
Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Myoglobin: Intrinsic Fenton Reactivity Reflects Co-Ordination Mode of the Haem Group
2.2. ADH: Highly Reactive Sulphur-Containing Side Chains and Surface-Selective Labelling
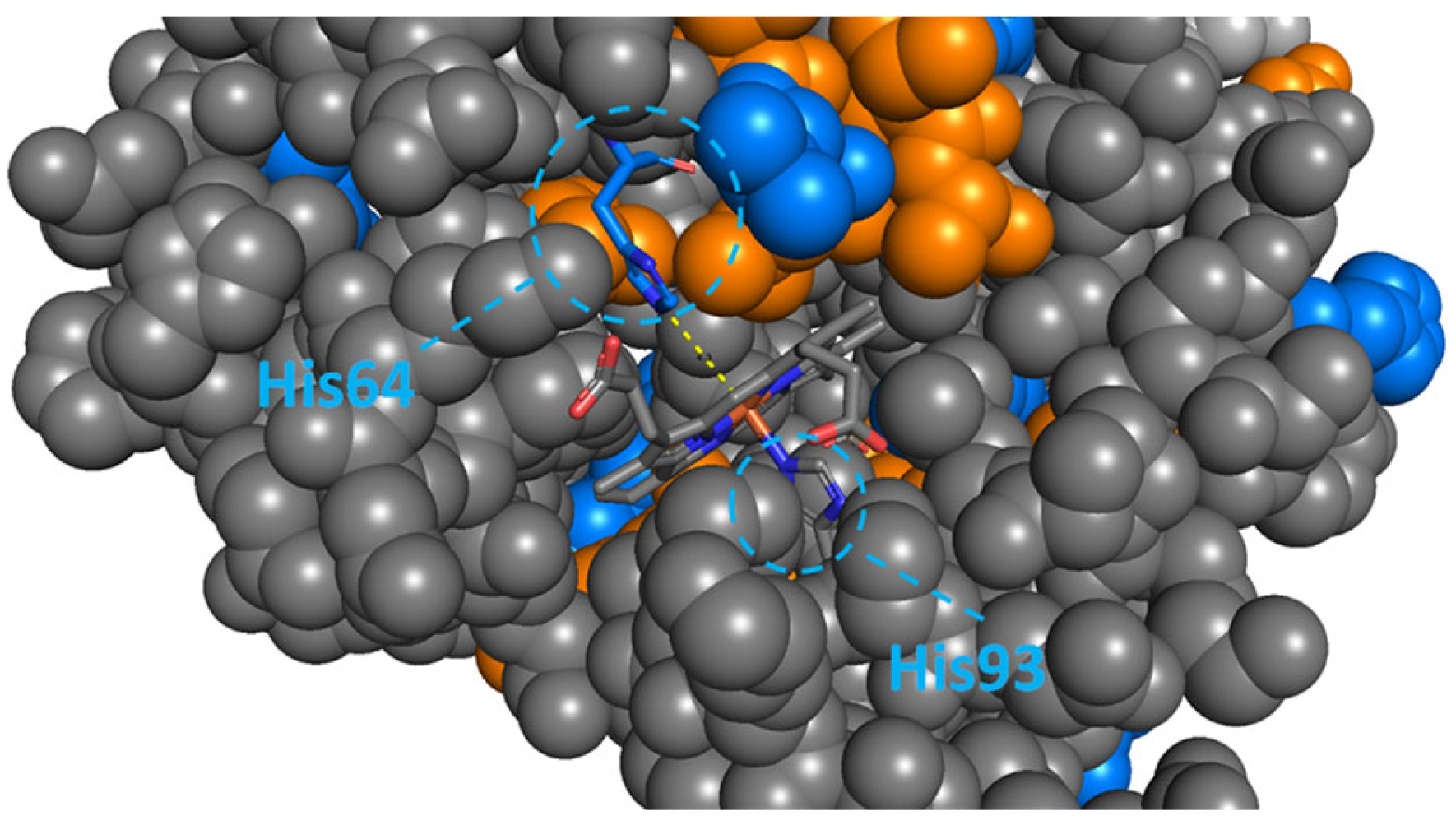
2.3. FKBP51 and FKBP12: Key Interactions Drive Remarkable Structural Stabilisation
3. Discussion
4. Materials and Methods
4.1. Proteins, Reagents and Solvents
4.2. Oxidative Footprinting
4.3. Tryptic Digest
4.4. LC-MS/MS Analysis
4.5. Data Analysis Using MaxQuant
4.6. Quantifying Peptides Using pepFoot
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konijnenberg, A.; Butterer, A.; Sobott, F. Native ion mobility-mass spectrometry and related methods in structural biology. Biochim. Biophys. Acta 2013, 1834, 1239–1256. [Google Scholar] [CrossRef]
- Politis, A.; Stengel, F.; Hall, Z.; Hernandez, H.; Leitner, A.; Walzthoeni, T.; Robinson, C.V.; Aebersold, R. A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat. Methods 2014, 11, 403–406. [Google Scholar] [CrossRef]
- Leney, A.C.; Heck, A.J. Native Mass Spectrometry: What is in the Name? J. Am. Soc. Mass Spectrom. 2017, 28, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.V. Mass spectrometry: From plasma proteins to mitochondrial membranes. Proc. Natl. Acad. Sci. USA 2019, 116, 2814–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, T.M.; Barran, P.; Cianferani, S.; Degiacomi, M.T.; Gabelica, V.; Grandori, R.; Marklund, E.G.; Menneteau, T.; Migas, L.G.; Politis, A.; et al. Computational Strategies and Challenges for Using Native Ion Mobility Mass Spectrometry in Biophysics and Structural Biology. Anal. Chem. 2020, 92, 10872–10880. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Lantz, C.; Brown, K.A.; Ge, Y.; Pasa Tolic, L.; Loo, J.A.; Lermyte, F. Higher-order structural characterisation of native proteins and complexes by top-down mass spectrometry. Chem. Sci. 2020, 11, 12918–12936. [Google Scholar] [CrossRef]
- Groves, K.; Ashcroft, A.E.; Cryar, A.; Sula, A.; Wallace, B.A.; Stocks, B.B.; Burns, C.; Cooper-Shepherd, D.; De Lorenzi, E.; Rodriguez, E.; et al. Reference Protocol to Assess Analytical Performance of Higher Order Structural Analysis Measurements: Results from an Interlaboratory Comparison. Anal. Chem. 2021, 93, 9041–9048. [Google Scholar] [CrossRef]
- Wang, L.; Chance, M.R. Protein Footprinting Comes of Age: Mass Spectrometry for Biophysical Structure Assessment. Mol. Cell Proteom. 2017, 16, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, U.; Johnson, D.T.; Chea, E.E.; Deredge, D.J.; Espino, J.A.; Jones, L.M. Evolution of Structural Biology through the Lens of Mass Spectrometry. Anal. Chem. 2019, 91, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.R.; Zhang, M.M.; Gross, M.L. Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev. 2020, 120, 4355–4454. [Google Scholar] [CrossRef]
- Pan, Y.; Konermann, L. Membrane protein structural insights from chemical labeling and mass spectrometry. Analyst 2010, 135, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Konermann, L.; Pan, Y.; Stocks, B.B. Protein folding mechanisms studied by pulsed oxidative labeling and mass spectrometry. Curr. Opin. Struct. Biol. 2011, 21, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Khanal, A.; Pan, Y.; Brown, L.S.; Konermann, L. Pulsed hydrogen/deuterium exchange mass spectrometry for time-resolved membrane protein folding studies. J. Mass Spectrom. 2012, 47, 1620–1626. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Piyadasa, H.; O’Neil, J.D.; Konermann, L. Conformational dynamics of a membrane transport protein probed by H/D exchange and covalent labeling: The glycerol facilitator. J. Mol. Biol. 2012, 416, 400–413. [Google Scholar] [CrossRef]
- Pan, Y.; Brown, L.; Konermann, L. Hydrogen exchange mass spectrometry of bacteriorhodopsin reveals light-induced changes in the structural dynamics of a biomolecular machine. J. Am. Chem. Soc. 2011, 133, 20237–20244. [Google Scholar] [CrossRef]
- Benesch, J.L.; Ruotolo, B.T. Mass spectrometry: Come of age for structural and dynamical biology. Curr. Opin. Struct. Biol. 2011, 21, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Karch, K.R.; Coradin, M.; Zandarashvili, L.; Kan, Z.Y.; Gerace, M.; Englander, S.W.; Black, B.E.; Garcia, B.A. Hydrogen-Deuterium Exchange Coupled to Top- and Middle-Down Mass Spectrometry Reveals Histone Tail Dynamics before and after Nucleosome Assembly. Structure 2018, 26, 1651–1663.e1653. [Google Scholar] [CrossRef] [Green Version]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Strutzenberg, T.; Pascal, B.D.; Griffin, P.R. Protein dynamics and conformational changes explored by hydrogen/deuterium exchange mass spectrometry. Curr. Opin. Struct. Biol. 2019, 58, 305–313. [Google Scholar] [CrossRef]
- Zehl, M.; Rand, K.D.; Jensen, O.N.; Jorgensen, T.J. Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. J. Am. Chem. Soc. 2008, 130, 17453–17459. [Google Scholar] [CrossRef]
- Wang, Q.; Borotto, N.B.; Hakansson, K. Gas-Phase Hydrogen/Deuterium Scrambling in Negative-Ion Mode Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2019, 30, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Manzi, L.; Barrow, A.S.; Scott, D.; Layfield, R.; Wright, T.G.; Moses, J.E.; Oldham, N.J. Carbene footprinting accurately maps binding sites in protein-ligand and protein-protein interactions. Nat. Commun. 2016, 7, 13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzi, L.; Barrow, A.S.; Hopper, J.T.S.; Kaminska, R.; Kleanthous, C.; Robinson, C.V.; Moses, J.E.; Oldham, N.J. Carbene Footprinting Reveals Binding Interfaces of a Multimeric Membrane-Spanning Protein. Angew. Chem. Int. Ed. Engl. 2017, 56, 14873–14877. [Google Scholar] [CrossRef]
- Barth, M.; Bender, J.; Kundlacz, T.; Schmidt, C. Evaluation of NHS-Acetate and DEPC labelling for determination of solvent accessible amino acid residues in protein complexes. J. Proteom. 2020, 222, 103793. [Google Scholar] [CrossRef]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambly, D.; Gross, M.L. Laser flash photochemical oxidation to locate heme binding and conformational changes in myoglobin. Int. J. Mass Spectrom. 2007, 259, 124–129. [Google Scholar] [CrossRef]
- Li, K.S.; Shi, L.; Gross, M.L. Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization. Acc. Chem. Res. 2018, 51, 736–744. [Google Scholar] [CrossRef]
- Vahidi, S.; Konermann, L. Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds. J. Am. Soc. Mass Spectrom. 2016, 27, 1156–1164. [Google Scholar] [CrossRef]
- Maleknia, S.D.; Brenowitz, M.; Chance, M.R. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal. Chem. 1999, 71, 3965–3973. [Google Scholar] [CrossRef]
- Xu, G.; Chance, M.R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef] [PubMed]
- Tullius, T.D.; Dombroski, B.A. Hydroxyl radical “footprinting”: High-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein. Proc. Natl. Acad. Sci. USA 1986, 83, 5469–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.S.; Tullius, T.D. Footprinting protein-DNA complexes using the hydroxyl radical. Nat. Protoc. 2008, 3, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Guo, T.; Park, J.E.; Li, X.; Meng, W.; Datta, A.; Bern, M.; Lim, S.K.; Sze, S.K. Elucidating in vivo structural dynamics in integral membrane protein by hydroxyl radical footprinting. Mol. Cell Proteom. 2009, 8, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leser, M.; Chapman, J.R.; Khine, M.; Pegan, J.; Law, M.; Makkaoui, M.E.; Ueberheide, B.M.; Brenowitz, M. Chemical Generation of Hydroxyl Radical for Oxidative ‘Footprinting’. Protein Pept. Lett. 2019, 26, 61–69. [Google Scholar] [CrossRef]
- Johnson, D.T.; Di Stefano, L.H.; Jones, L.M. Fast photochemical oxidation of proteins (FPOP): A powerful mass spectrometry-based structural proteomics tool. J. Biol. Chem. 2019, 294, 11969–11979. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chance, M.R. Structural mass spectrometry of proteins using hydroxyl radical based protein footprinting. Anal. Chem. 2011, 83, 7234–7241. [Google Scholar] [CrossRef] [Green Version]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands-Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [Green Version]
- Hahle, A.; Merz, S.; Meyners, C.; Hausch, F. The Many Faces of FKBP51. Biomolecules 2019, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Gaali, S.; Kirschner, A.; Cuboni, S.; Hartmann, J.; Kozany, C.; Balsevich, G.; Namendorf, C.; Fernandez-Vizarra, P.; Sippel, C.; Zannas, A.S.; et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 2015, 11, 33–37. [Google Scholar] [CrossRef]
- Pomplun, S.; Sippel, C.; Hahle, A.; Tay, D.; Shima, K.; Klages, A.; Unal, C.M.; Riess, B.; Toh, H.T.; Hansen, G.; et al. Chemogenomic Profiling of Human and Microbial FK506-Binding Proteins. J. Med. Chem. 2018, 61, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Han, J.; Borchers, C.H.; Konermann, L. Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J. Am. Chem. Soc. 2009, 131, 12801–12808. [Google Scholar] [CrossRef]
- Lim, J.; Vachet, R.W. Development of a methodology based on metal-catalyzed oxidation reactions and mass spectrometry to determine the metal binding sites in copper metalloproteins. Anal. Chem. 2003, 75, 1164–1172. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Fraczkiewicz, R.; Braun, W. Exact and Efficient Analytical Calculation of the Accessible Surface Areas and Their Gradients for Macromolecules. J. Comp. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Jagtap, P.K.A.; Asami, S.; Sippel, C.; Kaila, V.R.I.; Hausch, F.; Sattler, M. Selective Inhibitors of FKBP51 Employ Conformational Selection of Dynamic Invisible States. Angew. Chem. Int. Ed. Engl. 2019, 58, 9429–9433. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.W.; Goncz, E.J.; Krist, D.T.; Statsyuk, A.V.; Nesvizhskii, A.I.; Weiss, E.L. Mapping low-affinity/high-specificity peptide-protein interactions using ligand-footprinting mass spectrometry. Proc. Natl. Acad. Sci. USA 2019, 116, 21001–21011. [Google Scholar] [CrossRef] [Green Version]
- Voll, A.M.; Meyners, C.; Taubert, M.C.; Bajaj, T.; Heymann, T.; Merz, S.; Charalampidou, A.; Kolos, J.; Purder, P.L.; Geiger, T.M.; et al. Macrocyclic FKBP51 Ligands Define a Transient Binding Mode with Enhanced Selectivity. Angew. Chem. Int. Ed. Engl. 2021, 60, 13257–13263. [Google Scholar] [CrossRef]
- Schopper, S.; Kahraman, A.; Leuenberger, P.; Feng, Y.; Piazza, I.; Muller, O.; Boersema, P.J.; Picotti, P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc. 2017, 12, 2391–2410. [Google Scholar] [CrossRef]
- Leuenberger, P.; Ganscha, S.; Kahraman, A.; Cappelletti, V.; Boersema, P.J.; von Mering, C.; Claassen, M.; Picotti, P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 2017, 355, eaai7825. [Google Scholar] [CrossRef]
- Wilson, K.P.; Yamashita, M.M.; Sintchak, M.D.; Rotstein, S.H.; Murcko, M.A.; Boger, J.; Thomson, J.A.; Fitzgibbon, M.J.; Black, J.R.; Navia, M.A. Comparative X-ray structures of the major binding protein for the immunosuppressant FK506 (tacrolimus) in unliganded form and in complex with FK506 and rapamycin. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Gaali, S.; Feng, X.; Hahle, A.; Sippel, C.; Bracher, A.; Hausch, F. Rapid, Structure-Based Exploration of Pipecolic Acid Amides as Novel Selective Antagonists of the FK506-Binding Protein 51. J. Med. Chem. 2016, 59, 2410–2422. [Google Scholar] [CrossRef]
- Bauder, M.; Meyners, C.; Purder, P.L.; Merz, S.; Sugiarto, W.O.; Voll, A.M.; Heymann, T.; Hausch, F. Structure-Based Design of High-Affinity Macrocyclic FKBP51 Inhibitors. J. Med. Chem. 2021, 64, 3320–3349. [Google Scholar] [CrossRef] [PubMed]
- Resetca, D.; Wilson, D.J. Characterizing rapid, activity-linked conformational transitions in proteins via sub-second hydrogen deuterium exchange mass spectrometry. FEBS J. 2013, 280, 5616–5625. [Google Scholar] [CrossRef]
- Deng, B.; Zhu, S.; Macklin, A.M.; Xu, J.; Lento, C.; Sljoka, A.; Wilson, D.J. Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. MAbs 2017, 9, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Bellamy-Carter, J.; Oldham, N.J. PepFoot: A Software Package for Semiautomated Processing of Protein Footprinting Data. J. Proteome Res. 2019, 18, 2925–2930. [Google Scholar] [CrossRef] [PubMed]
- Kozany, C.; Marz, A.; Kress, C.; Hausch, F. Fluorescent probes to characterise FK506-binding proteins. Chembiochem 2009, 10, 1402–1410. [Google Scholar] [CrossRef]
- Bracher, A.; Kozany, C.; Thost, A.K.; Hausch, F. Structural characterization of the PPIase domain of FKBP51, a cochaperone of human Hsp90. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.T.; Baldwin, A.J.; Marklund, E.G.; Hochberg, G.K.; Benesch, J.L.; Robinson, C.V. Bayesian deconvolution of mass and ion mobility spectra: From binary interactions to polydisperse ensembles. Anal. Chem. 2015, 87, 4370–4376. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nehls, T.; Heymann, T.; Meyners, C.; Hausch, F.; Lermyte, F. Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation. Int. J. Mol. Sci. 2021, 22, 9927. https://doi.org/10.3390/ijms22189927
Nehls T, Heymann T, Meyners C, Hausch F, Lermyte F. Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation. International Journal of Molecular Sciences. 2021; 22(18):9927. https://doi.org/10.3390/ijms22189927
Chicago/Turabian StyleNehls, Thomas, Tim Heymann, Christian Meyners, Felix Hausch, and Frederik Lermyte. 2021. "Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation" International Journal of Molecular Sciences 22, no. 18: 9927. https://doi.org/10.3390/ijms22189927
APA StyleNehls, T., Heymann, T., Meyners, C., Hausch, F., & Lermyte, F. (2021). Fenton-Chemistry-Based Oxidative Modification of Proteins Reflects Their Conformation. International Journal of Molecular Sciences, 22(18), 9927. https://doi.org/10.3390/ijms22189927