
Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies




Abstract
:1. General Introduction
2. Plant Reproductive Strategies and Breeding Schemes in the Apiaceae Family
2.1. Plant Reproductive Systems in the Apiaceae Family
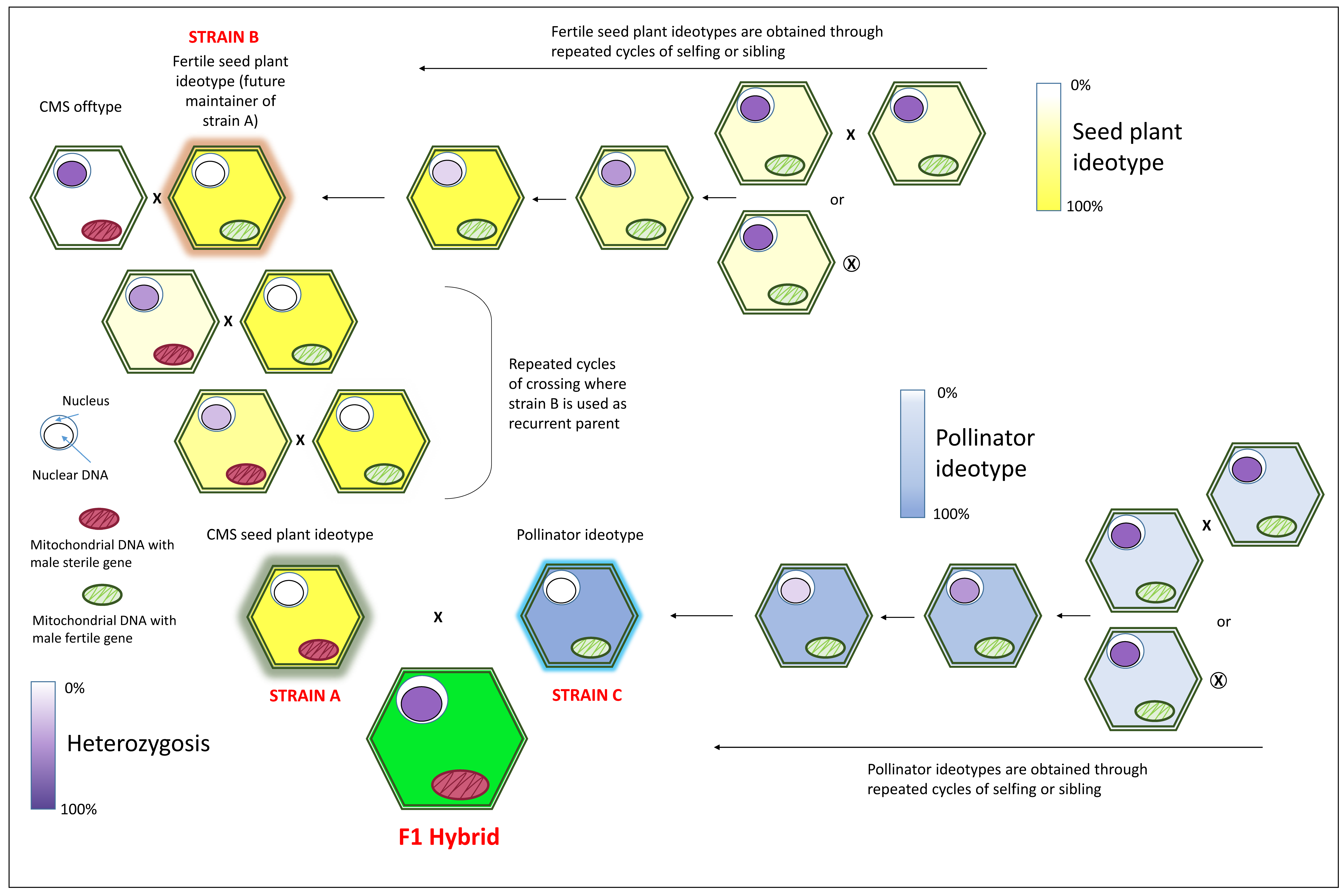
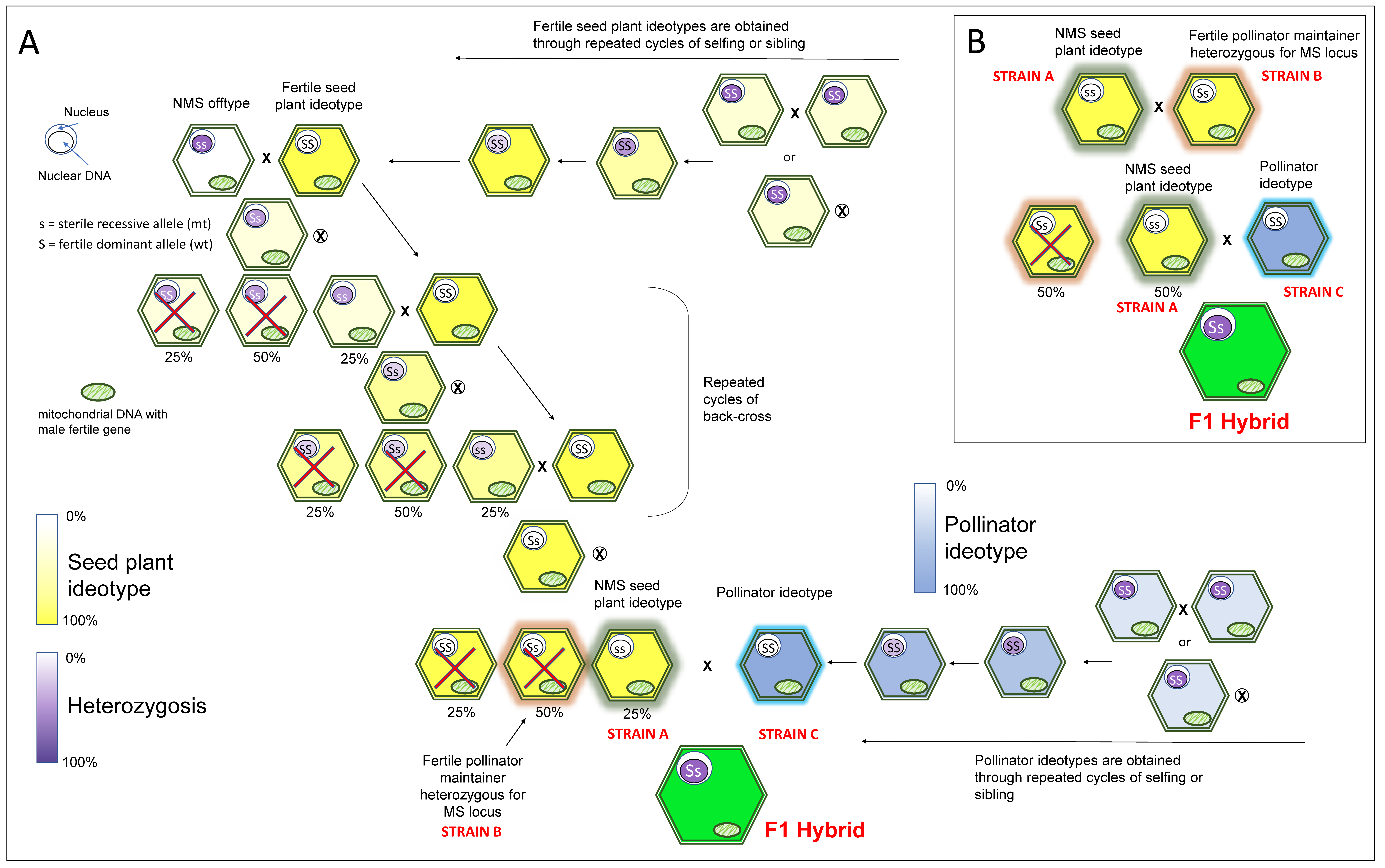
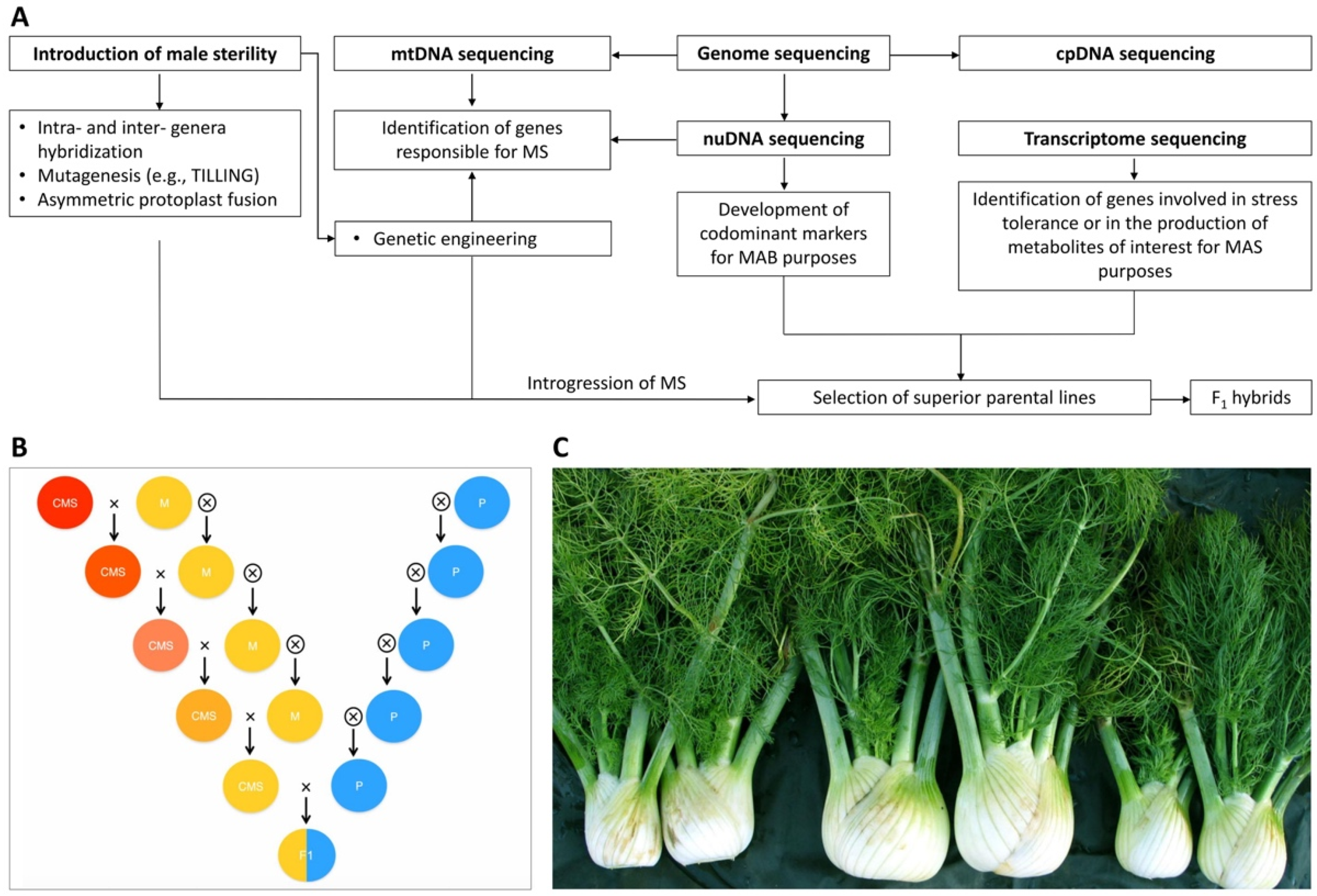
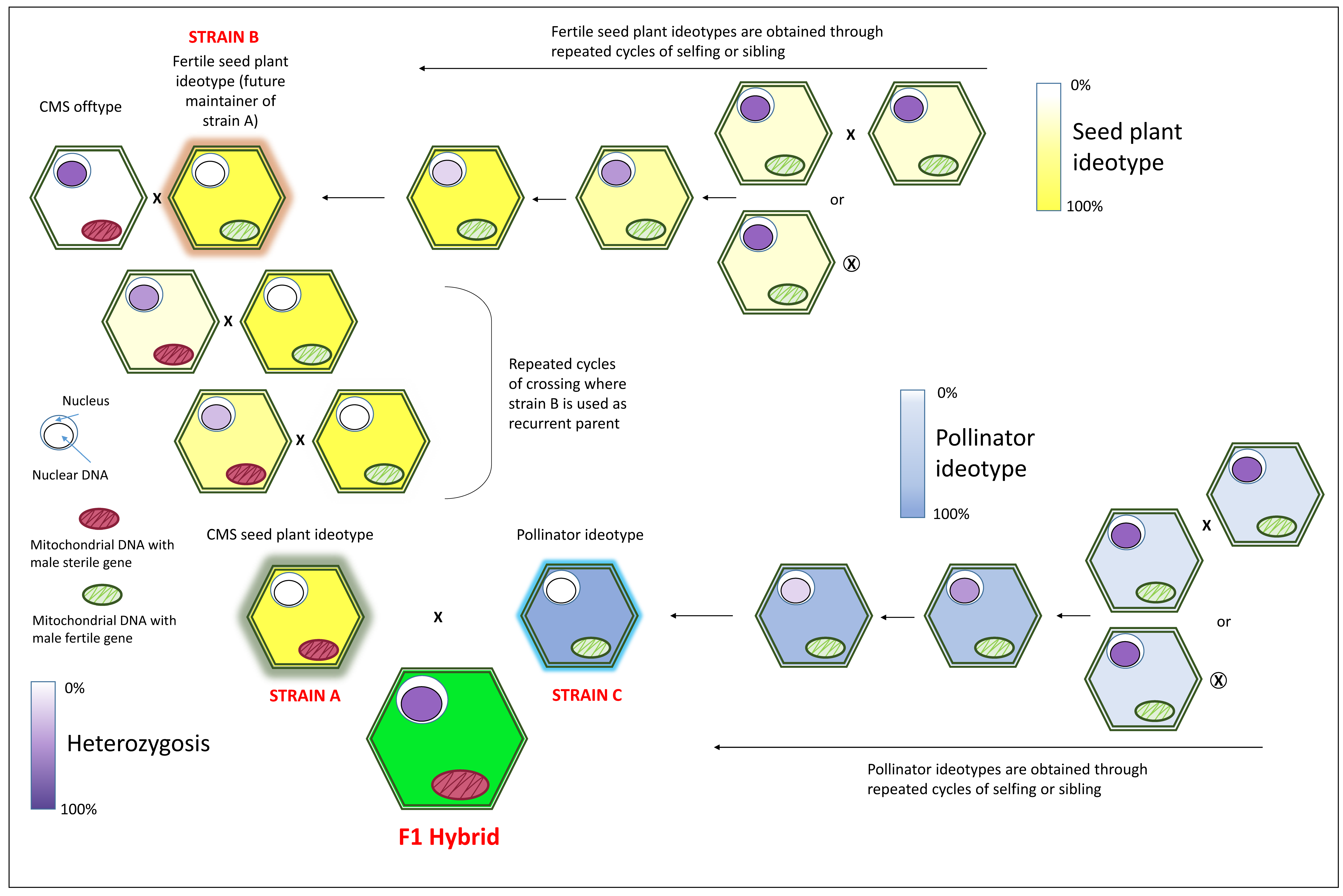
2.2. Using Plant Reproductive Barriers for Varietal Constitution
3. Genomic and Transcriptomic Resources for Breeding Varieties and Phylogenetic Analyses
3.1. Whole Genome Sequencing Provides Powerful Tools for Marker-Assisted Breeding
3.2. RNA-seq Analyses Are Starting Points for the Identification of Genes Responsible for Traits of Agronomic Interest
3.3. From Gene to Genome: cpDNA Is Improving Phylogenetic Analyses through Super-Barcoding
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plunkett, G.M.; Pimenov, M.; Reduron, J.-P.; Kljuykov, E.V.; Van Wyk, B.-E.; Ostroumova, T.A.; Henwood, M.J.; Tilney, P.M.; Spalik, K.; Watson, M.F.; et al. Flowering Plants. Eudicots; Kadereit, J.W., Bittrich, V., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Volume 15, ISBN 9783319936055. [Google Scholar]
- Nicolas, A.N.; Plunkett, G.M. Diversification times and biogeographic patterns in Apiales. Bot. Rev. 2014, 80, 30–58. [Google Scholar] [CrossRef]
- Downie, S.R.; Spalik, K.; Katz-Downie, D.S.; Reduron, J.-P. Major clades within Apiaceae subfamily Apioideae as inferred by phylogenetic analysis of nrDNA ITS sequences. Plant Divers. Evol. 2010, 128, 111–136. [Google Scholar] [CrossRef] [Green Version]
- Baczyński, J.; Miłobȩdzka, A.; Banasiak, Ł. Morphology of Pollen in Apiales (Asterids, Eudicots). Phytotaxa 2021, 478, 1–32. [Google Scholar] [CrossRef]
- Magee, A.R.; Calviño, C.I.; Liu, M.R.; Downie, S.R.; Tilney, P.M.; van Wyk, B.E. New tribal delimitations for the early diverging lineages of Apiaceae subfamily Apioideae. Taxon 2010, 59, 567–580. [Google Scholar] [CrossRef]
- Dueholm, B.; Krieger, C.; Drew, D.; Olry, A.; Kamo, T.; Taboureau, O.; Weitzel, C.; Bourgaud, F.; Hehn, A.; Simonsen, H.T. Evolution of substrate recognition sites (SRSs) in cytochromes P450 from Apiaceae exemplified by the CYP71AJ subfamily. BMC Evol. Biol. 2015, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, R.A.T. Apiaceae (formerly Umbelliferae). In Vegetable Seed Production; George, R.A.T., Ed.; CABI: Wallingford, UK, 2009; pp. 226–250. [Google Scholar]
- Geoffriau, E.; Simon, P.W. Carrots and Related Apiaceae Crops, 2nd ed.; Geoffriau, E., Simon, P.W., Eds.; CABI: Wallingford, UK, 2021. [Google Scholar]
- Davidson, A. The Oxford Companion to Food, 2nd ed.; Davidson, A., Ed.; OUP Oxford: Oxford, UK, 2006. [Google Scholar]
- Anastasopoulou, E.; Graikou, K.; Ganos, C.; Calapai, G.; Chinou, I. Pimpinella anisum seeds essential oil from Lesvos island: Effect of hydrodistillation time, comparison of its aromatic profile with other samples of the Greek market. Safe use. Food Chem. Toxicol. 2020, 135, 110875. [Google Scholar] [CrossRef] [PubMed]
- Morata, A.; Vaquero, C.; Palomero, F.; Loira, I.; Bañuelos, M.A.; Suárez-Lepe, J.A. Technology of Vermouth Wines; Elsevier Inc.: Amsterdam, The Netherlands, 2019; ISBN 9780128152690. [Google Scholar]
- Keshavarz, A.; Minaiyan, M.; Ghannadi, A.; Mahzouni, P. Effects of Carum carvi L. (Caraway) extract and essential oil on TNBS-induced colitis in rats. Res. Pharm. Sci. 2013, 8, 1–8. [Google Scholar]
- Ahmad, B.S.; Talou, T.; Saad, Z.; Hijazi, A.; Ahmad, B.S.; Talou, T.; Saad, Z.; Hijazi, A.; Merah, O.; Apiaceae, T. The Apiaceae: Ethnomedicinal family as source for industrial uses. Ind. Crops Prod. 2017, 109, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Mimica-Dukic, N.; Popovic, M. Apiaceae species: A promising sources of pharmacologically active compounds. Part I: Petrosellinum crispum, Apium graveolens and Pastinaca sativa. In Phytopharmacology and Therapeutic Values III; Singh, V.K., Govil, J.N., Eds.; Studium Press LLC: Houston, TX, USA, 2008; pp. 147–163. [Google Scholar]
- Zengin, G.; Sinan, K.I.; Ak, G.; Mahomoodally, M.F.; Paksoy, M.Y.; Picot-Allain, C.; Glamocilja, J.; Sokovic, M.; Jekő, J.; Cziáky, Z.; et al. Chemical profile, antioxidant, antimicrobial, enzyme inhibitory, and cytotoxicity of seven Apiaceae species from Turkey: A comparative study. Ind. Crops Prod. 2020, 153, 112572. [Google Scholar] [CrossRef]
- Trifan, A.; Bostănaru, A.C.; Luca, S.V.; Grădinaru, A.C.; Jităreanu, A.; Aprotosoaie, A.C.; Miron, A.; Cioancă, O.; Hăncianu, M.; Ochiuz, L.; et al. Antifungal potential of Pimpinella anisum, Carum carvi and Coriandrum sativum extracts. A comparative study with focus on the phenolic composition. Farmacia 2020, 68, 22–27. [Google Scholar] [CrossRef]
- Mottaghipisheh, J.; Vitalini, S.; Pezzani, R.; Iriti, M. A Comprehensive Review on Ethnobotanical, Phytochemical and Pharmacological Aspects of the Genus Dorema; Springer International Publishing: Cham, Switzerland, 2021; ISBN 0123456789. [Google Scholar]
- Sahebkar, A.; Iranshahi, M. Volatile constituents of the genus Ferula (apiaceae): A review. J. Essent. Oil-Bear. Plants 2011, 14, 504–531. [Google Scholar] [CrossRef]
- Wittstock, U.; Lichtnow, K.H.; Teuscher, E. Effects of cicutoxin and related polyacetylenes from Cicuta virosa on neuronal action potentials: A comparative study on the mechanism of the convulsive action. Planta Med. 1997, 63, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Radulović, N.; DorDević, N.; Denić, M.; Pinheiro, M.M.G.; Fernandes, P.D.; Boylan, F. A novel toxic alkaloid from poison hemlock (Conium maculatum L., Apiaceae): Identification, synthesis and antinociceptive activity. Food Chem. Toxicol. 2012, 50, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhang, W.; Su, J. Toxic polyacetylenes in the genus Bupleurum (Apiaceae)—Distribution, toxicity, molecular mechanism and analysis. J. Ethnopharmacol. 2016, 193, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Aćimović, M.G. Nutraceutical Potential of Apiaceae. In Bioactive Molecules in Food, Reference Series in Phytochemistry; Mérillon, J.-M., Ramawat, K.G., Eds.; Springer: New York, NY, USA, 2017; pp. 1–31. ISBN 9783319545288. [Google Scholar]
- Tian, Y.Q.; Zhang, Z.X.; Xu, H.H. Laboratory and field evaluations on insecticidal activity of Cicuta virosa L. var. latisecta Celak. Ind. Crops Prod. 2013, 41, 90–93. [Google Scholar] [CrossRef]
- Madaan, R.; Kumar, S. Screening of alkaloidal fraction of Conium maculatum L. aerial parts for analgesic and antiinflammatory activity. Indian J. Pharm. Sci. 2012, 74, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Simon, P.W.; Iorizzo, M.; Grzebelus, D.; Baranski, R. The Carrot Genome; Simon, P.W., Iorizzo, M., Grzebelus, D., Baranski, R., Eds.; Springer: New York, NY, USA, 2019. [Google Scholar]
- FAO Food and Agriculture Organization of the United Nations: Value of Agricultural Production. Available online: http://www.fao.org/faostat/ (accessed on 30 June 2020).
- Koul, P.; Sharma, N.; Koul, A.K. Pollination biology of Apiaceae. Curr. Sci. 1993, 65, 219–222. [Google Scholar]
- Koul, P.; Koul, A.K.; Hamal, I.A. Reproductive biology of wild and cultivated carrot (Daucus carota L.). New Phytol. 1989, 112, 437–443. [Google Scholar] [CrossRef]
- Gupta, S.K.; Hamal, I.A.; Koul, A.K. Reproductive biology of Coriandrum sativum L. Plant Sci. Res. 1986, 2, 81–95. [Google Scholar]
- Doust, J. Floral sex ratios in andromonoecious Umbelliferae. New Phytol. 1980, 85, 265–273. [Google Scholar] [CrossRef]
- Reuther, K.; Claßen-Bockhoff, R. Andromonoecy and developmental plasticity in Chaerophyllum bulbosum (Apiaceae-Apioideae). Ann. Bot. 2013, 112, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, A.; van den Berg, B.M.; Burg, H.C.J.; Görg, A. Genetic variability of carrot seed proteins analyzed by one- and two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 1995, 16, 1312–1316. [Google Scholar] [CrossRef]
- Yadav, S.K. Creation of Genetic Variation through Mutagenesis in Cumin (Cuminum cyminum L.); Rajasthan Agricultural University: Rajasthan, India, 2006. [Google Scholar]
- Shojaiefar, S.; Sabzalian, M.R.; Mirlohi, A.; Tajdivand, A. Evidence for self-compatibility and variation for inbreeding depression within breeding populations of fennel (Foeniculum vulgare Mill.). J. Appl. Res. Med. Aromat. Plants 2021, 22, 100299. [Google Scholar] [CrossRef]
- Chaudhary, P.; Ramkrishna, K. An analysis of polygenic variation in the M4 families of coriander (Coriandrum sativum L.). Indian J. Genet. Plant Breed. 2003, 63, 181–182. [Google Scholar]
- Gaudeul, M.; Till-bottraud, I. Low selfing in a mass-flowering, endangered perennial, Eryngium alpinum L. (Apiaceae). Am. J. Bot. 2003, 90, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Molano-Flores, B. Breeding systems of plants used for prairie restorations: A review. Trans. Ill. State Acad. Sci. 2004, 97, 95–102. [Google Scholar]
- Cruden, R.W. Temporal dioecism: Systematic breadth, associated traits, and temporal patterns. Bot. Gaz. 1988, 1, 1–15. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.-G. Male Sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Linke, B.; Alessandro, M.S.; Galmarini, C.R.; Nothnagel, T. Carrot floral development and reproductive biology. In The Carrot Genome, Compendium of Plant Genomes; Simon, P., Iorizzo, M., Dariusz, G., Baranski, R., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 27–57. ISBN 9783030033897. [Google Scholar]
- Palumbo, F.; Vitulo, N.; Vannozzi, A.; Magon, G.; Barcaccia, G. The mitochondrial genome assembly of fennel (Foeniculum vulgare) reveals two different ATP6 gene sequences in cytoplasmic male sterile accessions. Int. J. Mol. Sci. 2020, 21, 4664. [Google Scholar] [CrossRef]
- Li, M.Y.; Hou, X.L.; Wang, F.; Tan, G.F.; Xu, Z.S.; Xiong, A.S. Advances in the research of celery, an important Apiaceae vegetable crop. Crit. Rev. Biotechnol. 2018, 38, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Kalloo, G. Parsnip—Pastinaca sativa L. In Genetic Improvement of Vegetable Crops; Kalloo, G., Bergh, B., Eds.; Pergamon Press: Oxford, UK, 1993; pp. 485–486. [Google Scholar]
- Bruznican, S.; De Clercq, H.; Eeckhaut, T.; Van Huylenbroeck, J.; Geelen, D. Celery and celeriac: A critical view on present and future breeding. Front. Plant Sci. 2020, 10, 1699. [Google Scholar] [CrossRef]
- Pank, F.; Quilitzsch, R.; Krüger, H. Male sterility in selected fennel populations (Foeniculum vulgare Mill.) and the influence of the fertility types on economically important traits. J. Med. Spice Plants 2007, 12, 88–94. [Google Scholar]
- Palumbo, F.; Galla, G.; Vitulo, N.; Barcaccia, G. First draft genome sequencing of fennel (Foeniculum vulgare Mill.): Identification of simple sequence repeats and their application in marker-assisted breeding. Mol. Breed. 2018, 38, 122. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Eric Schranz, M.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualberto, J.M.; Newton, K.J. Plant mitochondrial genomes: Dynamics and mechanisms of mutation. Annu. Rev. Plant Biol. 2017, 68, 225–252. [Google Scholar] [CrossRef]
- Smýkalová, I.; Šmirous, P.; Kubošiová, M.; Gasmanová, N.; Griga, M. Doubled haploid production via anther culture in annual, winter type of caraway (Carum carvi L.). Acta Physiol. Plant. 2009, 31, 21–31. [Google Scholar] [CrossRef]
- Ferrie, A.M.R.; Bethune, T.D.; Mykytyshyn, M. Microspore embryogenesis in Apiaceae. Plant Cell Tissue Organ Cult. 2011, 104, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Kalia, P. Root vegetable crops. J. New Seeds 2004, 6, 247–275. [Google Scholar] [CrossRef]
- Dhall, R.K. Status of male sterility in vegetables for hybrid development. A Review. Adv. Hortic. Sci. 2010, 24, 263–279. [Google Scholar] [CrossRef]
- Kumar, S.; Banerjee, M.K.; Kalloo, G. Male sterility: Mechanisms and current status on identification, characterization and utilization in vegetables. Veg. Sci. 2000, 27, 1–24. [Google Scholar]
- Honma, S.; Lacy, M.L. Hybridization between pascal celery and parsley. Euphytica 1980, 29, 801–805. [Google Scholar] [CrossRef]
- Magnussen, L.S.; Hauser, T.P. Hybrids between cultivated and wild carrots in natural populations in Denmark. Heredity 2007, 99, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camadro, E.L.; Cauhépé, M.A.; Simon, P.W. Compatibility relations between the edible carrot Daucus carota and D. pusillus, a related wild species from the Argentinian Pampas. Euphytica 2008, 159, 103–109. [Google Scholar] [CrossRef]
- Kalidasu, G.; Sarada, C.; Reddy, P.V.; Reddy, T.Y. Use of male gametocide: An alternative to cumbersome emasculation in coriander (Coriandrum sativum L.). J. Hortic. For. 2009, 1, 126–132. [Google Scholar]
- Bruznican, S.; Eeckhaut, T.; Van Huylenbroeck, J.; De Keyser, E.; De Clercq, H.; Geelen, D. An asymmetric protoplast fusion and screening method for generating celeriac cybrids. Sci. Rep. 2021, 11, 4553. [Google Scholar] [CrossRef]
- Bruznican, S.; Eeckhaut, T.; Van Huylenbroeck, J.; De Clercq, H.; Geelen, D. Regeneration of cell suspension derived Apium graveolens L. protoplasts. Plant Cell Tissue Organ Cult. 2017, 131, 163–174. [Google Scholar] [CrossRef]
- Grzebelus, E.; Maćkowska, K.; Macko-Podgórni, A.; Kiełkowska, A.; Szklarczyk, M.; Baranski, R.; Grzebelus, D. Application of protoplast technology to Apiaceae species. Acta Hortic. 2019, 1264, 67–74. [Google Scholar] [CrossRef]
- von Maydell, D.; Brandes, J.; Lehnert, H.; Junghanns, W.; Marthe, F. Breeding synthetic varieties in annual caraway: Observations on the outcrossing rate in a polycross using a high-throughput genotyping system. Euphytica 2021, 217, 1. [Google Scholar] [CrossRef]
- Kalloo, G. Parsley and turnip-rooted parsley Petrosellinum crispum Mill. Nym., P. crispum var. tuberosum. In Genetic Improvement of Vegetable Crops; Kalloo, G., Bergh, B.O., Eds.; Pergamon Press: Oxford, UK, 1993; pp. 573–577. ISBN 978-0-08-040826-2. [Google Scholar]
- Patella, A.; Palumbo, F.; Galla, G.; Barcaccia, G. The molecular determination of hybridity and homozygosity estimates in breeding populations of lettuce (Lactuca sativa L.). Genes 2019, 10, 916. [Google Scholar] [CrossRef] [Green Version]
- Patella, A.; Palumbo, F.; Ravi, S.; Stevanato, P.; Barcaccia, G. Genotyping by RAD sequencing analysis assessed the genetic distinctiveness of experimental lines and narrowed down the genomic region responsible for leaf shape in endive (Cichorium endivia L.). Genes 2020, 11, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwosisi, S.; Dhakal, K.; Nandwani, D.; Raji, J.I.; Krishnan, S.; Beovides-García, Y. Genetic diversity in vegetable and fruit crops. In Genetic Diversity in Horticultural Plants; Nandwani, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 87–125. ISBN 978-3-319-96454-6. [Google Scholar]
- Jaiswal, S.; Sheoran, S.; Arora, V.; Angadi, U.B.; Iquebal, M.A.; Raghav, N.; Aneja, B.; Kumar, D.; Singh, R.; Sharma, P.; et al. Putative microsatellite DNA marker-based wheat genomic resource for varietal improvement and management. Front. Plant Sci. 2017, 8, 2009. [Google Scholar] [CrossRef] [Green Version]
- Bostan, H.; Senalik, D.; Simon, P.W.; Iorizzo, M. Carrot genetics, omics and breeding toolboxes. In The Carrot Genome, Compendium of Plant Genomes; Simon, P., Iorizzo, M., Grzebelus, D., Baranski, R., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 225–245. ISBN 9783030033897. [Google Scholar]
- Mezghani, N.; Ruess, H.; Tarchoun, N.; Ben Amor, J.; Simon, P.W.; Spooner, D.M. Genotyping-by-sequencing reveals the origin of the Tunisian relatives of cultivated carrot (Daucus carota). Genet. Resour. Crop Evol. 2018, 65, 1359–1368. [Google Scholar] [CrossRef]
- Arbizu, C.I.; Ellison, S.L.; Senalik, D.; Simon, P.W.; Spooner, D.M. Genotyping-by-sequencing provides the discriminating power to investigate the subspecies of Daucus carota (Apiaceae). BMC Evol. Biol. 2016, 16, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellison, S.L.; Luby, C.H.; Corak, K.E.; Coe, K.M.; Senalik, D.; Iorizzo, M.; Goldman, I.L.; Simon, P.W.; Dawson, J.C. Carotenoid presence is associated with the or gene in domesticated carrot. Genetics 2018, 210, 1497–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, A.J.; Moore, C.L.; Schuette, S.; Martine, C.T. Population genomics and conservation of Erigenia bulbosa (Apiaceae), an edge-of-range species in Pennsylvania. Int. J. Plant Sci. 2021, 182, 344–355. [Google Scholar] [CrossRef]
- Piwczyński, M.; Trzeciak, P.; Popa, M.O.; Pabijan, M.; Corral, J.M.; Spalik, K.; Grzywacz, A. Using RAD seq for reconstructing phylogenies of highly diverged taxa: A test using the tribe Scandiceae (Apiaceae). J. Syst. Evol. 2021, 59, 58–72. [Google Scholar] [CrossRef]
- Von Maydell, D.; Lehnert, H.; Berner, T.; Klocke, E.; Junghanns, W.; Keilwagen, J.; Marthe, F. On genetic diversity in caraway: Genotyping of a large germplasm collection. PLoS ONE 2020, 15, e0244666. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Senalik, D.A.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.F.; Matvienko, M.; Ashrafi, H.; Van Deynze, A.; Simon, P.W. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genom. 2011, 12, 389. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, F.; Vannozzi, A.; Vitulo, N.; Lucchin, M.; Barcaccia, G. The leaf transcriptome of fennel (Foeniculum vulgare Mill.) enables characterization of the t-anethole pathway and the discovery of microsatellites and single-nucleotide variants. Sci. Rep. 2018, 8, 10459. [Google Scholar] [CrossRef]
- Iorizzo, M.; Ellison, S.; Senalik, D.; Zeng, P.; Satapoomin, P.; Huang, J.; Bowman, M.; Iovene, M.; Sanseverino, W.; Cavagnaro, P.; et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 2016, 48, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Cavagnaro, P.F.; Chung, S.M.; Szklarczyk, M.; Grzebelus, D.; Senalik, D.; Atkins, A.E.; Simon, P.W. Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol. Genet. Genom. 2009, 281, 273–288. [Google Scholar] [CrossRef]
- Aiello, D.; Ferradini, N.; Torelli, L.; Volpi, C.; Lambalk, J.; Russi, L.; Albertini, E. Evaluation of cross-species transferability of SSR markers in Foeniculum vulgare. Plants 2020, 9, 175. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Asamadi, M.H.; Fougat, R.S.; Sakure, A.A.; Mistry, J.G. Transferability of carrot (Daucus carota) microsatellite markers to cumin (Cuminum cyminum). Int. J. Seed Spices 2014, 4, 88–90. [Google Scholar]
- Cholin, S.S.; Poleshi, C.A.; Manikanta, D.S.; Christopher, C. Exploring the genomic resources of carrot for cross-genera transferability and phylogenetic assessment among orphan spices and vegetables of Apiaceae family. Hortic. Environ. Biotechnol. 2019, 60, 81–93. [Google Scholar] [CrossRef]
- Cavagnaro, P.F.; Chung, S.M.; Manin, S.; Yildiz, M.; Ali, A.; Alessandro, M.S.; Iorizzo, M.; Senalik, D.A.; Simon, P.W. Microsatellite isolation and marker development in carrot—Genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genom. 2011, 12, 386. [Google Scholar] [CrossRef] [Green Version]
- Morillo, E.; Second, G.; Pham, J.L.; Risterucci, A.M. Development of DNA microsatellite markers in the Andean root crop arracacha: Arracacia xanthorrhiza Banc. (Apiaceae). Mol. Ecol. Notes 2004, 4, 680–682. [Google Scholar] [CrossRef]
- Sui, C.; Wei, J.H.; Chen, S.L.; Chen, H.Q.; Yang, C.M. Development of genomic SSR and potential EST-SSR markers in Bupleurum chinense DC. Afr. J. Biotechnol. 2009, 8, 6233–6240. [Google Scholar] [CrossRef]
- Daemi-Saeidabad, M.; Shojaeiyan, A.; Vivian-Smith, A.; Stenøien, H.K.; Falahati-Anbaran, M. The taxonomic significance of ddRADseq based microsatellite markers in the closely related species of Heracleum (Apiaceae). PLoS ONE 2020, 15, e0232471. [Google Scholar] [CrossRef] [PubMed]
- Sahu, J.; Das Talukdar, A.; Devi, K.; Choudhury, M.D.; Barooah, M.; Modi, M.K.; Sen, P. E-microsatellite markers for Centella asiatica (gotu kola) genome: Validation and cross-transferability in Apiaceae family for plant omics research and development. Omics J. Integr. Biol. 2015, 19, 52–65. [Google Scholar] [CrossRef]
- Jia, Y.; Bai, J.Q.; Liu, M.L.; Jiang, Z.F.; Wu, Y.; Fang, M.F.; Li, Z.H. Transcriptome analysis of the endangered Notopterygium incisum: Cold-tolerance gene discovery and identification of EST-SSR and SNP markers. Plant Divers. 2019, 41, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, F.; Tan, H.W.; Li, M.Y.; Xu, Z.S.; Tan, G.F.; Xiong, A.S. De novo transcriptome assembly, gene annotation, marker development, and miRNA potential target genes validation under abiotic stresses in Oenanthe javanica. Mol. Genet. Genom. 2015, 290, 671–683. [Google Scholar] [CrossRef]
- Tulsani, N.J.; Hamid, R.; Jacob, F.; Umretiya, N.G.; Nandha, A.K.; Tomar, R.S.; Golakiya, B.A. Transcriptome landscaping for gene mining and SSR marker development in Coriander (Coriandrum sativum L.). Genomics 2020, 112, 1545–1553. [Google Scholar] [CrossRef]
- Chen, C.; Chen, Y.; Huang, W.; Jiang, Y.; Zhang, H.; Wu, W. Mining of simple sequence repeats (SSRs) loci and development of novel transferability-across EST-SSR markers from de novo transcriptome assembly of Angelica dahurica. PLoS ONE 2019, 14, e0221040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Hu, X.; Wang, X.; Zhang, J.; Peng, X.; Hu, Z.; Liu, Y. Constructing a core collection of the medicinal plant Angelica biserrata using genetic and metabolic data. Front. Plant Sci. 2020, 11, 600249. [Google Scholar] [CrossRef]
- Fu, N.; Wang, Q.; Shen, H.L. De novo assembly, gene annotation and marker development using Illumina paired-end transcriptome sequences in celery (Apium graveolens L.). PLoS ONE 2013, 8, e57686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Zhao, Z.; Shi, Y.Y.; Yang, J.; Jia, Y.; Gong, L.L.; Li, Z.H. Novel microsatellite markers for Notopterygium oviforme (Apiaceae), an endangered herb endemic to China. Ann. Bot. Fenn. 2017, 54, 423–427. [Google Scholar] [CrossRef]
- Baldemir, A.; Topçu, H.; Paksoy, M.Y.; Motalebipour, E.Z.; Kafkas, S. First microsatellite markers for Scaligeria lazica Boiss. (Apiaceae) by next-generation sequencing: Population structure and genetic diversity analysis. Biotechnol. Biotechnol. Equip. 2017, 31, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Gandham, P.; Palve, A.; Rathore, A. Survey sequencing and in-silico development and validation of genomic SSR markers in Indian dill seed. J. King Saud Univ. Sci. 2020, 32, 862–866. [Google Scholar] [CrossRef]
- Bharti, R.; Kumar, S.; Parekh, M.J. Development of genomic simple sequence repeat (gSSR) markers in cumin and their application in diversity analyses and cross-transferability. Ind. Crops Prod. 2018, 111, 158–164. [Google Scholar] [CrossRef]
- Gil, J.; Um, Y.; Kim, S.; Kim, O.T.; Koo, S.C.; Reddy, C.S.; Kim, S.C.; Hong, C.P.; Park, S.G.; Kim, H.B.; et al. Development of genome-wide SSR markers from Angelica gigas nakai using next generation sequencing. Genes 2017, 8, 238. [Google Scholar] [CrossRef]
- Zhu, C.R.; Xu, J.; Du, M.L.; Wang, L.H.; Sui, C.; Wei, J.H. Genome survey analysis and SSR loci mining of Bupleurum falcatum. Zhongguo Zhong Yao Za Zhi 2019, 44, 3960–3966. [Google Scholar] [CrossRef]
- Song, X.; Sun, P.; Yuan, J.; Gong, K.; Li, N.; Meng, F.; Zhang, Z.; Li, X.; Hu, J.; Wang, J.; et al. The celery genome sequence reveals sequential paleo-polyploidizations, karyotype evolution and resistance gene reduction in apiales. Plant Biotechnol. J. 2021, 19, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Pootakham, W.; Naktang, C.; Kongkachana, W.; Sonthirod, C.; Yoocha, T.; Sangsrakru, D.; Jomchai, N.; U-thoomporn, S.; Romyanon, K.; Toojinda, T.; et al. De novo chromosome-level assembly of the Centella asiatica genome. Genomics 2021, 113, 2221–2228. [Google Scholar] [CrossRef]
- Song, X.; Wang, J.; Li, N.; Yu, J.; Meng, F.; Wei, C.; Liu, C.; Chen, W.; Nie, F.; Zhang, Z.; et al. Deciphering the high-quality genome sequence of coriander that causes controversial feelings. Plant Biotechnol. J. 2020, 18, 1444–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Xiong, A. Oenanthe Javanica Genome. Available online: https://www.ncbi.nlm.nih.gov/genome/?term=oenanthe+javanica (accessed on 15 May 2021).
- Xu, Z.S.; Tan, H.W.; Wang, F.; Hou, X.L.; Xiong, A.S. CarrotDB: A genomic and transcriptomic database for carrot. Database 2014, 2014, bau096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Nie, F.; Chen, W.; Ma, X.; Gong, K.; Yang, Q.; Wang, J.; Li, N.; Sun, P.; Pei, Q.; et al. Coriander Genomics Database: A genomic, transcriptomic, and metabolic database for coriander. Hortic. Res. 2020, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.; Hou, X.L.; Li, M.Y.; Jiang, Q.; Xu, Z.S.; Liu, J.X.; Xiong, A.S. CeleryDB: A genomic database for celery. Database 2018, 2018, bay070. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Carvalho, A.B.; Dupim, E.G.; Goldstein, G. Improved assembly of noisy long reads by k-mer validation. Genome Res. 2016, 26, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Avvaru, A.K.; Sowpati, D.T.; Mishra, R.K. Patterns of microsatellite distribution across eukaryotic genomes. BMC Genom. 2019, 20, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Kodama, M.; Brinch-Pedersen, H.; Sharma, S.; Holme, I.B.; Joernsgaard, B.; Dzhanfezova, T.; Amby, D.B.; Vieira, F.G.; Liu, S.; Gilbert, M.T.P. Identification of transcription factor genes involved in anthocyanin biosynthesis in carrot (Daucus carota L.) using RNA-Seq. BMC Genom. 2018, 19, 811. [Google Scholar] [CrossRef]
- Meng, G.; Clausen, S.K.; Rasmussen, S.K. Transcriptome analysis reveals candidate genes related to anthocyanin biosynthesis in different carrot genotypes and tissues. Plants 2020, 9, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, B.; Koul, A.; Sharma, D.; Manzoor, M.M.; Kaul, S.; Gupta, S.; Dhar, M.K. Identification and expression profiling of miRNAs in two color variants of carrot (Daucus carota L.) using deep sequencing. PLoS ONE 2019, 14, e0212746. [Google Scholar] [CrossRef]
- Ma, J.; Li, J.; Xu, Z.; Wang, F.; Xiong, A. Transcriptome profiling of genes involving in carotenoid biosynthesis and accumulation between leaf and root of carrot (Daucus carota L.). Acta Biochim. Biophys. Sin. 2018, 50, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Bannoud, F.; Ellison, S.; Paolinelli, M.; Horejsi, T.; Senalik, D.; Fanzone, M.; Iorizzo, M.; Simon, P.W.; Cavagnaro, P.F. Dissecting the genetic control of root and leaf tissue-specific anthocyanin pigmentation in carrot (Daucus carota L.). Theor. Appl. Genet. 2019, 132, 2485–2507. [Google Scholar] [CrossRef] [PubMed]
- Sobhani Najafabadi, A.; Naghavi, M.R. Mining Ferula gummosa transcriptome to identify miRNAs involved in the regulation and biosynthesis of terpenes. Gene 2018, 645, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Sobhani Najafabadi, A.; Naghavi, M.R.; Farahmand, H.; Abbasi, A. Transcriptome and metabolome analysis of Ferula gummosa Boiss. to reveal major biosynthetic pathways of galbanum compounds. Funct. Integr. Genom. 2017, 17, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Amini, H.; Naghavi, M.R.; Shen, T.; Wang, Y.; Nasiri, J.; Khan, I.A.; Fiehn, O.; Zerbe, P.; Maloof, J.N. Tissue-specific transcriptome analysis reveals candidate genes for terpenoid and phenylpropanoid metabolism in the medicinal plant Ferula assafoetida. G3 Genes Genomes Genet. 2019, 9, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Soltani Howyzeh, M.; Sadat Noori, S.A.; Shariati, V.; Amiripour, M. Comparative transcriptome analysis to identify putative genes involved in thymol biosynthesis pathway in medicinal plant Trachyspermum ammi L. Sci. Rep. 2018, 8, 13405. [Google Scholar] [CrossRef]
- Amiripour, M.; Sadat Noori, S.A.; Shariati, V.; Soltani Howyzeh, M. Transcriptome analysis of Ajowan (Trachyspermum ammi L.) inflorescence. J. Plant Biochem. Biotechnol. 2019, 28, 496–508. [Google Scholar] [CrossRef]
- Munakata, R.; Olry, A.; Karamat, F.; Courdavault, V.; Sugiyama, A.; Date, Y.; Krieger, C.; Silie, P.; Foureau, E.; Papon, N.; et al. Molecular evolution of parsnip (Pastinaca sativa) membrane-bound prenyltransferases for linear and/or angular furanocoumarin biosynthesis. New Phytol. 2016, 211, 332–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, S.; Shan, C.; Shi, Y.; Wu, H.; Wu, J.; Peng, D. De novo transcriptome assembly of Angelica dahurica and characterization of coumarin biosynthesis pathway genes. Gene 2021, 791, 145713. [Google Scholar] [CrossRef] [PubMed]
- Sui, C.; Zhang, J.; Wei, J.; Chen, S.; Li, Y.; Xu, J.; Jin, Y.; Xie, C.; Gao, Z.; Chen, H.; et al. Transcriptome analysis of Bupleurum chinense focusing on genes involved in the biosynthesis of saikosaponins. BMC Genom. 2011, 12, 539. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.T.; Jin, M.L.; Lee, D.Y.; Jetter, R. Characterization of the asiatic acid glucosyltransferase, UGT73AH1, involved in asiaticoside biosynthesis in Centella asiatica (L.) Urban. Int. J. Mol. Sci. 2017, 18, 2630. [Google Scholar] [CrossRef] [Green Version]
- Sangwan, R.S.; Tripathi, S.; Singh, J.; Narnoliya, L.K.; Sangwan, N.S. De novo sequencing and assembly of Centella asiatica leaf transcriptome for mapping of structural, functional and regulatory genes with special reference to secondary metabolism. Gene 2013, 525, 58–76. [Google Scholar] [CrossRef] [PubMed]
- Galata, M.; Sarker, L.S.; Mahmoud, S.S. Transcriptome profiling, and cloning and characterization of the main monoterpene synthases of Coriandrum sativum L. Phytochemistry 2014, 102, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Feng, S.Y.; Yang, Q.H.; Bhetariya, P.J.; Gong, K.; Cui, C.L.; Song, J.; Ping, X.R.; Pei, Q.Y.; Yu, T.; et al. Integration of the metabolome and transcriptome reveals the metabolites and genes related to nutritional and medicinal value in Coriandrum sativum. J. Integr. Agric. 2021, 20, 1807–1818. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Jia, Q.; Zhao, C.; Taylor, D.C.; Li, D.; Zhang, M. Transcriptome analysis reveals candidate genes for petroselinic acid biosynthesis in fruits of Coriandrum sativum L. J. Agric. Food Chem. 2020, 68, 5507–5520. [Google Scholar] [CrossRef]
- Yu, M.; Chen, H.; Liu, Q.; Huang, J.; Semagn, K.; Liu, D.; Li, Y.; Yang, B.; He, Y.; Sui, C.; et al. Analysis of unigenes involved in lateral root development in Bupleurum chinense and B. scorzonerifolium. Planta 2021, 253, 128. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, G.; Gao, Z.; Sui, C.; Ji, H.; Jiang, J.; Xinwei, G.; Wei, J. Transcriptome profiling of Bupleurum chinense DC. root provides new insights into the continuous inflorescence removal induced improvements to root growth and saikosaponin biosynthesis. Ind. Crops Prod. 2021, 160, 113085. [Google Scholar] [CrossRef]
- Pei, Q.; Li, N.; Yang, Q.; Wu, T.; Feng, S.; Feng, X.; Jing, Z.; Zhou, R.; Gong, K.; Yu, T.; et al. Genome-wide identification and comparative analysis of ARF family genes in three Apiaceae species. Front. Genet. 2021, 11, 590535. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Tan, H.W.; Wang, F.; Jiang, Q.; Xu, Z.S.; Tian, C.; Xiong, A.S. De Novo transcriptome sequence assembly and identification of AP2/ERF transcription factor related to abiotic stress in parsley (Petroselinum crispum). PLoS ONE 2014, 9, e108977. [Google Scholar] [CrossRef] [Green Version]
- Li, M.Y.; Wang, F.; Jiang, Q.; Ma, J.; Xiong, A.S. Identification of SSRs and differentially expressed genes in two cultivars of celery (Apium graveolens L.) by deep transcriptome sequencing. Hortic. Res. 2014, 1, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, B.; Zhao, C.R.; Sun, J.; Lai, Q.; Yu, C. Comparative de novo transcriptomics and untargeted metabolomic analyses elucidate complicated mechanisms regulating celery (Apium graveolens L.) responses to selenium stimuli. PLoS ONE 2019, 14, e0226752. [Google Scholar] [CrossRef] [Green Version]
- Pei, Q.; Yu, T.; Wu, T.; Yang, Q.; Gong, K.; Zhou, R.; Cui, C.; Yu, Y.; Zhao, W.; Kang, X.; et al. Comprehensive identification and analyses of the Hsf gene family in the whole-genome of three Apiaceae species. Hortic. Plant J. 2021, 7, 457–468. [Google Scholar] [CrossRef]
- Choudhary, S.; Naika, M.B.N.; Meena, R.D. Identification and expression analysis of candidate genes associated with stem gall disease in Coriander (Coriandrum sativum L.) cultivars. Mol. Biol. Rep. 2020, 47, 5403–5409. [Google Scholar] [CrossRef]
- Choudhary, S.; Naika, M.B.N.; Sharma, R.; Meena, R.D.; Singh, R.; Lal, G. Transcriptome profiling of coriander: A dual purpose crop unravels stem gall resistance genes. J. Genet. 2019, 98, 19. [Google Scholar] [CrossRef]
- D’Antonio, V.; Falk, B.; Quiros, C.F. Inheritance of resistance to Celery Mosaic Virus in celery. Plant Dis. 2001, 85, 1276–1277. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.J.; Pico, B.; Li, G.; D’Antanio, V.; Falk, B.; Quiros, C.F. Identification of markers linked to a celery mosaic virus resistance gene in celery. J. Am. Soc. Hortic. Sci. 2001, 126, 432–435. [Google Scholar] [CrossRef]
- Li, M.Y.; Liu, J.X.; Hao, J.N.; Feng, K.; Duan, A.Q.; Yang, Q.Q.; Xu, Z.S.; Xiong, A.S. Genomic identification of AP2/ERF transcription factors and functional characterization of two cold resistance-related AP2/ERF genes in celery (Apium graveolens L.). Planta 2019, 250, 1265–1280. [Google Scholar] [CrossRef]
- Zhifang, G.; Loescher, W.H. Expression of a celery mannose 6-phosphate reductase in Arabidopsis thaliana enhances salt tolerance and induces biosynthesis of both mannitol and a glucosyl-mannitol dimer. Plant Cell Environ. 2003, 26, 275–283. [Google Scholar] [CrossRef]
- Noiraud, N.; Delrot, S.; Lemoine, R. The sucrose transporter of celery. Identification and expression during salt stress. Plant Physiol. 2000, 122, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Jennings, D.B.; Daub, M.E.; Pharr, D.M.; Williamson, J.D. Constitutive expression of a celery mannitol dehydrogenase in tobacco enhances resistance to the mannitol-secreting fungal pathogen Alternaria alternata. Plant J. 2002, 32, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Toit, L.J.; Le Clerc, V.; Briard, M. Genetics and genomics of carrot biotic stress. In The Carrot Genome, Compendium of Plant Genomes; Simon, P., Iorizzo, M., Grzebelus, D., Baranski, R., Eds.; Springer: Cham, Switzerland, 2019; pp. 317–362. ISBN 978-3-030-03389-7. [Google Scholar]
- Grzebelus, D. Genetics and genomics of carrot abiotic stress. In The Carrot Genome, Compendium of Plant Genomes; Simon, P., Iorizzo, M., Grzebelus, D., Baranski, R., Eds.; Springer: Cham, Switzerland, 2019; pp. 363–372. ISBN 978-3-030-03389-7. [Google Scholar]
- Baranski, R.; Lukasiewicz, A. Genetic engineering of carrot. In The Carrot Genome, Compendium of Plant Genomes; Simon, P., Iorizzo, M., Grzebelus, D., Baranski, R., Eds.; Springer: Cham, Switzerland, 2019; pp. 149–186. ISBN 9783030033897. [Google Scholar]
- CBOL Plant Working Group A DNA barcode for land plants. PNAS 2009, 106, 12794–12797. [CrossRef] [PubMed] [Green Version]
- Liu, J.; Shi, L.; Han, J.; Li, G.; Lu, H.; Hou, J.; Zhou, X.; Meng, F.; Downie, S.R. Identification of species in the angiosperm family Apiaceae using DNA barcodes. Mol. Ecol. Resour. 2014, 14, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Hansenia, S.; Gou, W.; Jia, S.; Price, M.; Guo, X.; Zhou, S.; He, X. Complete plastid genome sequencing of eight species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative analyses and phylogenetic implications. Plants 2020, 9, 1523. [Google Scholar]
- Wen, J.; Xie, D.; Price, M.; Ren, T.; Deng, Y.; Gui, L.; Guo, X.; He, X. Backbone phylogeny and evolution of Apioideae (Apiaceae): New insights from phylogenomic analyses of plastome data. Mol. Phylogenet. Evol. 2021, 161, 107183. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gao, Y.Z.; Wei, J.; Liu, Z.W.; Downie, S.R. Molecular phylogenetics of Ligusticum (Apiaceae) based on nrDNA ITS sequences: Rampant polyphyly, placement of the chinese endemic species, and a much-reduced circumscription of the genus. Int. J. Plant Sci. 2020, 181, 306–323. [Google Scholar] [CrossRef]
- Bao, Z.; Zhu, Z.; Zhang, H.; Zhong, Y.; Wang, W.; Zhang, J.; Wu, J. The complete chloroplast genome of Saposhnikovia divaricata. Mitochondrial DNA Part B Resour. 2020, 5, 360–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbagarova, B.M.; Shults, E.E.; Taraskin, V.V.; Radnaeva, L.D.; Petrova, T.N.; Rybalova, T.V.; Frolova, T.S.; Pokrovskii, A.G.; Ganbaatar, J. Chromones and coumarins from Saposhnikovia divaricata (Turcz.) Schischk. Growing in Buryatia and Mongolia and their cytotoxicity. J. Ethnopharmacol. 2020, 261, 112517. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Use | Examples | References |
|---|---|---|
| Food (root) | Daucus carota, Pastinaca sativa, Apium graveolens (var. rapaceum), Arracacia xanthorrhiza, Bunium bulbocastanum, Cryptotaenia canadensis, Sium sisarum | [1,7,8] |
| Food (leaf) | Apium graveolens, Foeniculum vulgare, Pimpinella anisum, Coriandrum sativum, Petroselinum crispum, Angelica spp., Anthriscus cerefolium, Levisticum officinale, Smyrnium olusatrum, Centella asiatica, Oenanthe javanica | [1,7,8,9] |
| Food (seed) | Apium graveolens, Pimpinella anisum, Anethum graveolens and Foeniculum vulgare, Coriandrum sativum, Cuminum cyminum, Smyrnium olusatrum | [1,7,8] |
| Beverages | Pimpinella anisum (for anisette, ouzo, and raki), Carum carvi (for akvavit and Kümmel), Angelica archangelica (for vermouth and Chartreuse) | [8,10,11,12] |
| Pharmacological use and folk remedies | Foeniculum vulgare, Petroselinum crispum, Pimpinella anisum, Carum carvi, Visnaga daucoides, Centella asiatica, Cuminum cyminum, Pastinaca sativa, Apium graveolens | [1,13,14] |
| Antibacterial and antifungal properties | Cuminum cyminum, Carum carvi, Foeniculum vulgare, Coriandrum sativum, Pimpinella anisum, Anethum graveolens, Petroselinum crispum, Ferula spp. | [13,15,16] |
| Ornamental plants | Eryngium spp., Heracleum spp., Angelica spp., Astrantia spp., Bupleurum spp. | [1] |
| Fragrance and cosmetics | Coriandrum sativum, Anethum graveolens | [13] |
| Gum and resins | Ferula spp., Dorema ammoniacum | [17,18] |
| Toxic | Cicuta spp., Conium maculatum, Bupleurum spp., Aethusa cynapium | [19,20,21] |
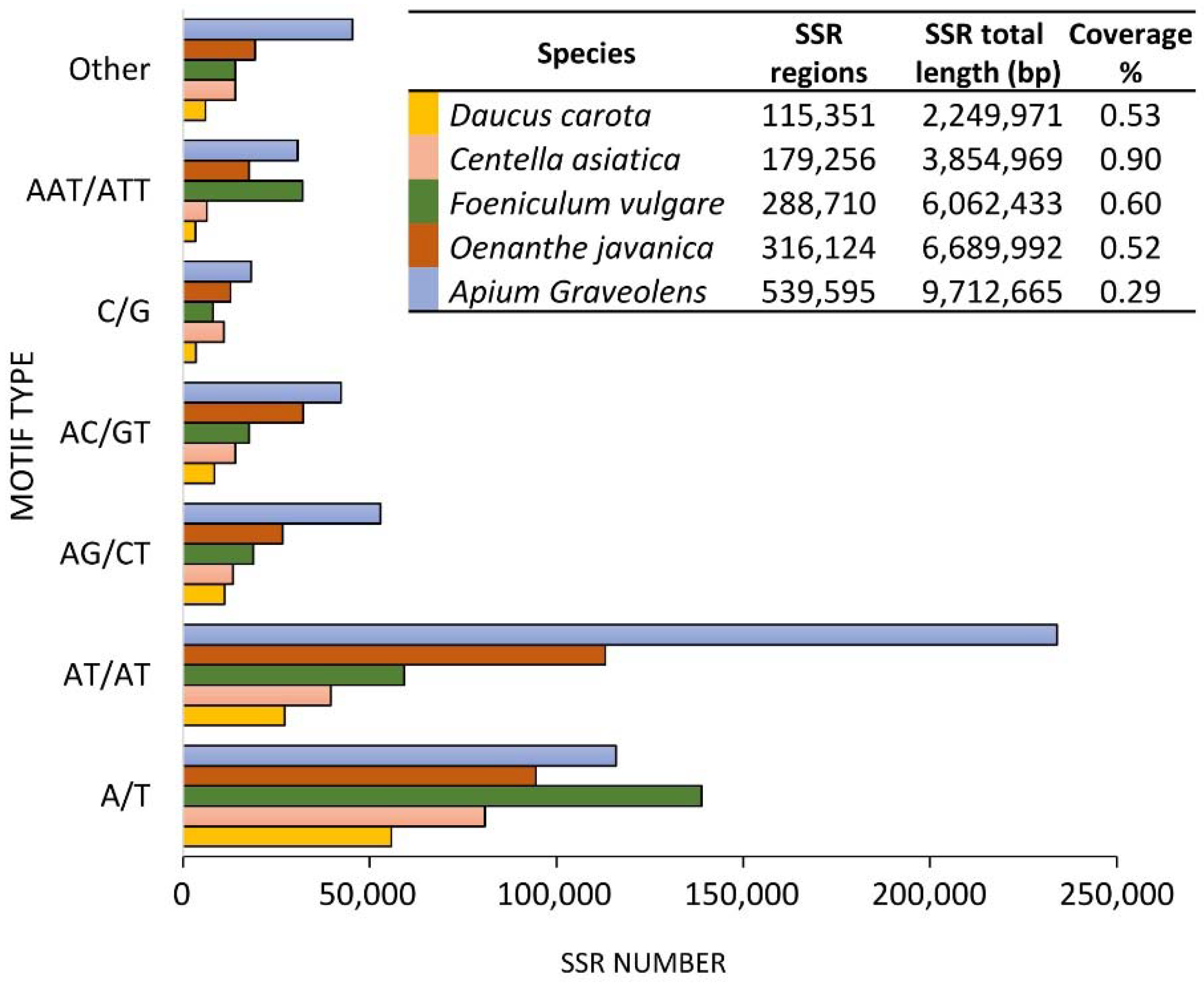
| Species | Methodology | SSR Identified | SSR Validated | Samples Tested | PIC | Ref |
|---|---|---|---|---|---|---|
| Anethum graveolens | SSR transfer from D. carota | 30 gSSR | 15 gSSR | 5 | n.s. | [80] |
| Anethum sowa | gDNA-seq | 48,951 gSSR | 10 gSSR | n.s. | n.s. | [94] |
| Angelica biserrata | RNA-seq | 8371 EST_SSR | 17 EST-SSR | 208 | 0.44–0.83 | [90] |
| Angelica dahurica | RNA-seq | 33,724 EST-SSR | 10 EST-SSR | 56 | 0.27–0.63 | [89] |
| Angelica gigas | gDNA-seq | 138,113 gSSR | 36 gSSR | 16 | 0.44–0.89 | [96] |
| Apium graveolens | RNA-seq | 80 EST-SSR | 28 EST-SSR | 31 | 0.06–0.72 | [91] |
| Arracacia xanthorrhiza | biotinylated SSR primer | 26 gSSR | 14 gSSR | 58 | 0.00–0.65 | [82] |
| Bupleurum chinense | I-SSR | 100 gSSR | 19 gSSR | 22 | 0.20–0.92 | [83] |
| Bupleurum falcatum | gDNA-seq | 91,377 EST-SSR | 21 gSSR | n.s. | n.s. | [97] |
| Centella asiatica | data mining from EST-db | 686 EST-SSR | 18 EST-SSR | n.s. | n.s. | [85] |
| Coriandrum sativum | RNA-seq | 9746 EST-SSR | 76 EST-SSR | 14 | 0.00–0.79 | [88] |
| Cuminum cyminum | gDNA-seq | 8086 gSSR | 23 gSSR | 30 | 0.03–0.70 | [95] |
| Daucus carota | hybridization-based library | n.s. | 196 gSSR | n.s. | n.s. | [81] |
| Foeniculum vulgare | gDNA-seq | 103,306 gSSR | 27 SSR | 100 | 0.03–0.92 | [46] |
| Heracleum spp. | ddRAD-seq | 54 gSSR | 19 gSSR | 48 | n.s. | [84] |
| Notopterygium incisum | RNA-seq | 13,149 EST-SSR | 19 EST-SSR | 24 | 0.53–0.83 | [86] |
| Notopterygium oviforme | gDNA-seq | 793 | 17 gSSR | 94 | 0.37–0.64 | [92] |
| Oenanthe javanica | RNA-seq | 1233 EST-SSR | n.s. | n.s. | n.s. | [87] |
| Pimpinella anisum | SSR transfer from D. carota | 30 gSSR | 16 gSSR | 5 | n.s. | [80] |
| Scaligeria lazica | gDNA-seq | 1982 | 40 gSSR | 40 | 0.37–0.84 | [93] |
| Common Name | Scientific Name | Chr Number | Genome Size (Mbp) | Assembly Level | Sequencing Platforms | Ref | |
|---|---|---|---|---|---|---|---|
| Estimated | Assembled | ||||||
| Celery | Apium graveolens | 2n = 22 | n.s. | 3332 | Chr | Illumina Hiseq4000; PacBio Seq I; Hi-C; 10x Genomics | [98] |
| Carrot | Daucus carota | 2n = 18 | 473 | 421 | Chr | Illumina Hiseq2000, Sanger (BAC libraries), 454 GS FLX | [76] |
| Asiatic pennywort | Centella asiatica | 2n = 18 | 430 | 430 | Chr | 10x Genomics, Hi-C, Illumina Hiseq X | [99] |
| Coriander | Coriandrum sativum | 2n = 22 | 2130 | 2119 | Chr | Illumina Miseq, PacBio Seq I, 10x Genomics, Hi-C | [100] |
| Korean angelica | Angelica gigas | 2n = 22 | 2670 | 804 | Scaff | Illumina Hiseq2500 | [96] |
| Java waterdropwort | Oenanthe javanica | 2n = 22 | n.s. | 1278 | Scaff | Illumina Hiseq2500 | [101] |
| Sickle hare’s-ear | Bupleurum falcatum | 2n = 16 | 2120 | 922 | Scaff | Illumina Hiseq2000 | [97] |
| Fennel | Foeniculum vulgare | 2n = 22 | 1320 | 1010 | Scaff | Illumina Hiseq2500 | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, F.; Vannozzi, A.; Barcaccia, G. Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies. Int. J. Mol. Sci. 2021, 22, 9713. https://doi.org/10.3390/ijms22189713
Palumbo F, Vannozzi A, Barcaccia G. Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies. International Journal of Molecular Sciences. 2021; 22(18):9713. https://doi.org/10.3390/ijms22189713
Chicago/Turabian StylePalumbo, Fabio, Alessandro Vannozzi, and Gianni Barcaccia. 2021. "Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies" International Journal of Molecular Sciences 22, no. 18: 9713. https://doi.org/10.3390/ijms22189713
APA StylePalumbo, F., Vannozzi, A., & Barcaccia, G. (2021). Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies. International Journal of Molecular Sciences, 22(18), 9713. https://doi.org/10.3390/ijms22189713