
Impairment of the Hypothalamus–Pituitary–Thyroid Axis Caused by Naturally Occurring GATA2 Mutations In Vitro

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Characteristics of GATA2 Mutations Studied
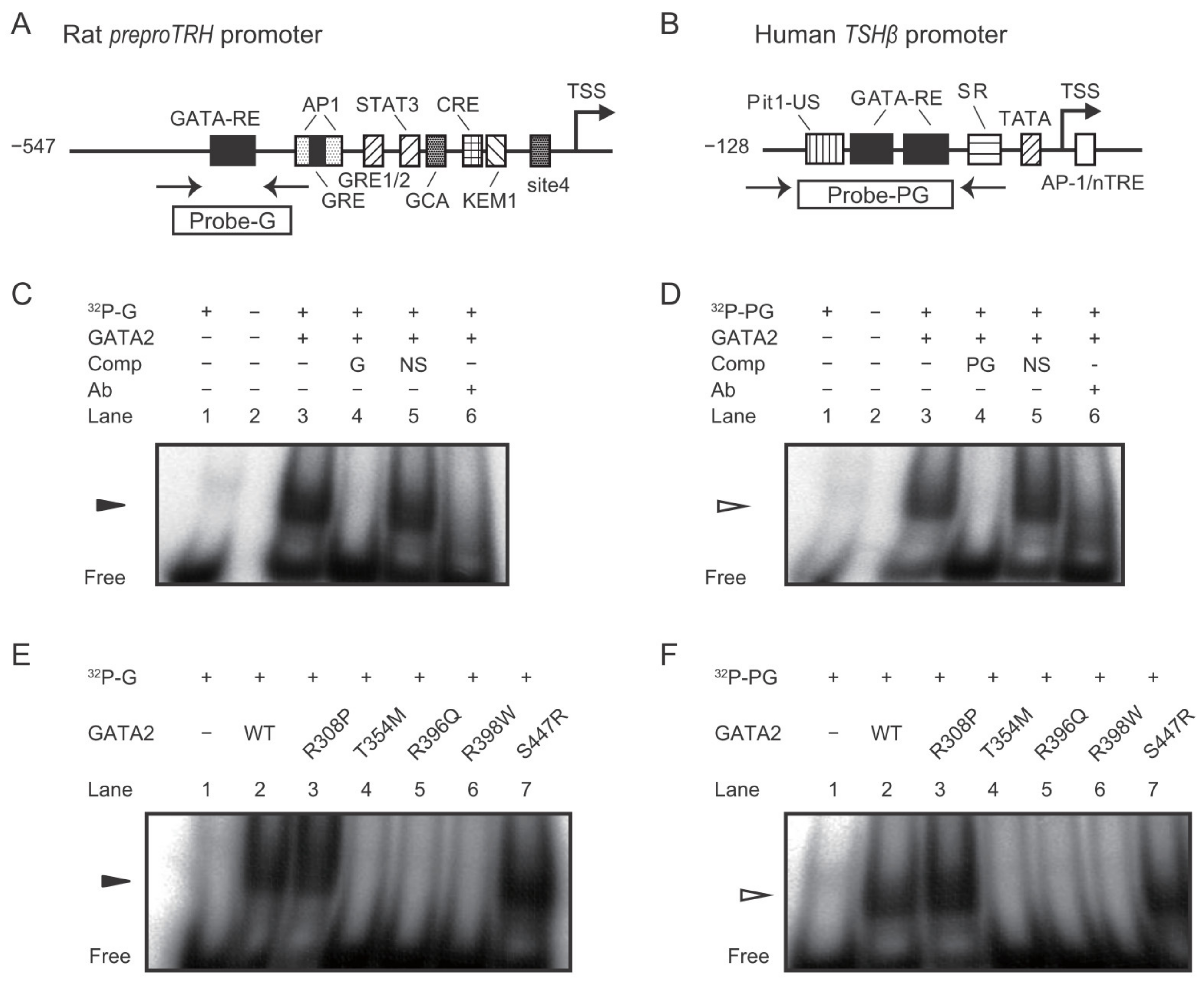
2.2. The GATA2 Mutants Have Reduced Transcriptional Activity on the preproTRH Promoter
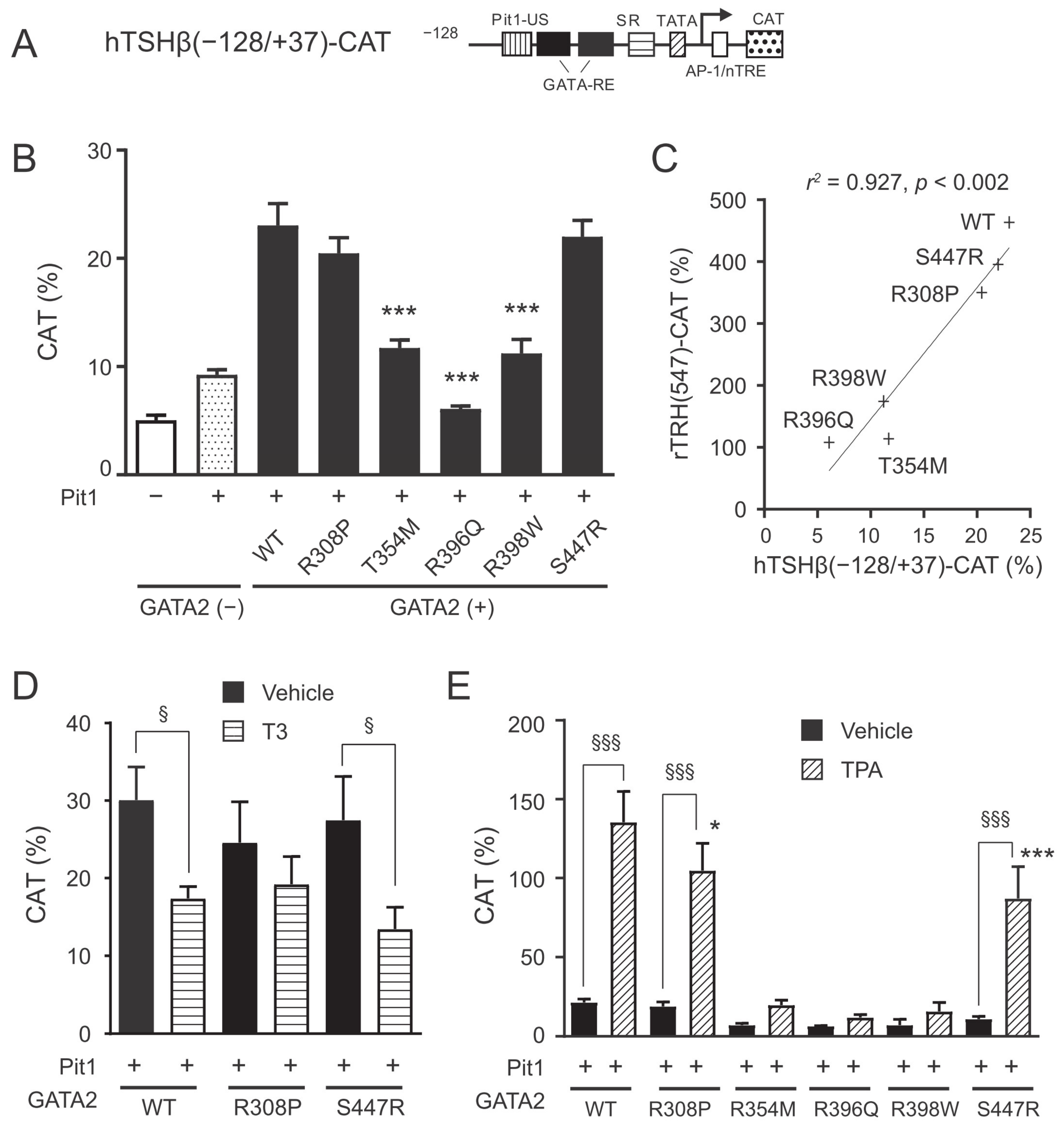
2.3. The GATA2 Mutants Have Reduced Transcriptional Activity on the TSHβ Promoter
2.4. DNA Binding Affinity of the GATA2 Mutants
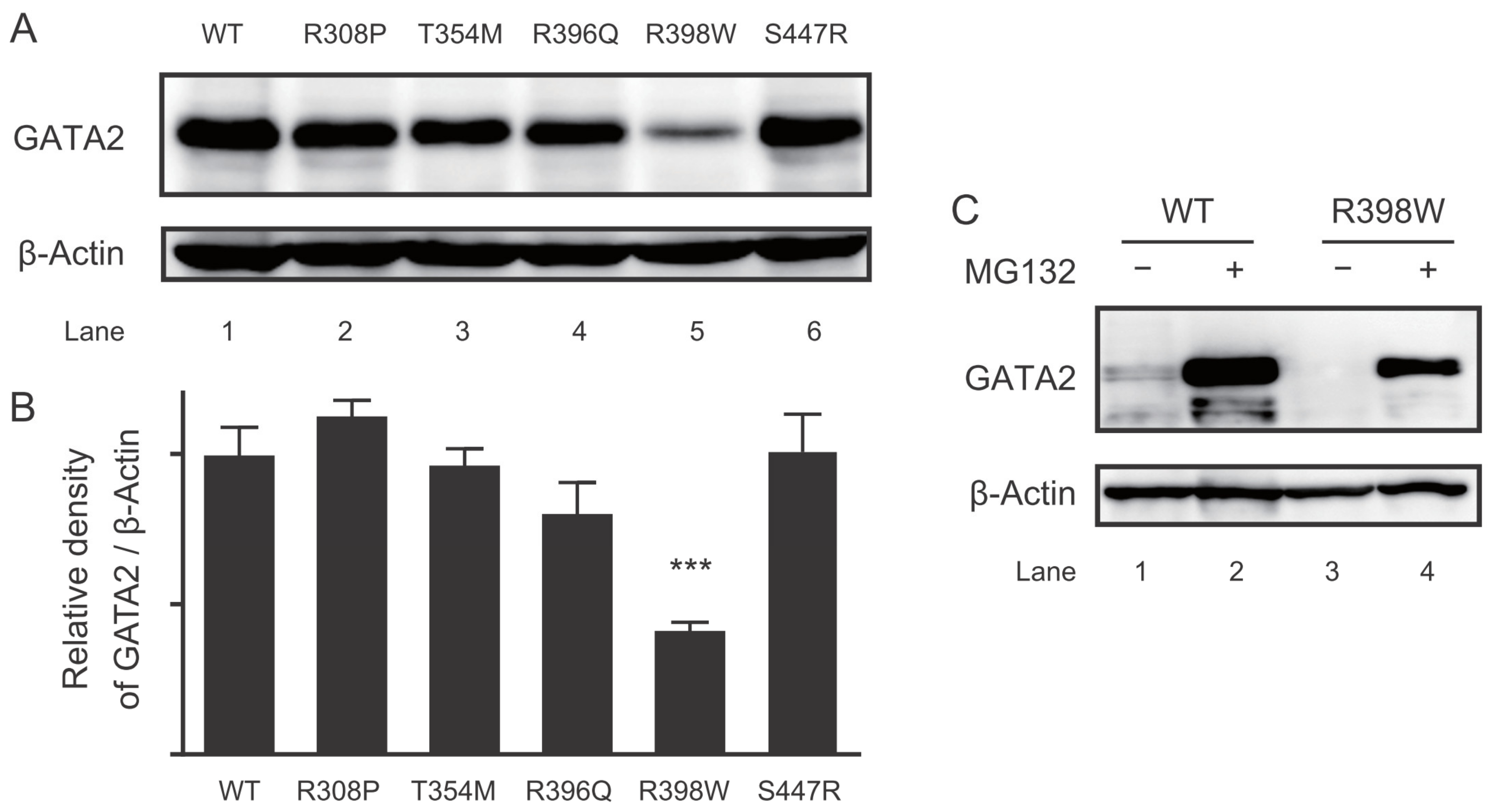
2.5. Protein Expression of the GATA2 Mutants
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructions
4.2. Cell Culture and Transient Transfection
4.3. Gel Shift Assay
4.4. Western Blotting Analysis
4.5. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| FCS | fetal calf serum |
| FOG | friend of GATA |
| GATA-RE | GATA responsive element |
| HPT axis | hypothalamus–pituitary–thyroid axis |
| PKC | protein kinase C |
| preproTRH | prepro-thyrotropin-releasing hormone |
| SDS-PAGE | sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| T3 | triiodothyronine |
| TPA | tetradecanoylphorbol acetate |
| TR | thyroid hormone receptor |
| TRH | thyrotropin-releasing hormone |
| TSH | thyroid-stimulating hormone |
| ZF | zinc-finger domain |
| ZF1 | N-terminal zinc-finger domain |
| ZF2 | C-terminal zinc finger domain |
References
- Fekete, C.; Lechan, R.M. Central regulation of hypothalamic-pituitary-thyroid axis under physiological and pathophysiological conditions. Endocr. Rev. 2014, 35, 159–194. [Google Scholar] [CrossRef]
- Hollenberg, A.N. Regulation of Thyrotropin Secretion. In Werner & Ingbar’s The Thyroid A Fundamental and Clinical Text, 10th ed.; Braverman, L.E., Cooper, D.S., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 169–182. [Google Scholar]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.F.; Lewis, S.R.; Haugen, B.R.; James, R.A.; McDermott, M.T.; Wood, W.M.; Ridgway, E.C. Pit-1 and GATA-2 interact and functionally cooperate to activate the thyrotropin beta-subunit promoter. J. Biol. Chem. 1997, 272, 24339–24347. [Google Scholar] [CrossRef] [PubMed]
- Dasen, J.S.; O’Connell, S.M.; Flynn, S.E.; Treier, M.; Gleiberman, A.S.; Szeto, D.P.; Hooshmand, F.; Aggarwal, A.K.; Rosenfeld, M.G. Reciprocal interactions of Pit1 and GATA2 mediate signaling gradient-induced determination of pituitary cell types. Cell 1999, 97, 587–598. [Google Scholar] [CrossRef]
- Charles, M.A.; Saunders, T.L.; Wood, W.M.; Owens, K.; Parlow, A.F.; Camper, S.A.; Ridgway, E.C.; Gordon, D.F. Pituitary-specific Gata2 knockout: Effects on gonadotrope and thyrotrope function. Mol. Endocrinol. 2006, 20, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Kashiwabara, Y.; Sasaki, S.; Matsushita, A.; Nagayama, K.; Ohba, K.; Iwaki, H.; Matsunaga, H.; Suzuki, S.; Misawa, H.; Ishizuka, K.; et al. Functions of PIT1 in GATA2-dependent transactivation of the thyrotropin beta promoter. J. Mol. Endocrinol. 2009, 42, 225–237. [Google Scholar] [CrossRef]
- Liu, C.; Goshu, E.; Wells, A.; Fan, C.M. Identification of the downstream targets of SIM1 and ARNT2, a pair of transcription factors essential for neuroendocrine cell differentiation. J. Biol. Chem. 2003, 278, 44857–44867. [Google Scholar] [CrossRef]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998, 12, 3264–3275. [Google Scholar] [CrossRef]
- Kuroda, G.; Sasaki, S.; Matsushita, A.; Ohba, K.; Sakai, Y.; Shinkai, S.; Nakamura, H.M.; Yamagishi, S.; Sato, K.; Hirahara, N.; et al. G ATA2 mediates the negative regulation of the prepro-thyrotropin-releasing hormone gene by liganded T3 receptor β2 in the rat hypothalamic paraventricular nucleus. PLoS ONE 2020, 15, e0242380. [Google Scholar] [CrossRef]
- Katsumura, K.R.; Bresnick, E.H. The GATA factor revolution in hematology. Blood 2017, 129, 2092–2102. [Google Scholar] [CrossRef]
- Lentjes, M.H.; Niessen, H.E.; Akiyama, Y.; de Bruïne, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef]
- Nardelli, J.; Thiesson, D.; Fujiwara, Y.; Tsai, F.Y.; Orkin, S.H. Expression and genetic interaction of transcription factors GATA-2 and GATA-3 during development of the mouse central nervous system. Dev. Biol. 1999, 210, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Groenen, P.; Nesbit, M.A.; Schuffenhauer, S.; Lichtner, P.; Vanderlinden, G.; Harding, B.; Beetz, R.; Bilous, R.W.; Holdaway, I.; et al. GATA3 haplo-insufficiency causes human HDR syndrome. Nature 2000, 406, 419–422. [Google Scholar] [CrossRef]
- Bresnick, E.H.; Johnson, K.D. Blood disease-causing and -suppressing transcriptional enhancers: General principles and GATA2 mechanisms. Blood Adv. 2019, 3, 2045–2056. [Google Scholar] [CrossRef]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and immune defects associated with GATA2 mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef]
- Matsushita, A.; Sasaki, S.; Kashiwabara, Y.; Nagayama, K.; Ohba, K.; Iwaki, H.; Misawa, H.; Ishizuka, K.; Nakamura, H. Essential role of GATA2 in the negative regulation of thyrotropin beta gene by thyroid hormone and its receptors. Mol. Endocrinol. 2007, 21, 865–884. [Google Scholar] [CrossRef]
- Sasaki, S.; Matsushita, A.; Kuroda, G.; Nakamura, H.M.; Oki, Y.; Suda, T. The Mechanism of Negative Transcriptional Regulation by Thyroid Hormone: Lessons From the Thyrotropin β Subunit Gene. Vitam. Horm. 2018, 106, 97–127. [Google Scholar]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef]
- Donadieu, J.; Lamant, M.; Fieschi, C.; de Fontbrune, F.S.; Caye, A.; Ouachee, M.; Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018, 103, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Hollowell, J.G.; Staehling, N.W.; Flanders, W.D.; Hannon, W.H.; Gunter, E.W.; Spencer, C.A.; Braverman, L.E. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 2002, 87, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Kiyoi, H.; Ishikawa, Y.; Hayakawa, F.; Kurahashi, S.; Kihara, R.; Tomita, A.; Naoe, T. GATA2 zinc finger 2 mutation found in acute myeloid leukemia impairs myeloid differentiation. Leuk. Res. Rep. 2013, 2, 21–25. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mir, M.A.; Kochuparambil, S.T.; Abraham, R.S.; Rodriguez, V.; Howard, M.; Hsu, A.P.; Jackson, A.E.; Holland, S.M.; Patnaik, M.M. Spectrum of myeloid neoplasms and immune deficiency associated with germline GATA2 mutations. Cancer Med. 2015, 4, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Lavaud, X.; Landecho, M.F.; Maicas, M.; Urquiza, L.; Merino, J.; Moreno-Miralles, I.; Odero, M.D. GATA2 germline mutations impair GATA2 transcription, causing haploinsufficiency: Functional analysis of the p.Arg396Gln mutation. J. Immunol. 2015, 194, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef]
- Chong, C.E.; Venugopal, P.; Stokes, P.H.; Lee, Y.K.; Brautigan, P.J.; Yeung, D.T.O.; Babic, M.; Engler, G.A.; Lane, S.W.; Klingler-Hoffmann, M.; et al. Differential effects on gene transcription and hematopoietic differentiation correlate with GATA2 mutant disease phenotypes. Leukemia 2018, 32, 194–202. [Google Scholar] [CrossRef]
- Tien, F.M.; Hou, H.A.; Tsai, C.H.; Tang, J.L.; Chiu, Y.C.; Chen, C.Y.; Kuo, Y.Y.; Tseng, M.H.; Peng, Y.L.; Liu, M.C.; et al. GATA2 zinc finger 1 mutations are associated with distinct clinico-biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer J. 2018, 8, 87. [Google Scholar] [CrossRef]
- Ohba, K.; Sasaki, S.; Matsushita, A.; Iwaki, H.; Matsunaga, H.; Suzuki, S.; Ishizuka, K.; Misawa, H.; Oki, Y.; Nakamura, H. GATA2 mediates thyrotropin-releasing hormone-induced transcriptional activation of the thyrotropin β gene. PLoS ONE 2011, 6, e18667. [Google Scholar] [CrossRef]
- Minegishi, N.; Suzuki, N.; Kawatani, Y.; Shimizu, R.; Yamamoto, M. Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes Cells 2005, 10, 693–704. [Google Scholar] [CrossRef]
- Nakajima, T.; Kitagawa, K.; Ohhata, T.; Sakai, S.; Uchida, C.; Shibata, K.; Minegishi, N.; Yumimoto, K.; Nakayama, K.I.; Masumoto, K.; et al. Regulation of GATA-binding protein 2 levels via ubiquitin-dependent degradation by Fbw7: Involvement of cyclin B-cyclin-dependent kinase 1-mediated phosphorylation of THR176 in GATA-binding protein 2. J. Biol. Chem. 2015, 290, 10368–10381. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.; Conchillo, A.; García-Sánchez, M.A.; Odero, M.D. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 2012, 82, 1–17. [Google Scholar] [CrossRef]
- Haddox, C.L.; Carr, R.M.; Abraham, R.S.; Perez Botero, J.; Rodriguez, V.; Pardanani, A.; Patnaik, M.M. Phenotypic heterogeneity associated with germline GATA2 haploinsufficiency: A comprehensive kindred study. Leuk. Lymp. 2019, 60, 3282–3286. [Google Scholar] [CrossRef]
- Greif, P.A.; Dufour, A.; Konstandin, N.P.; Ksienzyk, B.; Zellmeier, E.; Tizazu, B.; Sturm, J.; Benthaus, T.; Herold, T.; Yaghmaie, M.; et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood 2012, 120, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Ma, L.Y.; Huang, Q.H.; Li, G.; Gu, B.W.; Gao, X.D.; Shi, J.Y.; Wang, Y.Y.; Gao, L.; Cai, X.; et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 2076–2081. [Google Scholar] [CrossRef]
- Trainor, C.D.; Ghirlando, R.; Simpson, M.A. GATA zinc finger interactions modulate DNA binding and transactivation. J. Biol. Chem. 2000, 275, 28157–28166. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Liu, Y.Y.; Karthikarj, R.; Kannan, K.; Jiang, J.; Abe, K.; Milanesi, A.; Brent, G.A. Thyroid hormone receptor beta sumoylation is required for thyrotropin regulation and thyroid hormone production. JCI Insight 2021, 6, e149425. [Google Scholar] [CrossRef]
- Persani, L.; Beck-Peccoz, P. Central Hypothyroidis. In Werner & Ingbar’s The Thyroid A Fundamental and Clinical Text, 10th ed.; Braverman, L.E., Cooper, D.S., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 560–568. [Google Scholar]
- Schoenmakers, N.; Alatzoglou, K.S.; Chatterjee, V.K.; Dattani, M.T. Recent advances in central congenital hypothyroidism. J. Endocrinol. 2015, 227, R51–R71. [Google Scholar] [CrossRef]
- Persani, L.; Bonomi, M. The multiple genetic causes of central hypothyroidism. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Persani, L.; Brabant, G.; Dattani, M.; Bonomi, M.; Feldt-Rasmussen, U.; Fliers, E.; Gruters, A.; Maiter, D.; Schoenmakers, N.; van Trotsenburg, A.S.P. 2018 European Thyroid Association (ETA) Guidelines on the Diagnosis and Management of Central Hypothyroidism. Eur. Thyroid J. 2018, 7, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Sugisawa, C.; Takamizawa, T.; Abe, K.; Hasegawa, T.; Shiga, K.; Sugawara, H.; Ohsugi, K.; Muroya, K.; Asakura, Y.; Adachi, M.; et al. Genetics of Congenital Isolated TSH Deficiency: Mutation Screening of the Known Causative Genes and a Literature Review. J. Clin. Endocrinol. Metab. 2019, 104, 6229–6237. [Google Scholar] [CrossRef]
- Misawa, H.; Sasaki, S.; Matsushita, A.; Ohba, K.; Iwaki, H.; Matsunaga, H.; Suzuki, S.; Ishizuka, K.; Oki, Y.; Nakamura, H. Liganded thyroid hormone receptor inhibits phorbol 12-O-tetradecanoate-13-acetate-induced enhancer activity via firefly luciferase cDNA. PLoS ONE 2012, 7, e28916. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F.C.; Girardi, A.J.; Gilden, R.V.; Koprowski, H. Infection of human and simian tissue cultures with Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 1964, 52, 53–59. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakai, Y.; Ohba, K.; Sasaki, S.; Matsushita, A.; Nakamura, H.M.; Kuroda, G.; Tsuriya, D.; Yamashita, M.; Suda, T. Impairment of the Hypothalamus–Pituitary–Thyroid Axis Caused by Naturally Occurring GATA2 Mutations In Vitro. Int. J. Mol. Sci. 2021, 22, 10015. https://doi.org/10.3390/ijms221810015
Sakai Y, Ohba K, Sasaki S, Matsushita A, Nakamura HM, Kuroda G, Tsuriya D, Yamashita M, Suda T. Impairment of the Hypothalamus–Pituitary–Thyroid Axis Caused by Naturally Occurring GATA2 Mutations In Vitro. International Journal of Molecular Sciences. 2021; 22(18):10015. https://doi.org/10.3390/ijms221810015
Chicago/Turabian StyleSakai, Yuki, Kenji Ohba, Shigekazu Sasaki, Akio Matsushita, Hiroko Misawa Nakamura, Go Kuroda, Daisuke Tsuriya, Miho Yamashita, and Takafumi Suda. 2021. "Impairment of the Hypothalamus–Pituitary–Thyroid Axis Caused by Naturally Occurring GATA2 Mutations In Vitro" International Journal of Molecular Sciences 22, no. 18: 10015. https://doi.org/10.3390/ijms221810015
APA StyleSakai, Y., Ohba, K., Sasaki, S., Matsushita, A., Nakamura, H. M., Kuroda, G., Tsuriya, D., Yamashita, M., & Suda, T. (2021). Impairment of the Hypothalamus–Pituitary–Thyroid Axis Caused by Naturally Occurring GATA2 Mutations In Vitro. International Journal of Molecular Sciences, 22(18), 10015. https://doi.org/10.3390/ijms221810015