Pandey, A.; Beach, J.A.; Fereday, S.; Berchuck, A.; et al. Going to extremes: Determinants of extraordinary response and survival in patients with cancer. Nat. Rev. Cancer 2019, 19, 339–348. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Wgcna: An r package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Jackson, N.; Menon, B.S.; Zarina, W.; Zawawi, N.; Naing, N.N. Why is acute leukemia more common in males? A possible sex-determined risk linked to the abo blood group genes. Ann. Hematol. 1999, 78, 233–236. [Google Scholar] [CrossRef]
- Almeida, A.M.; Ramos, F. Acute myeloid leukemia in the older adults. Leuk. Res. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seer Cancer Statistics Review, 1975–2000. Available online: http://seer.cancer.gov/csr/1975_2000/sections.html (accessed on 7 June 2021).
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Morton, L.M.; Dores, G.M.; Schonfeld, S.J.; Linet, M.S.; Sigel, B.S.; Lam, C.J.K.; Tucker, M.A.; Curtis, R.E. Association of chemotherapy for solid tumors with development of therapy-related myelodysplastic syndrome or acute myeloid leukemia in the modern era. JAMA Oncol. 2019, 5, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the acute leukaemias. French-american-british (fab) co-operative group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Bussel, J.B.; Frelinger, A.L.; Babula, B.; Linden, M.D.; Li, Y.; Barnard, M.R.; Tate, C.; Feldman, E.J.; Michelson, A.D. Differences in platelet function in patients with acute myeloid leukemia and myelodysplasia compared to equally thrombocytopenic patients with immune thrombocytopenia. J. Thromb. Haemost. JTH 2011, 9, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Saultz, J.N.; Garzon, R. Acute myeloid leukemia: A concise review. J. Clin. Med. 2016, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K.; National Cancer Research Institute Adult Leukaemia Working, G. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the united kingdom medical research council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Mahapatra, M.; Saxena, R. Cytogenetics’ impact on the prognosis of acute myeloid leukemia. J. Lab. Physicians 2019, 11, 133–137. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Garcia-Manero, G.; Pierce, S.; Nazha, A.; Bueso-Ramos, C.; Jabbour, E.; Ravandi, F.; Cortes, J.; Kantarjian, H. Interactions and relevance of blast percentage and treatment strategy among younger and older patients with acute myeloid leukemia (aml) and myelodysplastic syndrome (mds). Am. J. Hematol. 2016, 91, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Holtzman, N.G.; El Chaer, F.; Baer, M.R.; Ali, O.; Patel, A.; Duong, V.H.; Sausville, E.A.; Singh, Z.N.; Koka, R.; Zou, Y.S.; et al. Peripheral blood blast rate of clearance is an independent predictor of clinical response and outcomes in acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 881–887. [Google Scholar] [CrossRef]
- Weinkauff, R.; Estey, E.H.; Starostik, P.; Hayes, K.; Huh, Y.O.; Hirsch-Ginsberg, C.; Andreeff, M.; Keating, M.; Kantarjian, H.M.; Freireich, E.J.; et al. Use of peripheral blood blasts vs. bone marrow blasts for diagnosis of acute leukemia. Am. J. Clin. Pathol. 1999, 111, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Oran, B.; Weisdorf, D.J. Survival for older patients with acute myeloid leukemia: A population-based study. Haematologica 2012, 97, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Acute Myeloid Leukaemia (Aml)—Survival. Available online: https://www.cancerresearchuk.org/about-cancer/acute-myeloid-leukaemia-aml/survival (accessed on 7 June 2021).
- Cheng, M.J.; Hourigan, C.S.; Smith, T.J. Adult acute myeloid leukemia long-term survivors. J. Leuk. 2014, 2, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Vasu, S.; Kohlschmidt, J.; Mrozek, K.; Eisfeld, A.K.; Nicolet, D.; Sterling, L.J.; Becker, H.; Metzeler, K.H.; Papaioannou, D.; Powell, B.L.; et al. Ten-year outcome of patients with acute myeloid leukemia not treated with allogeneic transplantation in first complete remission. Blood Adv. 2018, 2, 1645–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, C.D.; Shuma, C.; Regal, L.; Philip, P.P.; Hossfeld, D.K.; Hagemeijer, A.M.; Garson, O.M.; Peterson, B.A.; Sakurai, M.; Alimena, G.; et al. Long-term survival of patients with acute myeloid leukemia: A third follow-up of the fourth international workshop on chromosomes in leukemia. Cancer 1997, 80, 2191–2198. [Google Scholar] [CrossRef]
- Arndt, V.; Koch-Gallenkamp, L.; Jansen, L.; Bertram, H.; Eberle, A.; Holleczek, B.; Schmid-Hopfner, S.; Waldmann, A.; Zeissig, S.R.; Brenner, H. Quality of life in long-term and very long-term cancer survivors versus population controls in germany. Acta Oncol. 2017, 56, 190–197. [Google Scholar] [CrossRef]
- Roussa, E.; Krieglstein, K. Gdnf promotes neuronal differentiation and dopaminergic development of mouse mesencephalic neurospheres. Neurosci. Lett. 2004, 361, 52–55. [Google Scholar] [CrossRef]
- Holland, P.W.; Booth, H.A.; Bruford, E.A. Classification and nomenclature of all human homeobox genes. BMC Biol. 2007, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Adès, L.; Guerci, A.; Raffoux, E.; Sanz, M.; Chevallier, P.; Lapusan, S.; Recher, C.; Thomas, X.; Rayon, C.; Castaigne, S.; et al. Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: The European APL Group experience. Blood J. Am. Soc. Hematol. 2010, 115, 1690–1696. [Google Scholar] [CrossRef] [Green Version]
- Logotheti, S.; Marquardt, S.; Richter, C.; Sophie Hain, R.; Murr, N.; Takan, I.; Pavlopoulou, A.; Putzer, B.M. Neural networks recapitulation by cancer cells promotes disease progression: A novel role of p73 isoforms in cancer-neuronal crosstalk. Cancers 2020, 12, 3789. [Google Scholar] [CrossRef]
- Friedenson, B. The brca1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer 2007, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Chen, G.; Cai, Z.D.; Wang, C.; Liu, Z.Z.; Lin, Z.Y.; Wu, Y.D.; Liang, Y.X.; Han, Z.D.; Liu, J.C.; et al. Overexpression of bub1b contributes to progression of prostate cancer and predicts poor outcome in patients with prostate cancer. Onco Targets Ther. 2016, 9, 2211–2220. [Google Scholar] [PubMed] [Green Version]
- Zhu, L.J.; Pan, Y.; Chen, X.Y.; Hou, P.F. Bub1 promotes proliferation of liver cancer cells by activating smad2 phosphorylation. Oncol. Lett. 2020, 19, 3506–3512. [Google Scholar] [CrossRef]
- Ding, N.; Li, R.; Shi, W.; He, C. Cenpi is overexpressed in colorectal cancer and regulates cell migration and invasion. Gene 2018, 674, 80–86. [Google Scholar] [CrossRef]
- Thangavelu, P.U.; Lin, C.Y.; Vaidyanathan, S.; Nguyen, T.H.M.; Dray, E.; Duijf, P.H.G. Overexpression of the e2f target gene cenpi promotes chromosome instability and predicts poor prognosis in estrogen receptor-positive breast cancer. Oncotarget 2017, 8, 62167–62182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Li, T.; Sheng, M.; Yang, B. Exonuclease 1 (exo1) participates in mammalian non-homologous end joining and contributes to drug resistance in ovarian cancer. Med Sci. Monit. 2020, 26, e918751. [Google Scholar] [CrossRef]
- Keijzers, G.; Bakula, D.; Petr, M.A.; Madsen, N.G.K.; Teklu, A.; Mkrtchyan, G.; Osborne, B.; Scheibye-Knudsen, M. Human exonuclease 1 (exo1) regulatory functions in DNA replication with putative roles in cancer. Int. J. Mol. Sci. 2018, 20, 74. [Google Scholar] [CrossRef] [Green Version]
- Bai, T.; Zhao, Y.; Liu, Y.; Cai, B.; Dong, N.; Li, B. Effect of knl1 on the proliferation and apoptosis of colorectal cancer cells. Technol. Cancer Res. Treat. 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tromer, E.; Snel, B.; Kops, G.J. Widespread recurrent patterns of rapid repeat evolution in the kinetochore scaffold knl1. Genome Biol. Evol. 2015, 7, 2383–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Stauffer, M.E.; Eichman, B.F. Structural biology of replication initiation factor mcm10. Sub-Cell. Biochem. 2012, 62, 197–216. [Google Scholar]
- Yang, W.D.; Wang, L. Mcm10 facilitates the invaded/migrated potentials of breast cancer cells via wnt/beta-catenin signaling and is positively interlinked with poor prognosis in breast carcinoma. J. Biochem. Mol. Toxicol. 2019, 33, e22330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, C.; Liang, H.; Han, L. Polo-like kinase 4’s critical role in cancer development and strategies for plk4-targeted therapy. Front. Oncol. 2021, 11, 587554. [Google Scholar]
- de Oliveira Lisboa, M.; Brofman, P.R.S.; Schmid-Braz, A.T.; Rangel-Pozzo, A.; Mai, S. Chromosomal instability in acute myeloid leukemia. Cancers 2021, 13, 2655. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Schnerch, D.; Yalcintepe, J.; Schmidts, A.; Becker, H.; Follo, M.; Engelhardt, M.; Wasch, R. Cell cycle control in acute myeloid leukemia. Am. J. Cancer Res. 2012, 2, 508–528. [Google Scholar]
- Yang, Y.; Zang, Y.; Zheng, C.; Li, Z.; Gu, X.; Zhou, M.; Wang, Z.; Xiang, J.; Chen, Z.; Zhou, Y. Cd3d is associated with immune checkpoints and predicts favorable clinical outcome in colon cancer. Immunotherapy 2020, 12, 25–35. [Google Scholar] [CrossRef]
- Garcia-Cuesta, E.M.; Santiago, C.A.; Vallejo-Diaz, J.; Juarranz, Y.; Rodriguez-Frade, J.M.; Mellado, M. The role of the cxcl12/cxcr4/ackr3 axis in autoimmune diseases. Front. Endocrinol. 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.S.; Kim, H.J.; Konopleva, M. Targeting the cxcl12/cxcr4 axis in acute myeloid leukemia: From bench to bedside. Korean J. Intern. Med. 2017, 32, 248–257. [Google Scholar] [CrossRef]
- Ali, R.A.; Wuescher, L.M.; Worth, R.G. Platelets: Essential components of the immune system. Curr. Trends Immunol. 2015, 16, 65–78. [Google Scholar] [PubMed]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Wen-jun, L. Platelet changes in acute leukemia. Cell Biochem. Biophys. 2013, 67, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Bautista, C.; Torres, C.; Pena, G.; Guevara, C.; Palacios, C.; Guevara, A.; Gavilanes, A.W.D. Insights from the clinical phenotype of subjects with laron syndrome in ecuador. Rev. Endocr. Metab. Disord. 2021, 22, 59–70. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Luksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Deak, D.; Gorcea-Andronic, N.; Sas, V.; Teodorescu, P.; Constantinescu, C.; Iluta, S.; Pasca, S.; Hotea, I.; Turcas, C.; Moisoiu, V.; et al. A narrative review of central nervous system involvement in acute leukemias. Ann. Transl. Med. 2021, 9, 68. [Google Scholar] [CrossRef]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Dolgin, E. Cancer-neuronal crosstalk and the startups working to silence it. Nat. Biotechnol. 2020, 38, 115–117. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Peterson, B.; Dodge, R.K.; Mayer, R.J.; Moore, J.O.; Lee, E.J.; Kolitz, J.; Baer, M.R.; Schiffer, C.A.; Carroll, A.J.; et al. Differences in prognostic factors and outcomes in african americans and whites with acute myeloid leukemia. Blood 2004, 103, 4036–4042. [Google Scholar] [CrossRef] [Green Version]
- R core team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. Tcgabiolinks: An r/bioconductor package for integrative analysis of tcga data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the tcgabiolinks package for the study and integration of cancer data from gdc and gtex. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [Green Version]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. Tcga workflow: Analyze cancer genomics and epigenomics data using bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Nellore, A.; Jaffe, A.E. Recount workflow: Accessing over 70,000 human rna-seq samples with bioconductor. F1000Research 2017, 6, 1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado-Torres, L.; Nellore, A.; Kammers, K.; Ellis, S.E.; Taub, M.A.; Hansen, K.D.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Reproducible rna-seq analysis using recount2. Nat. Biotechnol. 2017, 35, 319–321. [Google Scholar] [CrossRef]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. Gc-content normalization for rna-seq data. BMC Bioinform. 2011, 12, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Society. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Kontou, P.I.; Pavlopoulou, A.; Dimou, N.L.; Pavlopoulos, G.A.; Bagos, P.G. Network analysis of genes and their association with diseases. Gene 2016, 590, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The string database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Albert, R.; Jeong, H.; Barabasi, A.L. Error and attack tolerance of complex networks. Nature 2000, 406, 378–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. Cytohubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. Ncbi geo: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Clough, E.; Barrett, T. The gene expression omnibus database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar]
- Stirewalt, D.L.; Meshinchi, S.; Kopecky, K.J.; Fan, W.; Pogosova-Agadjanyan, E.L.; Engel, J.H.; Cronk, M.R.; Dorcy, K.S.; McQuary, A.R.; Hockenbery, D.; et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer 2008, 47, 8–20. [Google Scholar] [CrossRef]
- Celik, H.; Lindblad, K.E.; Popescu, B.; Gui, G.; Goswami, M.; Valdez, J.; DeStefano, C.; Lai, C.; Thompson, J.; Ghannam, J.Y.; et al. Highly multiplexed proteomic assessment of human bone marrow in acute myeloid leukemia. Blood Adv. 2020, 4, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Toy, H.I.; Karakulah, G.; Kontou, P.I.; Alotaibi, H.; Georgakilas, A.G.; Pavlopoulou, A. Investigating molecular determinants of cancer cell resistance to ionizing radiation through an integrative bioinformatics approach. Front. Cell Dev. Biol. 2021, 9, 620248. [Google Scholar] [CrossRef]
- Kontou, P.; Pavlopoulou, A.; Braliou, G.; Bogiatzi, S.; Dimou, N.; Bangalore, S.; Bagos, P. Identification of gene expression profiles in myocardial infarction: A systematic review and meta-analysis. BMC Med. Genom. 2018, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- StataCorp, Stata Statistical Software: Release 13; StataCorp LP: College Station, TX, USA, 2013.
- Alnasir, J.; Shanahan, H.P. Investigation into the annotation of protocol sequencing steps in the sequence read archive. GigaScience 2015, 4, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yılmaz, H.; Toy, H.I.; Marquardt, S.; Karakülah, G.; Küçük, C.; Kontou, P.I.; Logotheti, S.; Pavlopoulou, A. In Silico Methods for the Identification of Diagnostic and Favorable Prognostic Markers in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 9601. https://doi.org/10.3390/ijms22179601
Yılmaz H, Toy HI, Marquardt S, Karakülah G, Küçük C, Kontou PI, Logotheti S, Pavlopoulou A. In Silico Methods for the Identification of Diagnostic and Favorable Prognostic Markers in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2021; 22(17):9601. https://doi.org/10.3390/ijms22179601
Chicago/Turabian StyleYılmaz, Hande, Halil Ibrahim Toy, Stephan Marquardt, Gökhan Karakülah, Can Küçük, Panagiota I. Kontou, Stella Logotheti, and Athanasia Pavlopoulou. 2021. "In Silico Methods for the Identification of Diagnostic and Favorable Prognostic Markers in Acute Myeloid Leukemia" International Journal of Molecular Sciences 22, no. 17: 9601. https://doi.org/10.3390/ijms22179601
APA StyleYılmaz, H., Toy, H. I., Marquardt, S., Karakülah, G., Küçük, C., Kontou, P. I., Logotheti, S., & Pavlopoulou, A. (2021). In Silico Methods for the Identification of Diagnostic and Favorable Prognostic Markers in Acute Myeloid Leukemia. International Journal of Molecular Sciences, 22(17), 9601. https://doi.org/10.3390/ijms22179601