
The Central PXXP Motif Is Crucial for PMAP-23 Translocation across the Lipid Bilayer

Abstract
:1. Introduction
2. Results and Discussion
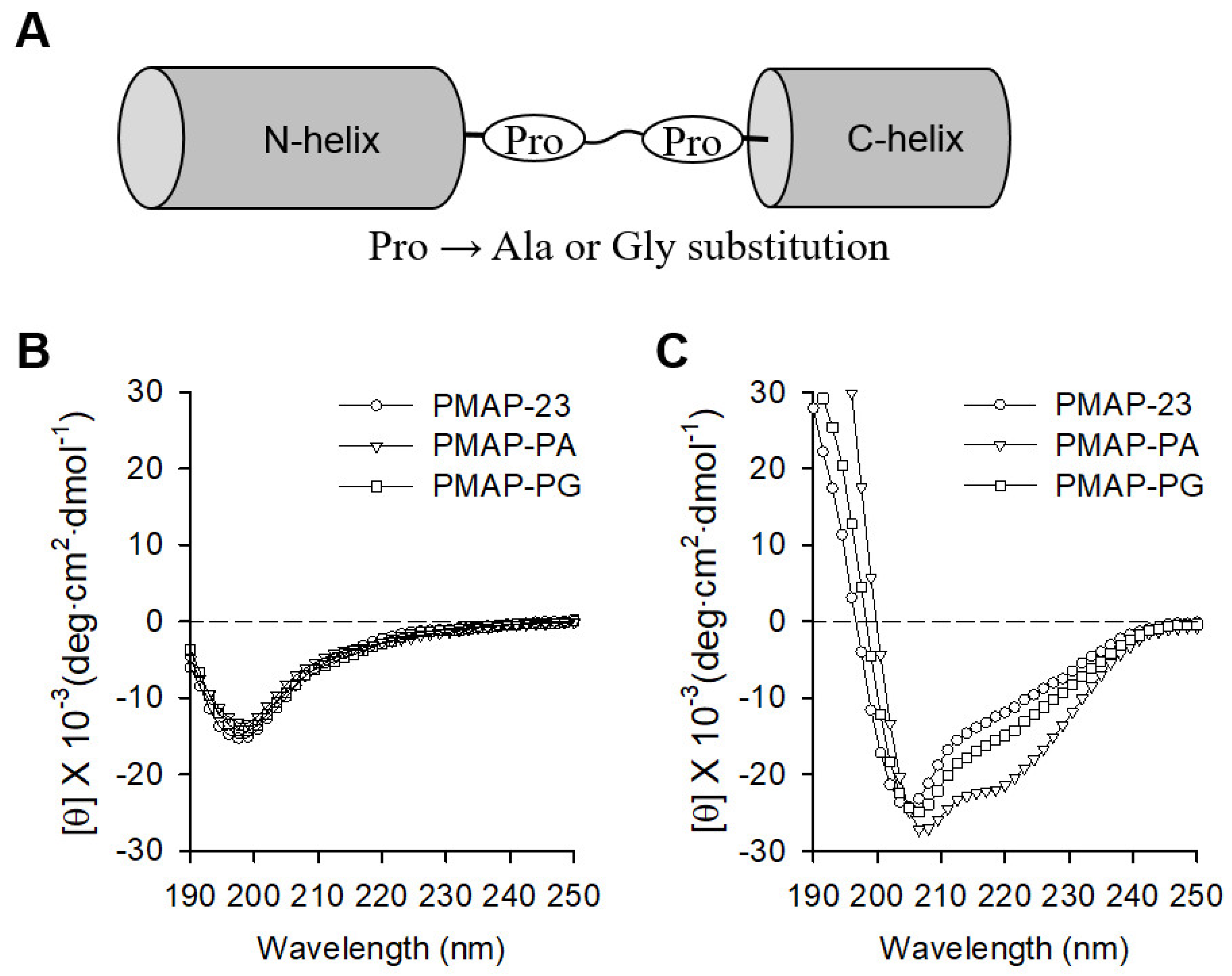
2.1. Peptide Design and Secondary Structure Analysis
2.2. Antimicrobial and Hemolytic Activities
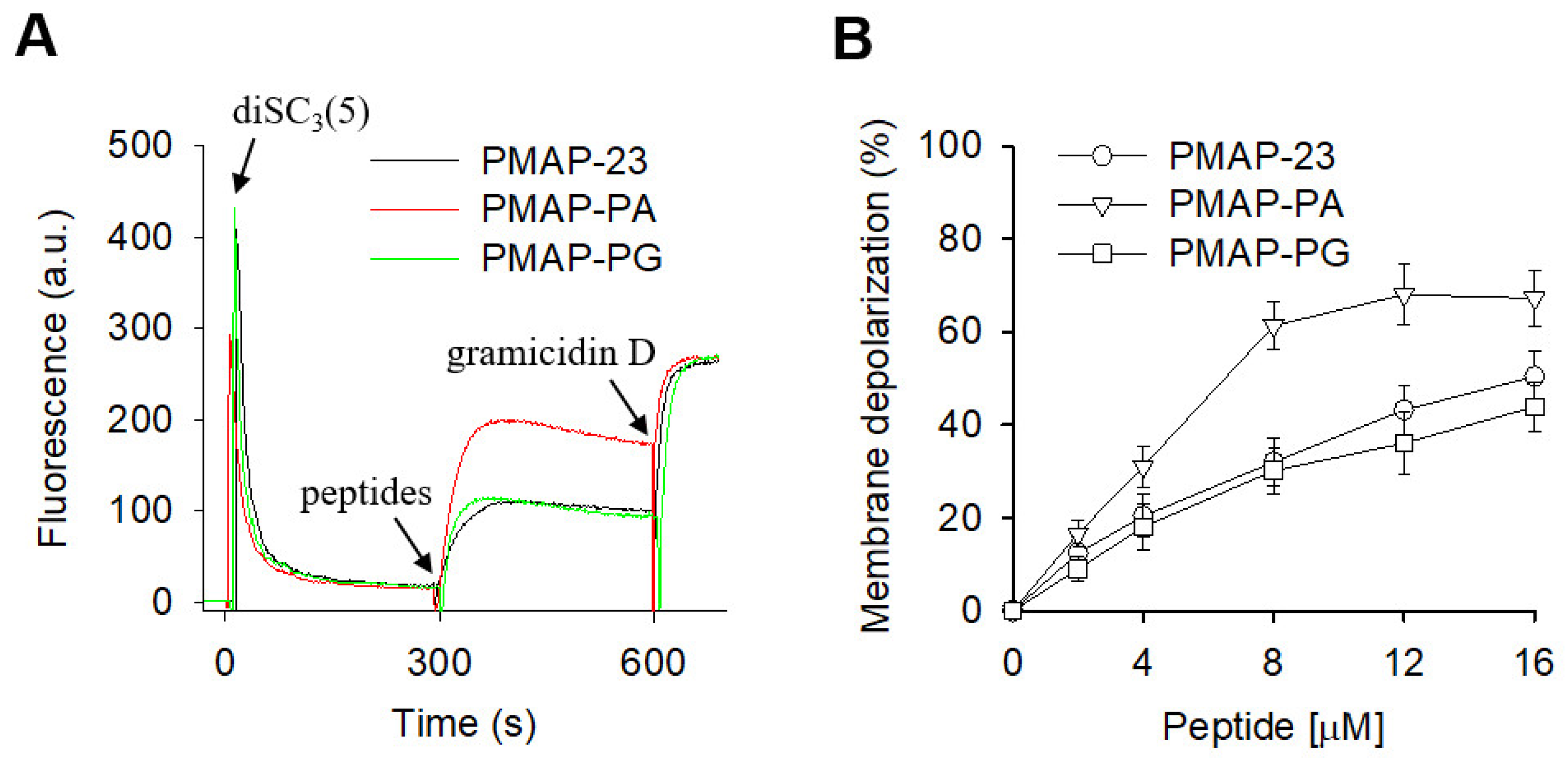
2.3. Membrane Depolarization by PMAP-23 and Its Analogs
2.4. Membrane Integration of Peptides by Trp Fluorescence Analysis
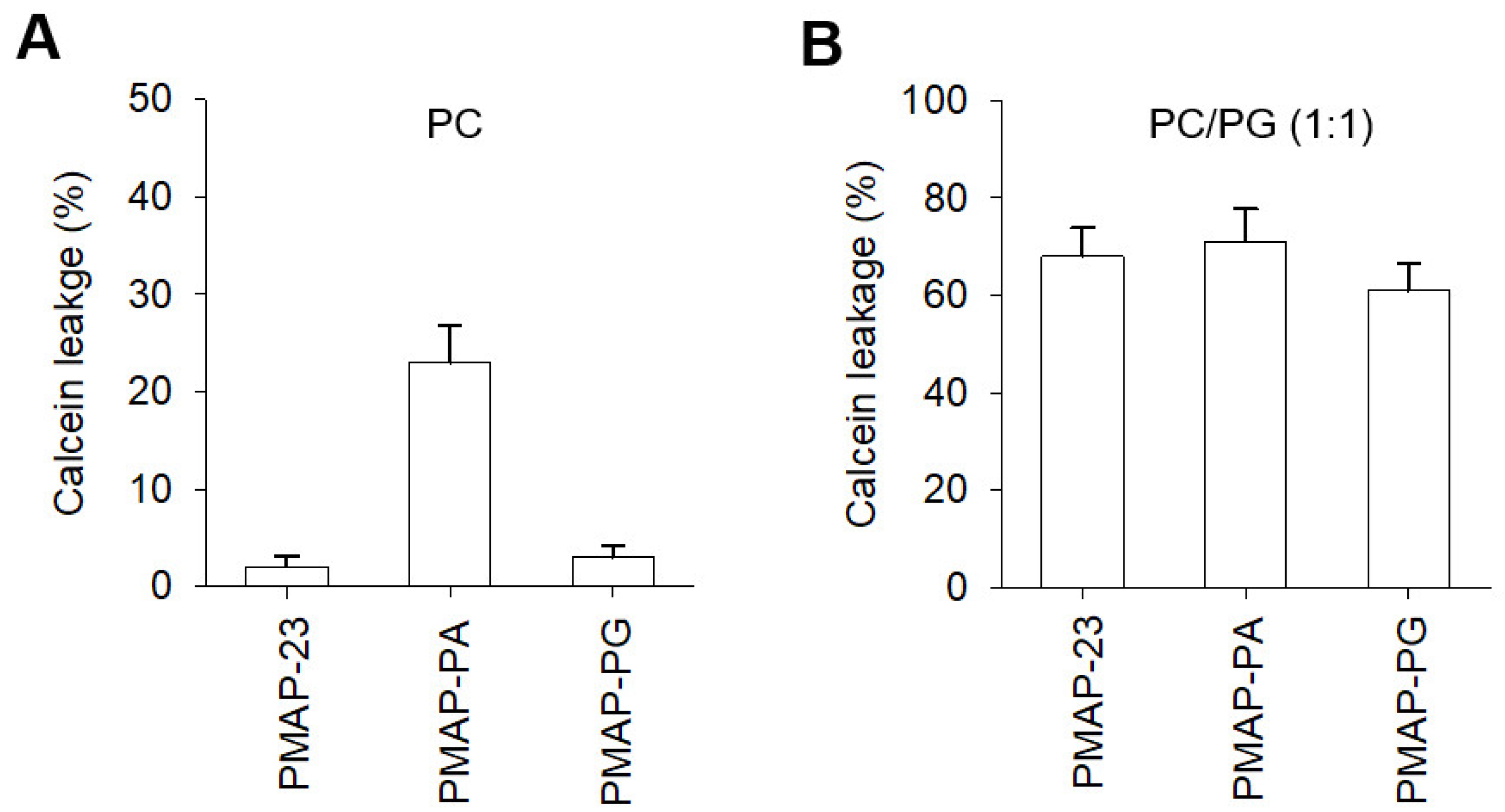
2.5. Membrane Disruption by PMAP-23 and Its Analogs
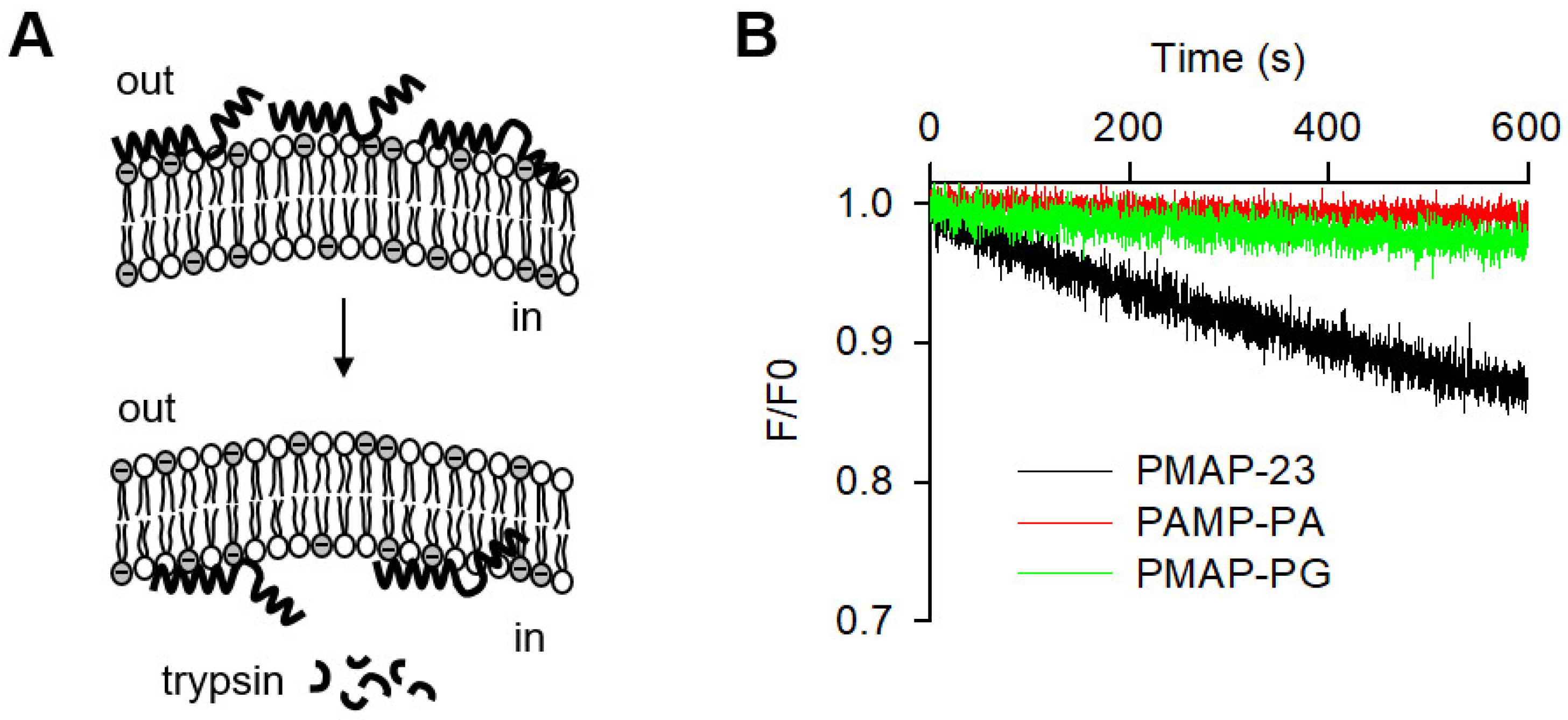
2.6. Peptide Translocation across Lipid Bilayers
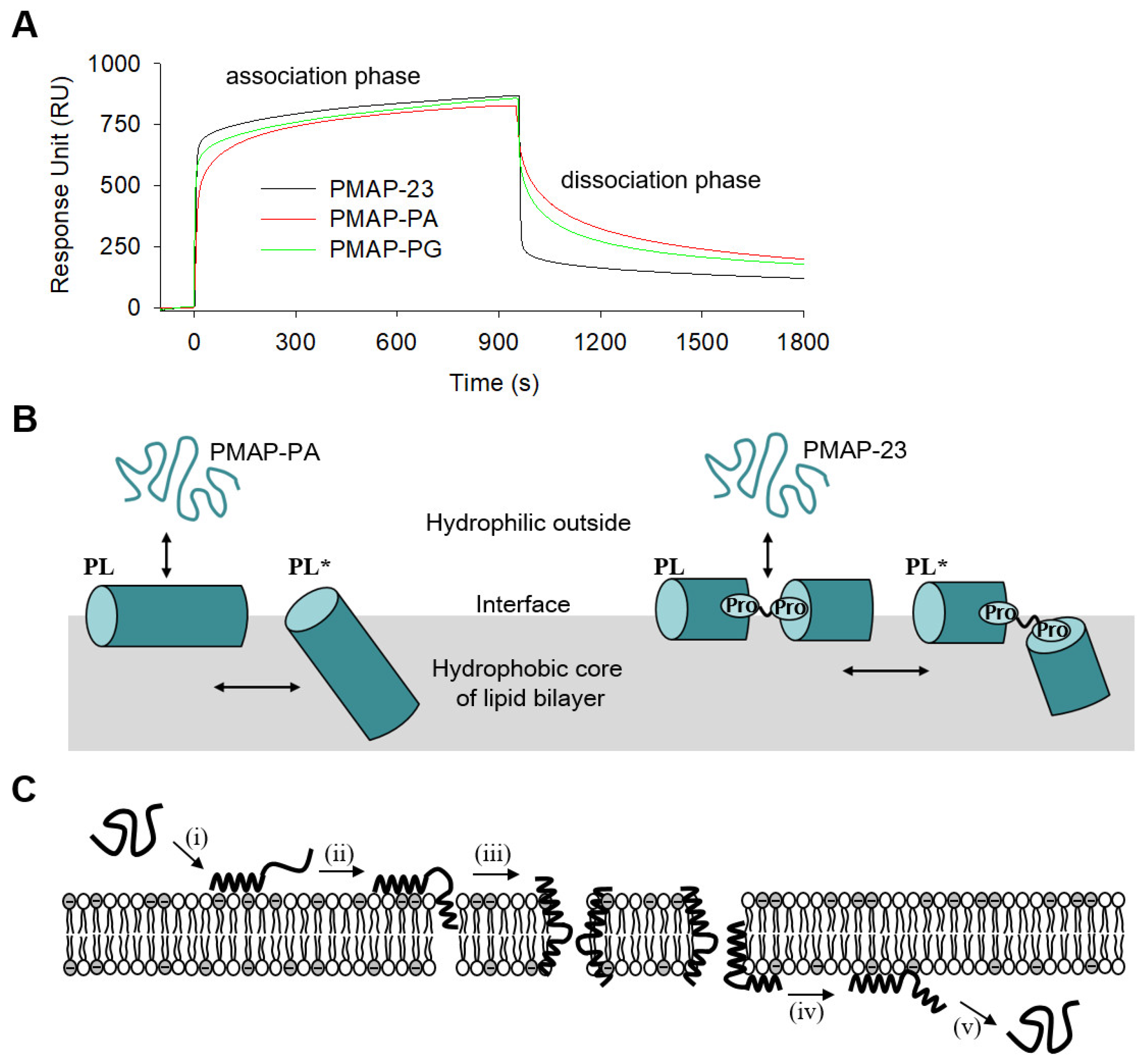
2.7. Surface Plasmon Resonance (SPR) Analysis
2.8. Model Elucidating the Translocation Mechanism of PMAP23
3. Materials and Methods
3.1. Chemicals, Peptides and Microorganisms
3.2. Circular Dichroism (CD) Spectroscopy
3.3. Antimicrobial Activity
3.4. Cytotoxic and Hemolytic Activity
3.5. Membrane Depolarization
3.6. Preparation of Vesicles
3.7. Calcein Leakage from LUVs
3.8. Tryptophan (Trp) Fluorescence
3.9. Peptide Translocation
3.10. Kinetic Analysis of Molecular Interaction Using Surface Plasmon Resonance (SPR)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Lohner, K. Membrane-active Antimicrobial Peptides as Template Structures for Novel Antibiotic Agents. Curr. Top. Med. Chem. 2017, 17, 508–519. [Google Scholar] [CrossRef]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial Peptides and Nanotechnology, Recent Advances and Challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef] [Green Version]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, K.; Chakraborty, S.; Chen, R.X.; Willcox, M.D.P.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- Stone, T.A.; Cole, G.B.; Ravamehr-Lake, D.; Nguyen, H.Q.; Khan, F.; Sharpe, S.; Deber, C.M. Positive Charge Patterning and Hydrophobicity of Membrane-Active Antimicrobial Peptides as Determinants of Activity, Toxicity, and Pharmacokinetic Stability. J. Med. Chem. 2019, 62, 6276–6286. [Google Scholar] [CrossRef]
- Guha, S.; Ghimire, J.; Wu, E.; Wimley, W.C. Mechanistic Landscape of Membrane-Permeabilizing Peptides. Chem Rev. 2019, 119, 6040–6085. [Google Scholar] [CrossRef]
- Sani, M.A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef]
- Matsuzaki, K. Membrane Permeabilization Mechanisms. Adv. Exp. Med. Biol. 2019, 1117, 9–16. [Google Scholar]
- Rounds, T.; Straus, S.K. Lipidation of Antimicrobial Peptides as a Design Strategy for Future Alternatives to Antibiotics. Int. J. Mol. Sci. 2020, 21, 9692. [Google Scholar] [CrossRef]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Lda, S.D.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.T.; Jeon, J.H.; Kim, Y.; Shin, S.Y.; Hahm, K.S.; Kim, J.I. Possible role of a PXXP central hinge in the antibacterial activity and membrane interaction of PMAP-23, a member of cathelicidin family. Biochemistry 2006, 45, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Lim, S.I.; Shin, S.H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [Green Version]
- Marin-Medina, N.; Mescola, A.; Alessandrini, A. Effects of the peptide Magainin H2 on Supported Lipid Bilayers studied by different biophysical techniques. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2635–2643. [Google Scholar] [CrossRef]
- Lorenzon, E.N.; Nobre, T.M.; Caseli, L.; Cilli, E.M.; da Hora, G.C.A.; Soares, T.A.; Oliveira, O.N., Jr. The “pre-assembled state” of magainin 2 lysine-linked dimer determines its enhanced antimicrobial activity. Colloids Surf. B Biointerfaces 2018, 167, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Mescola, A.; Marin-Medina, N.; Ragazzini, G.; Accolla, M.; Alessandrini, A. Magainin-H2 effects on the permeabilization and mechanical properties of giant unilamellar vesicles. J. Colloid Interface Sci. 2019, 553, 247–258. [Google Scholar] [CrossRef]
- Huang, H.W. DAPTOMYCIN, its membrane-active mechanism vs. that of other antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183395. [Google Scholar] [CrossRef]
- Huang, H.W. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim. Biophys. Acta 2006, 1758, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Howe, A.; Sofou, S. Daptomycin-Induced Lipid Phases on Model Lipid Bilayers: Effect of Lipid Type and of Lipid Leaflet Order on Membrane Permeability. J. Phys. Chem. B 2021, 125, 5775–5785. [Google Scholar] [CrossRef]
- Veldhuizen, E.J.A.; Scheenstra, M.R.; Bokhoven, J.L.M.T.; Coorens, M.; Schneider, V.A.F.; Bikker, F.J.; van Dijk, A.; Haagsman, H.P. Antimicrobial and Immunomodulatory Activity of PMAP-23 Derived Peptides. Protein Pept. Lett. 2017, 24, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shen, T.; Chen, L.; Zhou, J.; Wang, C. Analogs of the Cathelicidin-Derived Antimicrobial Peptide PMAP-23 Exhibit Improved Stability and Antibacterial Activity. Probiot. Antimicrob. Proteins 2021, 13, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Oh, D.; Shin, S.Y.; Hahm, K.S.; Kim, Y. Structural studies of porcine myeloid antibacterial peptide PMAP-23 and its analogues in DPC micelles by NMR spectroscopy. Biochem. Biophys. Res. Commun. 2002, 290, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.; Demoulins, T.; Python, S.; Summerfield, A. Porcine cathelicidins efficiently complex and deliver nucleic acids to plasmacytoid dendritic cells and can thereby mediate bacteria-induced IFN-alpha responses. J. Immunol. 2014, 193, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lee, C.W.; Kim, H.J.; Jung, H.H.; Kim, J.I.; Shin, S.Y.; Shin, S.H. Structural analysis and mode of action of BMAP-27, a cathelicidin-derived antimicrobial peptide. Peptides 2019, 118, 170106. [Google Scholar] [CrossRef]
- Yang, S.T.; Kim, J.I.; Shin, S.Y. Effect of dimerization of a beta-turn antimicrobial peptide, PST13-RK, on antimicrobial activity and mammalian cell toxicity. Biotechnol. Lett. 2009, 31, 233–237. [Google Scholar] [CrossRef]
- Yang, S.T.; Lee, J.Y.; Kim, H.J.; Eu, Y.J.; Shin, S.Y.; Hahm, K.S.; Kim, J.I. Contribution of a central proline in model amphipathic alpha-helical peptides to self-association, interaction with phospholipids, and antimicrobial mode of action. FEBS J. 2006, 273, 4040–4054. [Google Scholar] [CrossRef]
- Yang, S.T.; Shin, S.Y.; Hahm, K.S.; Kim, J.I. Different modes in antibiotic action of tritrpticin analogs, cathelicidin-derived Trp-rich and Pro/Arg-rich peptides. Biochim. Biophys. Acta 2006, 1758, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Takeshima, K.; Park, C.B.; Kim, S.C.; Matsuzaki, K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry 2000, 39, 8648–8654. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yang, S. Dimerization of cell-penetrating buforin II enhances antimicrobial properties. J. Anal. Sci. Technol. 2021, 12, 9. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptides | Amino Acid Sequences | Helicity a | MIC (μM) | Cytotoxicity b | Hemolysis c | |
|---|---|---|---|---|---|---|
| S. aureus | E. coli | |||||
| PMAP-23 | RIIDLLWRVRRPQKPKFVTVWVR | 27% | 2–4 | 2–4 | 0% | 0% |
| PMAP-PA | RIIDLLWRVRRAQKAKFVTVWVR | 63% | 8–16 | 8–16 | 37% | 14% |
| PMAP-PG | RIIDLLWRVRRGQKGKFVTVWVR | 31% | 8 | 8–16 | 3% | 0% |
| Peptide | PC | PC/PG (1:1) | ||
|---|---|---|---|---|
| λmax | KSV | λmax | KSV | |
| PMAP-23 | 352 nm (0 nm) a | 15.26 | 343 nm (9 nm) | 7.55 |
| PMAP-PA | 347 nm (5 nm) | 11.73 | 343 nm (9 nm) | 7.17 |
| PMAP-PG | 351 nm (1 nm) | 15.12 | 344 nm (8 nm) | 7.42 |
| Peptide | First Step | Second Step | ||
|---|---|---|---|---|
| ka1 (1/Ms) | kd1 (1/s) | ka2 (1/s) | kd2 (1/s) | |
| PMAP-23 | 164 | 8.7 × 10−3 | 82.3 × 10−3 | 732.5 × 10−3 |
| PMAP-PA | 375 | 9.0 × 10−3 | 2.1 × 10−3 | 73.8 × 10−3 |
| PMAP-PG | 284 | 9.4 × 10−3 | 2.7 × 10−3 | 68.2 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-T.; Shin, S.-Y.; Shin, S.-H. The Central PXXP Motif Is Crucial for PMAP-23 Translocation across the Lipid Bilayer. Int. J. Mol. Sci. 2021, 22, 9752. https://doi.org/10.3390/ijms22189752
Yang S-T, Shin S-Y, Shin S-H. The Central PXXP Motif Is Crucial for PMAP-23 Translocation across the Lipid Bilayer. International Journal of Molecular Sciences. 2021; 22(18):9752. https://doi.org/10.3390/ijms22189752
Chicago/Turabian StyleYang, Sung-Tae, Song-Yub Shin, and Sung-Heui Shin. 2021. "The Central PXXP Motif Is Crucial for PMAP-23 Translocation across the Lipid Bilayer" International Journal of Molecular Sciences 22, no. 18: 9752. https://doi.org/10.3390/ijms22189752