
The Role of Microglia in Modulating Neuroinflammation after Spinal Cord Injury



Abstract
:1. Introduction
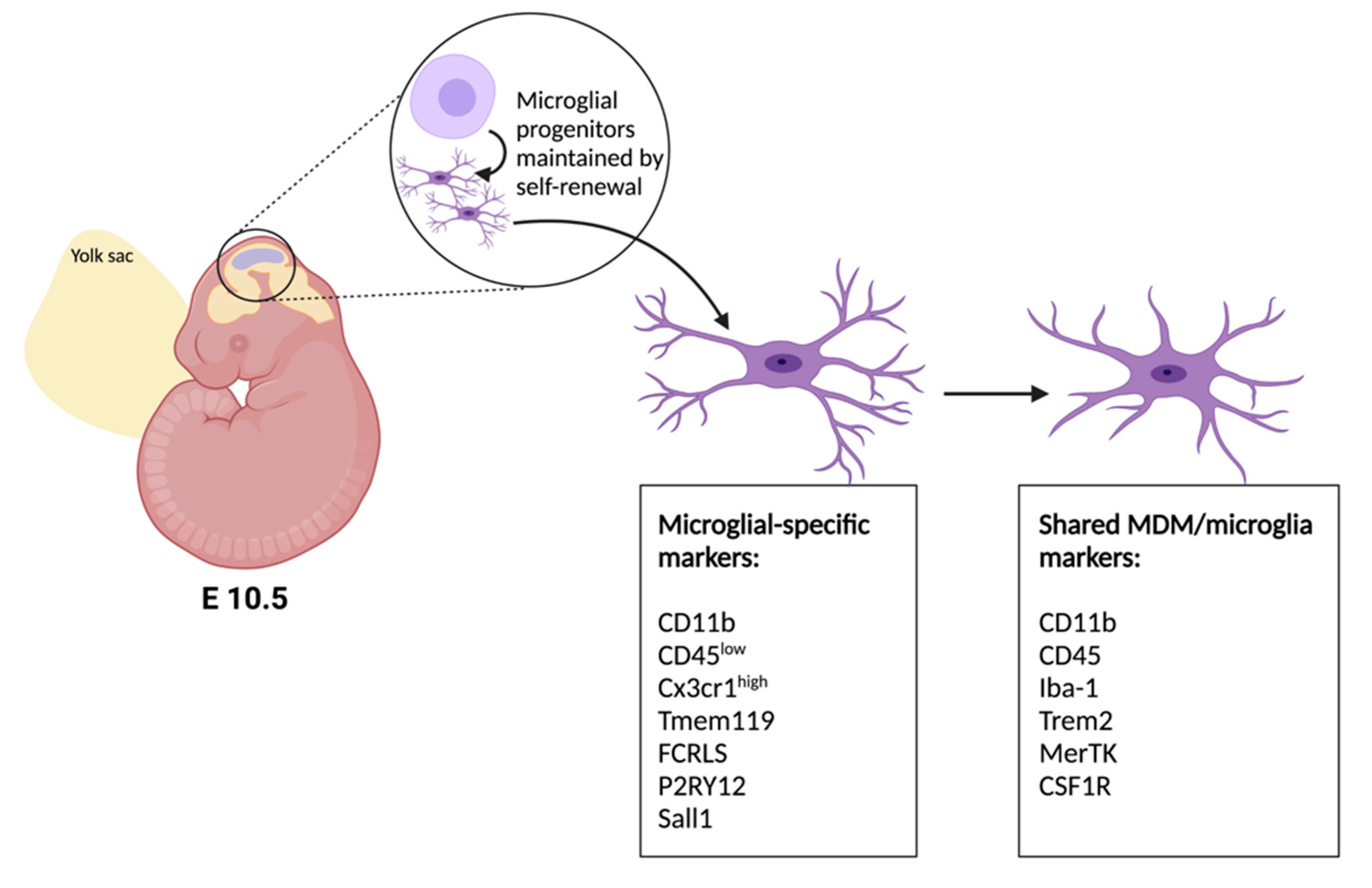
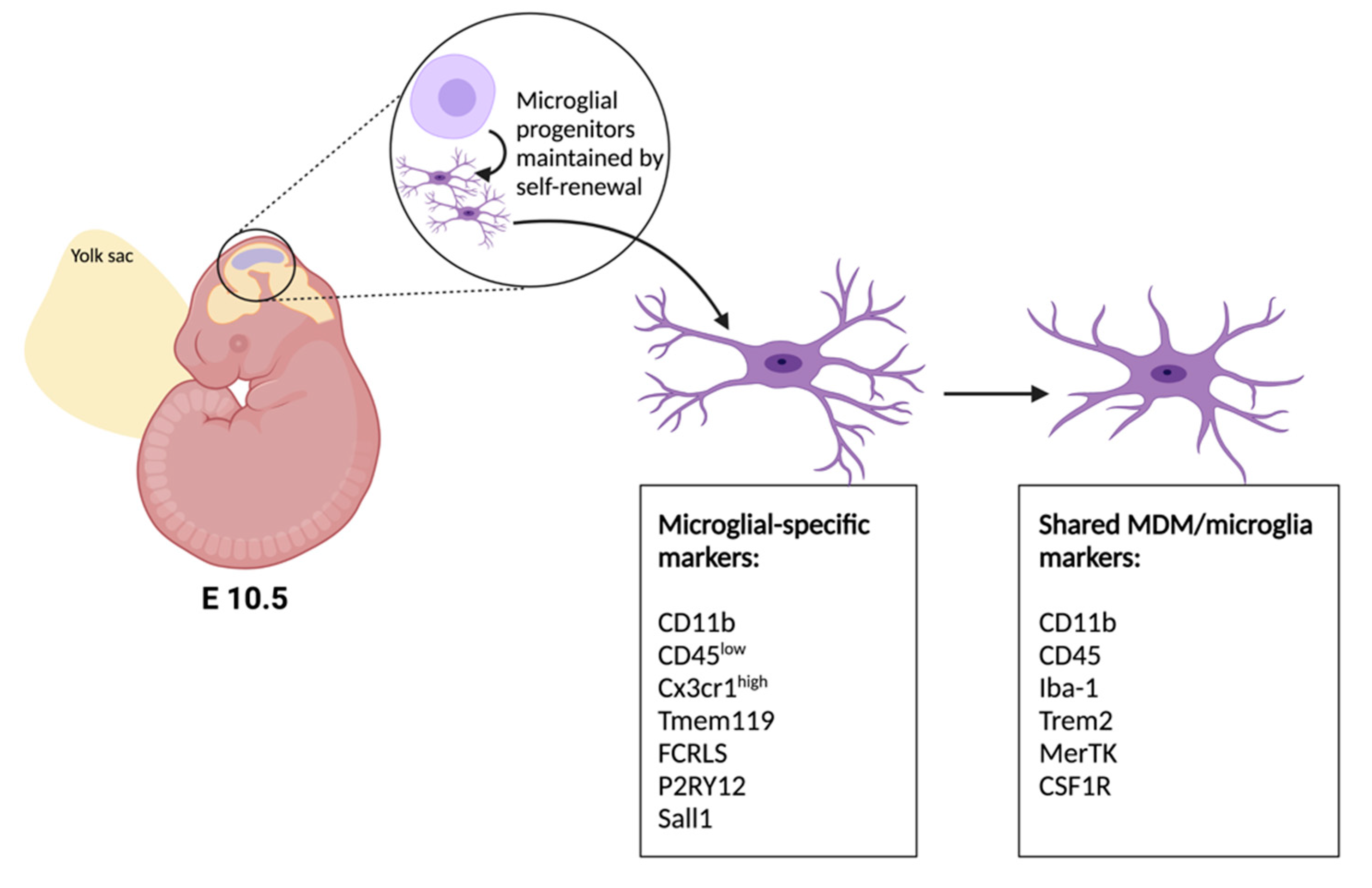
2. Developmental Origins of Microglia and Advances in Genetic Tools
3. Heterogeneity of Microglia in the Spinal Cord
4. Role of Microglia Following SCI
5. Chronic Inflammation Following SCI
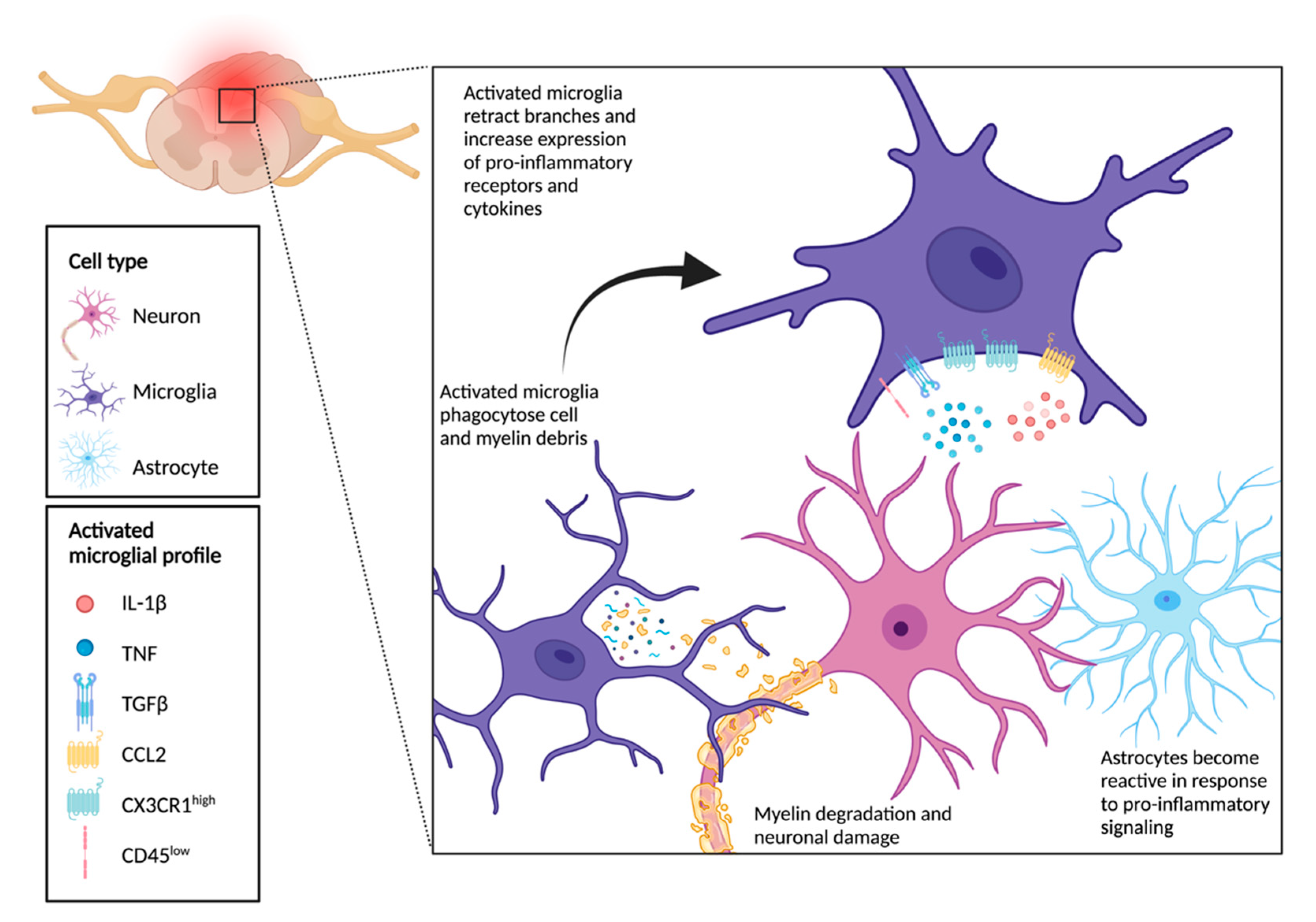
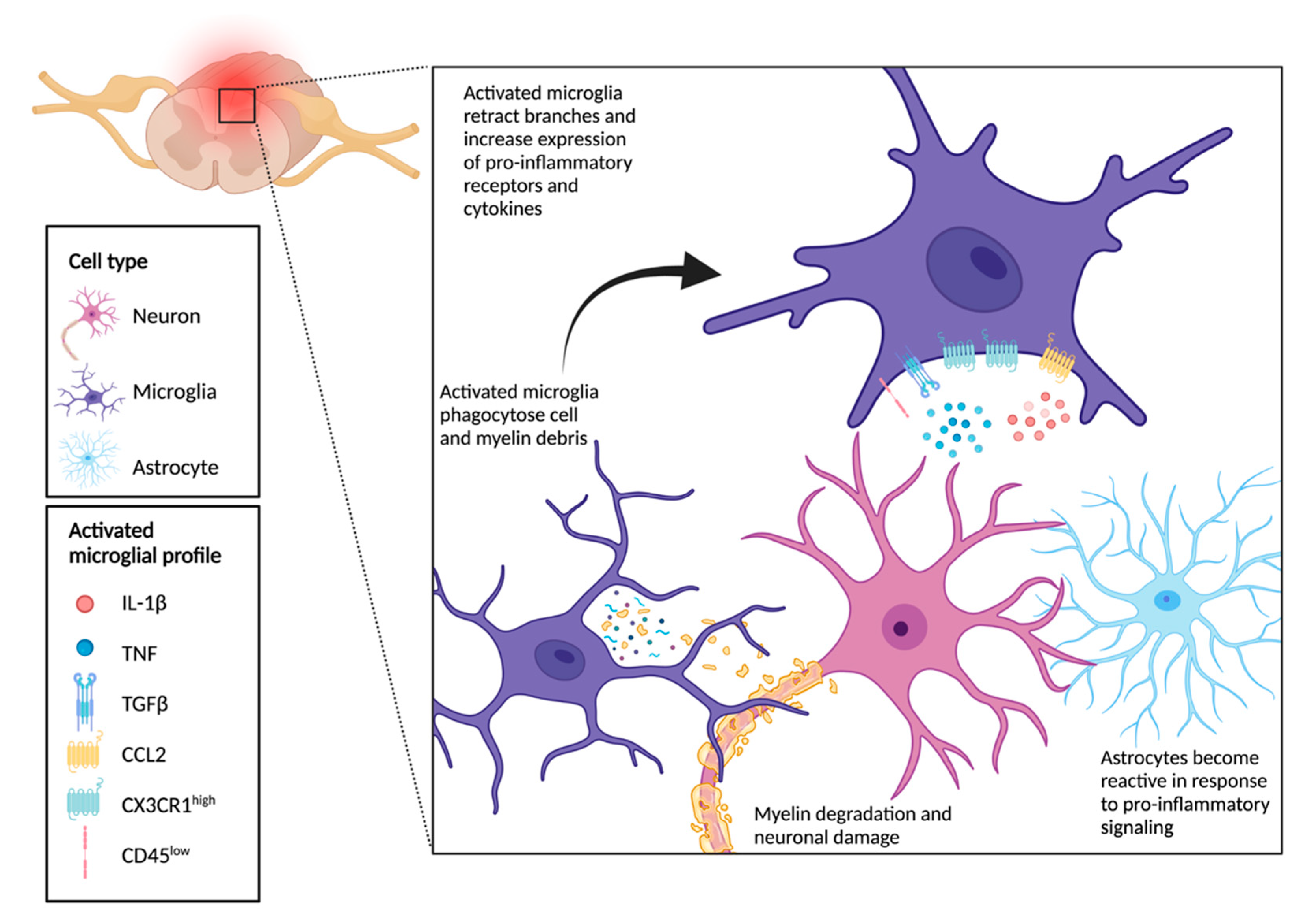
6. Crosstalk between Microglia, Neurons, and Astrocytes Drives Functional Outcomes Following SCI
7. Therapeutic Targeting of Microglia after SCI
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Druschel, C.; Curt, A.; Fehlings, M.G. Traumatic Spinal Cord Injury. Nat. Rev. Dis. Primers 2017, 3, 17018. [Google Scholar] [CrossRef]
- Badhiwala, J.H.; Ahuja, C.S.; Akbar, M.A.; Witiw, C.D.; Nassiri, F.; Furlan, J.C.; Curt, A.; Wilson, J.R.; Fehlings, M.G. Degenerative Cervical Myelopathy—Update and Future Directions. Nat. Rev. Neurol. 2020, 16, 108–124. [Google Scholar] [CrossRef]
- Katz, J.N. Lumbar Spinal Stenosis. N. Engl. J. Med. 2008, 358, 818–825. [Google Scholar] [CrossRef]
- Fehlings, M.G.; Tetreault, L.A.; Riew, K.D.; Middleton, J.W.; Aarabi, B.; Arnold, P.M.; Brodke, D.S.; Burns, A.S.; Carette, S.; Chen, R.; et al. A Clinical Practice Guideline for the Management of Patients with Degenerative Cervical Myelopathy: Recommendations for Patients with Mild, Moderate, and Severe Disease and Nonmyelopathic Patients with Evidence of Cord Compression. Glob. Spine J. 2017, 7, 70S–83S. [Google Scholar] [CrossRef] [Green Version]
- Vidal, P.M.; Karadimas, S.K.; Ulndreaj, A.; Laliberte, A.M.; Tetreault, L.; Forner, S.; Wang, J.; Foltz, W.D.; Fehlings, M.G. Delayed Decompression Exacerbates Ischemia-Reperfusion Injury in Cervical Compressive Myelopathy. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinz, M.; Priller, J. Microglia and Brain Macrophages in the Molecular Age: From Origin to Neuropsychiatric Disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia Development and Function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [Green Version]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.-È. Microglia across the Lifespan: From Origin to Function in Brain Development, Plasticity and Cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef] [Green Version]
- Kroner, A.; Rosas Almanza, J. Role of Microglia in Spinal Cord Injury. Neurosci. Lett. 2019, 709, 134370. [Google Scholar] [CrossRef]
- Li, K.; Tan, Y.-H.; Light, A.R.; Fu, K.-Y. Different Peripheral Tissue Injury Induces Differential Phenotypic Changes of Spinal Activated Microglia. Clin. Dev. Immunol. 2013, 2013, e901420. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New Tools for Studying Microglia in the Mouse and Human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kros, J.M.; van der Weiden, M.; Zheng, P.; Cheng, C.; Mustafa, D.A.M. Expression Site of P2RY12 in Residential Microglial Cells in Astrocytomas Correlates with M1 and M2 Marker Expression and Tumor Grade. Acta Neuropathol. Commun. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 Is a Transcriptional Regulator Defining Microglia Identity and Function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar] [CrossRef]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallières, N.; Lessard, M.; Janelle, M.-E.; Vernoux, N.; Tremblay, M.-È.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia Are an Essential Component of the Neuroprotective Scar That Forms after Spinal Cord Injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef] [Green Version]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of “homeostatic” Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; López-Vales, R. Myeloid Cell Responses after Spinal Cord Injury. J. Neuroimmunol. 2018, 321, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinsey, G.L.; Lizama, C.O.; Keown-Lang, A.E.; Niu, A.; Santander, N.; Larpthaveesarp, A.; Chee, E.; Gonzalez, F.F.; Arnold, T.D. A New Genetic Strategy for Targeting Microglia in Development and Disease. Elife 2020, 9, e54590. [Google Scholar] [CrossRef]
- Inoue, S.; Inoue, M.; Fujimura, S.; Nishinakamura, R. A Mouse Line Expressing Sall1-Driven Inducible Cre Recombinase in the Kidney Mesenchyme. Genesis 2010, 48, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Cătălin, B.; Mitran, S.; Albu, C.; Iancău, M. Comparative Aspects of Microglia Reaction in White and Gray Matter. Curr. Health Sci. J. 2013, 39, 151–154. [Google Scholar] [PubMed]
- David, G.; Mohammadi, S.; Martin, A.R.; Cohen-Adad, J.; Weiskopf, N.; Thompson, A.; Freund, P. Traumatic and Nontraumatic Spinal Cord Injury: Pathological Insights from Neuroimaging. Nat. Rev. Neurol. 2019, 15, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Tator, C.H.; Koyanagi, I. Vascular Mechanisms in the Pathophysiology of Human Spinal Cord Injury. J. Neurosurg. 1997, 86, 483–492. [Google Scholar] [CrossRef]
- Losey, P.; Young, C.; Krimholtz, E.; Bordet, R.; Anthony, D.C. The Role of Hemorrhage Following Spinal-Cord Injury. Brain Res. 2014, 1569, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Van der Poel, M.; Ulas, T.; Mizee, M.R.; Hsiao, C.-C.; Miedema, S.S.M.; Adelia; Schuurman, K.G.; Helder, B.; Tas, S.W.; Schultze, J.L.; et al. Transcriptional Profiling of Human Microglia Reveals Grey–White Matter Heterogeneity and Multiple Sclerosis-Associated Changes. Nat. Commun. 2019, 10, 1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Radaideh, A.M.; Wharton, S.J.; Lim, S.-Y.; Tench, C.R.; Morgan, P.S.; Bowtell, R.W.; Constantinescu, C.S.; Gowland, P.A. Increased Iron Accumulation Occurs in the Earliest Stages of Demyelinating Disease: An Ultra-High Field Susceptibility Mapping Study in Clinically Isolated Syndrome. Mult. Scler. J. 2013, 19, 896–903. [Google Scholar] [CrossRef]
- McKay, S.M.; Brooks, D.J.; Hu, P.; McLachlan, E.M. Distinct Types of Microglial Activation in White and Grey Matter of Rat Lumbosacral Cord after Mid-Thoracic Spinal Transection. J. Neuropathol. Exp. Neurol. 2007, 66, 698–710. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hamanaka, G.; Lo, E.H.; Arai, K. Heterogeneity of Microglia and Their Differential Roles in White Matter Pathology. CNS Neurosci. Ther. 2019, 25, 1290–1298. [Google Scholar] [CrossRef]
- Totoiu, M.O.; Keirstead, H.S. Spinal Cord Injury Is Accompanied by Chronic Progressive Demyelination. J. Comp. Neurol. 2005, 486, 373–383. [Google Scholar] [CrossRef]
- Dai, X.; Chen, J.; Xu, F.; Zhao, J.; Cai, W.; Sun, Z.; Hitchens, T.K.; Foley, L.M.; Leak, R.K.; Chen, J.; et al. TGFα Preserves Oligodendrocyte Lineage Cells and Improves White Matter Integrity after Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2020, 40, 639–655. [Google Scholar] [CrossRef]
- Dillenburg, A.; Ireland, G.; Holloway, R.K.; Davies, C.L.; Evans, F.L.; Swire, M.; Bechler, M.E.; Soong, D.; Yuen, T.J.; Su, G.H.; et al. Activin Receptors Regulate the Oligodendrocyte Lineage in Health and Disease. Acta Neuropathol. 2018, 135, 887–906. [Google Scholar] [CrossRef] [Green Version]
- Hlavica, M.; Delparente, A.; Good, A.; Good, N.; Plattner, P.S.; Seyedsadr, M.S.; Schwab, M.E.; Figlewicz, D.P.; Ineichen, B.V. Intrathecal Insulin-like Growth Factor 1 but Not Insulin Enhances Myelin Repair in Young and Aged Rats. Neurosci. Lett. 2017, 648, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Cardona, A.E. The Myeloid Cells of the Central Nervous System Parenchyma. Nature 2010, 468, 253–262. [Google Scholar] [CrossRef]
- Staszewski, O.; Hagemeyer, N. Unique Microglia Expression Profile in Developing White Matter. BMC Res. Notes 2019, 12, 367. [Google Scholar] [CrossRef]
- Chertoff, M.; Shrivastava, K.; Gonzalez, B.; Acarin, L.; Giménez-Llort, L. Differential Modulation of TREM2 Protein during Postnatal Brain Development in Mice. PLoS ONE 2013, 8, e72083. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.-K. Microglia as a Source and Target of Cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Pineau, I.; Lacroix, S. Proinflammatory Cytokine Synthesis in the Injured Mouse Spinal Cord: Multiphasic Expression Pattern and Identification of the Cell Types Involved. J. Comp. Neurol. 2007, 500, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Ling, E.A.; Wong, W.C. The Origin and Nature of Ramified and Amoeboid Microglia: A Historical Review and Current Concepts. Glia 1993, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 Microglia: The Good, the Bad, and the Inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. A Polarizing Question: Do M1 and M2 Microglia Exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.; Kroner, A. Repertoire of Microglial and Macrophage Responses after Spinal Cord Injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune Function of Microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S.; Sessa, W.C. Inflammation and the Blood Microvascular System. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; McGaughy, V.M.; Popovich, P.G. Comparative Analysis of Lesion Development and Intraspinal Inflammation in Four Strains of Mice Following Spinal Contusion Injury. J. Comp. Neurol. 2006, 494, 578–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shechter, R.; Miller, O.; Yovel, G.; Rosenzweig, N.; London, A.; Ruckh, J.; Kim, K.-W.; Klein, E.; Kalchenko, V.; Bendel, P.; et al. Recruitment of Beneficial M2 Macrophages to Injured Spinal Cord Is Orchestrated by Remote Brain Choroid Plexus. Immunity 2013, 38, 555–569. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, A.D.; David, S. Differences in the Phagocytic Response of Microglia and Peripheral Macrophages after Spinal Cord Injury and Its Effects on Cell Death. J. Neurosci. 2014, 34, 6316–6322. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally Derived Macrophages Modulate Microglial Function to Reduce Inflammation after CNS Injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [Green Version]
- Umebayashi, D.; Natsume, A.; Takeuchi, H.; Hara, M.; Nishimura, Y.; Fukuyama, R.; Sumiyoshi, N.; Wakabayashi, T. Blockade of Gap Junction Hemichannel Protects Secondary Spinal Cord Injury from Activated Microglia-Mediated Glutamate Exitoneurotoxicity. J. Neurotrauma 2014, 31, 1967–1974. [Google Scholar] [CrossRef] [Green Version]
- Cronin, M.; Anderson, P.N.; Cook, J.E.; Green, C.R.; Becker, D.L. Blocking Connexin43 Expression Reduces Inflammation and Improves Functional Recovery after Spinal Cord Injury. Mol. Cell. Neurosci. 2008, 39, 152–160. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Gorrie, C.A.; Velamoor, S.; Green, C.R.; Nicholson, L.F.B. Connexin43 Mimetic Peptide Is Neuroprotective and Improves Function Following Spinal Cord Injury. Neurosci. Res. 2013, 75, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. Mild Hypoxia Triggers Transient Blood-Brain Barrier Disruption: A Fundamental Protective Role for Microglia. Acta Neuropathol. Commun. 2020, 8, 175. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. A Critical Role for Microglia in Maintaining Vascular Integrity in the Hypoxic Spinal Cord. Proc. Natl. Acad. Sci. USA 2019, 116, 26029–26037. [Google Scholar] [CrossRef]
- Tator, C.H.; Fehlings, M.G. Review of the Secondary Injury Theory of Acute Spinal Cord Trauma with Emphasis on Vascular Mechanisms. J. Neurosurg. 1991, 75, 15–26. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, X.; Gong, L.; Cui, L.; Zhang, H.; Zhao, W.; Jiang, P.; Hou, G.; Hou, Y. Tetramethylpyrazine Protects Blood-Spinal Cord Barrier Integrity by Modulating Microglia Polarization Through Activation of STAT3/SOCS3 and Inhibition of NF-KB Signaling Pathways in Experimental Autoimmune Encephalomyelitis Mice. Cell. Mol. Neurobiol. 2021, 41, 717–731. [Google Scholar] [CrossRef]
- Allison, D.J.; Ditor, D.S. Immune Dysfunction and Chronic Inflammation Following Spinal Cord Injury. Spinal Cord 2015, 53, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Cruse, J.M.; Keith, J.C.; Bryant, M.L.; Lewis, R.E. Immune System-Neuroendocrine Dysregulation in Spinal Cord Injury. Immunol. Res. 1996, 15, 306–314. [Google Scholar] [CrossRef]
- Lucin, K.M.; Sanders, V.M.; Jones, T.B.; Malarkey, W.B.; Popovich, P.G. Impaired Antibody Synthesis after Spinal Cord Injury Is Level-Dependent and Is Due to Sympathetic Nervous System Dysregulation. Exp. Neurol. 2007, 207, 75–84. [Google Scholar] [CrossRef] [Green Version]
- Ankeny, D.P.; Lucin, K.M.; Sanders, V.M.; McGaughy, V.M.; Popovich, P.G. Spinal Cord Injury Triggers Systemic Autoimmunity: Evidence for Chronic B Lymphocyte Activation and Lupus-like Autoantibody Synthesis. J. Neurochem. 2006, 99, 1073–1087. [Google Scholar] [CrossRef]
- Campagnolo, D.I.; Dixon, D.; Schwartz, J.; Bartlett, J.A.; Keller, S.E. Altered Innate Immunity Following Spinal Cord Injury. Spinal Cord 2008, 46, 477–481. [Google Scholar] [CrossRef]
- Iversen, P.O.; Hjeltnes, N.; Holm, B.; Flatebø, T.; Strøm-Gundersen, I.; Rønning, W.; Stanghelle, J.; Benestad, H.B. Depressed Immunity and Impaired Proliferation of Hematopoietic Progenitor Cells in Patients with Complete Spinal Cord Injury. Blood 2000, 96, 2081–2083. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wahane, S.; Friedl, M.-S.; Kluge, M.; Friedel, C.C.; Avrampou, K.; Zachariou, V.; Guo, L.; Zhang, B.; He, X.; et al. Microglia and Macrophages Promote Corralling, Wound Compaction and Recovery after Spinal Cord Injury via Plexin-B2. Nat. Neurosci. 2020, 23, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.-H.; Hazell, A.S. Excitotoxic Mechanisms and the Role of Astrocytic Glutamate Transporters in Traumatic Brain Injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Velumian, A.A.; Fehlings, M.G. The Role of Excitotoxicity in Secondary Mechanisms of Spinal Cord Injury: A Review with an Emphasis on the Implications for White Matter Degeneration. J. Neurotrauma 2004, 21, 754–774. [Google Scholar] [CrossRef]
- Matute, C.; Sánchez-Gómez, M.V.; Martínez-Millán, L.; Miledi, R. Glutamate Receptor-Mediated Toxicity in Optic Nerve Oligodendrocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 8830–8835. [Google Scholar] [CrossRef] [Green Version]
- Mcdonald, J.W.; Althomsons, S.P.; Hyrc, K.L.; Choi, D.W.; Goldberg, M.P. Oligodendrocytes from Forebrain Are Highly Vulnerable to AMPA/Kainate Receptor-Mediated Excitotoxicity. Nat. Med. 1998, 4, 291–297. [Google Scholar] [CrossRef]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of Fractalkine Receptor CX(3)CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for Neuronally Derived Fractalkine in Mediating Interactions between Neurons and CX3CR1-Expressing Microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Soriano, S.G.; Amaravadi, L.S.; Wang, Y.F.; Zhou, H.; Yu, G.X.; Tonra, J.R.; Fairchild-Huntress, V.; Fang, Q.; Dunmore, J.H.; Huszar, D.; et al. Mice Deficient in Fractalkine Are Less Susceptible to Cerebral Ischemia-Reperfusion Injury. J. Neuroimmunol. 2002, 125, 59–65. [Google Scholar] [CrossRef]
- Sokolowski, J.D.; Chabanon-Hicks, C.N.; Han, C.Z.; Heffron, D.S.; Mandell, J.W. Fractalkine Is a “Find-Me” Signal Released by Neurons Undergoing Ethanol-Induced Apoptosis. Front. Cell. Neurosci. 2014, 8, 360. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of Microglial Neurotoxicity by the Fractalkine Receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Yona, S.; Kim, K.-W.; Jung, S. Microglia, Seen from the CX3CR1 Angle. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dénes, A.; Ferenczi, S.; Halász, J.; Környei, Z.; Kovács, K.J. Role of CX3CR1 (Fractalkine Receptor) in Brain Damage and Inflammation Induced by Focal Cerebral Ischemia in Mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-L.; Xu, B.; Li, S.-S.; Zhang, W.-S.; Xu, H.; Deng, X.-M.; Zhang, Y.-Q. Gabapentin Reduces CX3CL1 Signaling and Blocks Spinal Microglial Activation in Monoarthritic Rats. Mol. Brain 2012, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Pixley, F.J.; Stanley, E.R. CSF-1 Regulation of the Wandering Macrophage: Complexity in Action. Trends Cell Biol. 2004, 14, 628–638. [Google Scholar] [CrossRef]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Gerber, Y.N.; Saint-Martin, G.P.; Bringuier, C.M.; Bartolami, S.; Goze-Bac, C.; Noristani, H.N.; Perrin, F.E. CSF1R Inhibition Reduces Microglia Proliferation, Promotes Tissue Preservation and Improves Motor Recovery After Spinal Cord Injury. Front. Cell. Neurosci. 2018, 12, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Shi, X.Q.; Fan, A.; West, B.; Zhang, J. Targeting Macrophage and Microglia Activation with Colony Stimulating Factor 1 Receptor Inhibitor Is an Effective Strategy to Treat Injury-Triggered Neuropathic Pain. Mol. Pain 2018, 14, 1744806918764979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, N.S.; Brown, J.P.; Dissanayake, V.U.K.; Offord, J.; Thurlow, R.; Woodruff, G.N. The Novel Anticonvulsant Drug, Gabapentin (Neurontin), Binds to the A2δ Subunit of a Calcium Channel. J. Biol. Chem. 1996, 271, 5768–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenthal, M.; Mueller, M.; Olson, W.L.; Priebe, M.M.; Sherwood, A.M.; Olson, W.H. Gabapentin for the Treatment of Spasticity in Patients with Spinal Cord Injury. Spinal Cord 1997, 35, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Levendoglu, F.; Ögün, C.Ö.; Özerbil, Ö.; Ögün, T.C.; Ugurlu, H. Gabapentin Is a First Line Drug for the Treatment of Neuropathic Pain in Spinal Cord Injury. Spine 2004, 29, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Tzellos, T.G.; Papazisis, G.; Amaniti, E.; Kouvelas, D. Efficacy of Pregabalin and Gabapentin for Neuropathic Pain in Spinal-Cord Injury: An Evidence-Based Evaluation of the Literature. Eur. J. Clin. Pharmacol. 2008, 64, 851. [Google Scholar] [CrossRef]
- Brennan, F.H.; Noble, B.T.; Wang, Y.; Guan, Z.; Davis, H.; Mo, X.; Harris, C.; Eroglu, C.; Ferguson, A.R.; Popovich, P.G. Acute Post-Injury Blockade of A2δ-1 Calcium Channel Subunits Prevents Pathological Autonomic Plasticity after Spinal Cord Injury. Cell Rep. 2021, 34, 108667. [Google Scholar] [CrossRef]
- Kobashi, S.; Terashima, T.; Katagi, M.; Nakae, Y.; Okano, J.; Suzuki, Y.; Urushitani, M.; Kojima, H. Transplantation of M2-Deviated Microglia Promotes Recovery of Motor Function after Spinal Cord Injury in Mice. Mol. Ther. 2020, 28, 254–265. [Google Scholar] [CrossRef]
- Wieghofer, P.; Knobeloch, K.-P.; Prinz, M. Genetic Targeting of Microglia. Glia 2015, 63, 1–22. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Marker | Strain | Original Publication |
|---|---|---|
| Tmem119 | C57BL/6-Tmem119em1(cre/ERT2)Gfng/J | [19] |
| P2ry12 | B6(129S6)-P2ry12em1(icre/ERT2)Tda/J | [20] |
| Sall1 | Sall1tm5(cre/ERT2)Ryn | [21] |
| Target | Therapeutic Intervention | Original Publications |
|---|---|---|
| CX3CR1/CX3CL1 | Genetic deletion | [71,77] |
| CSF1R | Pharmacologic inhibition (GW2580, PLX5622) | [80,81] |
| Ca2+ channel α2δ subunit | Pharmacologic inhibition (Gabapentin) | [83,84,85,86] |
| M2 microglia | IL-4 biasing of M2 cells for transplant | [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brockie, S.; Hong, J.; Fehlings, M.G. The Role of Microglia in Modulating Neuroinflammation after Spinal Cord Injury. Int. J. Mol. Sci. 2021, 22, 9706. https://doi.org/10.3390/ijms22189706
Brockie S, Hong J, Fehlings MG. The Role of Microglia in Modulating Neuroinflammation after Spinal Cord Injury. International Journal of Molecular Sciences. 2021; 22(18):9706. https://doi.org/10.3390/ijms22189706
Chicago/Turabian StyleBrockie, Sydney, James Hong, and Michael G. Fehlings. 2021. "The Role of Microglia in Modulating Neuroinflammation after Spinal Cord Injury" International Journal of Molecular Sciences 22, no. 18: 9706. https://doi.org/10.3390/ijms22189706
APA StyleBrockie, S., Hong, J., & Fehlings, M. G. (2021). The Role of Microglia in Modulating Neuroinflammation after Spinal Cord Injury. International Journal of Molecular Sciences, 22(18), 9706. https://doi.org/10.3390/ijms22189706